Introduction
Differentiation therapy using all-trans retinoic acid (ATRA) for acute promyelocytic leukemia (APL) was established in the 1990s and has dramatically improved the clinical outcome of APL. For non-APL, intensive chemotherapy with allogenic stem cell transplantation is a standard therapy, whereas the optimization of intensive chemotherapy regimens and the use of nonmyeloablative regimens are alternative choices for older patients. Several attempts have been made to improve antileukemic activity in these patients by employing hypomethylating agents (HMAs) with low-dose cytarabine (Ara-C) (LDAC), with encouraging results []. These agents mainly serve as differentiation inducers [, ].
In this review, I attempt to summarize recent findings about inducers of myeloid differentiation. These include glycosylation modifiers, epigenetic modifiers, micro (mi)RNAs, vitamin derivatives, cytokines, and chemotherapeutic agents. Of these, glycosylation and epigenetic modifiers are attracting particular attention because hematopoietic malignancies have tumor-specific glycosylation and epigenomic modifications that can be used as tumor targets. These inducers may enable the development of efficient differentiation therapy against non-APL, AML.
Glycosylation Modifiers
Cossu et al. [] previously reported that the synthesis of high-molecular-weight glycopeptides is a property of human myeloid leukemia cell lines, including HL-60 cells, and that the proportionate decrease in the synthesis of these large glycopeptides is part of monocytic differentiation. Several reports describe differences in cell surface glycosylation changes that occur during hematopoietic cell differentiation, suggesting that glycosylation modifications may play a role in differentiation [, ]. The change of cell surface glycosylation and its effect on myeloid differentiation, discussed in this section, is summarized in Figure 1.
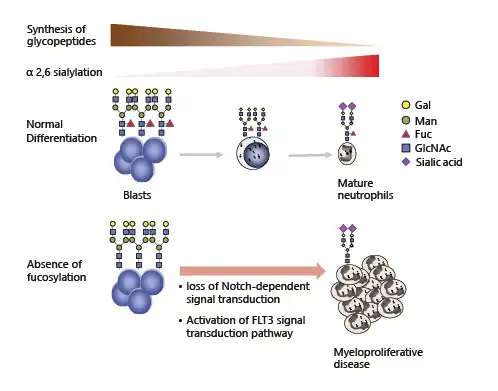
Fig. 1
The change of cell surface glycosylation and its effect on myeloid differentiation.
Statins were developed to lower cholesterol and triglyceride levels. They act by blocking 3-hydroxy-3-methylglutaryl coenzyme A reductase, which is a rate-limiting step in the mevalonate pathway []. This pathway produces dolichol, which is responsible for the co-translational transfer of oligosaccharides to nascent polypeptides that undergo N-linked glycosylation []. Interestingly, statins, atorvastatin, and fluvastatin were found to be potent inducers of cell differentiation and apoptosis of the APL cell line, NB4 []. This statin-dependent leukemic cell differentiation was found to require activation of the c-Jun NH2-terminal kinase pathway [] and protein kinase C δ pathways [].
The most widely occurring cancer-associated changes in protein glycosylation are increased sialylation, an increased number of branched-glycan structures, and the overexpression of core fucosylation []. A dramatic increase in cell surface alpha2,6 sialylation was previously reported during the late stage of myeloid maturation []. This is restricted to specific glycoproteins including CD11b and CD18. Changes in mature bone marrow myeloid cells are associated with reduced cell binding to fibronectin and cultured bone marrow stroma, which may play a role in the myeloid cell release from the bone marrow.
Notch-dependent control of myelopoiesis has been shown to be regulated by fucosylation []. FX is a GDP-4-keto-6-deoxy-mannose-3, 5-epimerase-4-reductase, which is a rate-limiting enzyme for GDP-fucose synthesis. FX knockout mice have a myeloproliferative phenotype and are conditionally deficient in cellular fucosylation through the loss of Notch-dependent signal transduction, which suppresses myeloid differentiation, in myeloid progenitor cells. FLT3 is a type III receptor tyrosine kinase that plays a pivotal role in hematopoietic cell proliferation and differentiation []. My group recently discovered that the deletion of core fucosylation by fucosyltransferase 8 (FUT8) knock out in the wild-type FLT3 receptor resulted in the potent activation of its downstream pathway []. FUT8 KO led to dimerization of the FLT3 receptor in the absence of FLT3 ligand, leading to the factor-independent growth of Ba/F3 cells, a mouse interleukin-3-dependent hematopoietic cell line. Because the activation of FLT3, by internal tandem duplication mutation-induced myeloproliferative disease in a transgenic mouse model [], we discovered that the deficiency of fucosylation, which activates FLT3, has the potential to function in myeloid cell proliferation. We further revealed that the combination of fucosylation inhibitor 2-fluorofucose, with FLT3 inhibitor PKC412, efficiently suppresses the growth of FLT3 expressed Ba/F3 cells []. To date, several fucosylation inhibitors have been developed [-], and there are many good examples of combination therapy with FLT3 inhibitors for AML [-]. Therefore, the modulation of FLT3 glycosylation, combined with FLT3 inhibition, could provide ideas for the development of new therapies for FLT3-mediated leukemias.
Modification of granulocyte colony-stimulating factor (G-CSF) receptor at W318 by C-mannosylation during myeloid differentiation [] is required for myeloid cell differentiation through activation of the Janus kinase-signal transducer and activator of transcription (STAT) pathway. Previously, the O-GlcNAcylation of STAT5 was found to control tyrosine phosphorylation and oncogenic transcription in STAT5-dependent malignancies [], while O-GlcNAcylation and tyrosine phosphorylation function together to trigger pYSTAT5 levels and oncogenic transcription in neoplastic cells.
Although the modification of hematopoietic progenitor cell surface glycosylation plays an important role in myeloid cell differentiation, the development of glycosylation-targeted therapy has been limited, possibly because of the complexity of the glycosylation process. However, glycosylation targeting may have the potential to start a new era for leukemia therapy.
Epigenetic Modifiers
Timely myeloid-specific gene expression is required for myeloid cell differentiation, and the lineage-specific transcription factor PU.1 and CCAAT/enhancer-binding protein alpha (C/EBPα) play a pivotal role in this together with DNA methyltransferase (DNMT) and histone deacetylases (HDAC) [, ]. Heterozygous somatic mutations in genes encoding epigenetic regulators have been found in all subtypes of myeloid malignancies [], while epigenetic modifiers such as HDAC inhibitors and HMAs such as 5-aza-2′-deoxycytidine (decitabine) were shown to induce growth arrest, cell death, and the terminal differentiation of myeloid cells [, ]. Already approved related therapeutics include HMAs (azacitidine and decitabine) and isocitrate dehydrogenase (IDH1 or IDH2) inhibitors (ivosidenib and enasidenib) []. Kantarijian et al. [] have reported the efficacy of decitabine, compared to supportive care or LDAC, for the treatment of older (≥65 years) patients with newly diagnosed AML and poor- or intermediate-risk cytogenetics. Since then, the use of HMAs represents an effective alternative for AML patients who are unfit for intensive chemotherapy. In the AML001 study, 488 patients aged 65 years and above with newly diagnosed AML were randomly assigned to receive azacitidine or conventional care regimens (including LDAC, intensive chemotherapy, or best supportive care) and resulted in improved median overall survival of 10.4 months with azacitidine versus 6.5 months for conventional care regimen [].
The mechanisms of the action of HMAs and HDAC inhibitors are still in debate; however, it was recently reported that the combination of these drugs in AML cell lines results in the massive downregulation of genes coding oncogenes (e.g., MYC) and epigenetic modifiers []. These genes are often overexpressed in cancer, and this downregulation was associated predominantly with gene body DNA demethylation and changes in chromatin marks acH3K9/27 []. Recently, another mechanism of action of decitabine, related to nucleophosmin (NPM) 1, was reported from Gu et al. []. NPM1 is one of the most frequently mutated genes in de novo AML (∼30% of cases) []. Mutant NPM1 represses monocyte and granulocyte terminal differentiation by disrupting myeloid master transcriptional regulators such as PU.1/CEBPα/RUNX1 collaboration in nucleus, leading to their accumulation in cytoplasm, resulting in the aberrant proliferation and block of differentiation []. Mutant NPM1 transforming action can be reversed by pharmacologically directed dosing of decitabine, which depletes the corepressor DNMT1 from CEBPα/RUNX1 protein interactome []. Although several epigenetic therapies have only limited efficacy when used as single agents, combination therapies using conventional chemotherapy that targets AML pathogenesis that exhibit synergistic mechanisms are also undergoing clinical trials []. In this context, it was recently shown that the addition of ATRA to decitabine resulted in a higher remission rate and a clinically meaningful survival extension []. Another example is venetoclax. Venetoclax is a small molecule inhibitor of the antiapoptotic protein B-cell lymphoma 2 (BCL2). BCL2 is a mitochondrial protein and functions as an antiapoptotic protein, and AML stem cells are dependent on BCL2 for survival. BCL2 inhibitor was first developed for single agents for AML, but clear synergy with venetoclax and both HMAs and LDAC was identified preclinically [-].
Approximately, 15–23% of AML genomes harbor mutations in 1 of 2 isoforms of IDH1 or IDH2 []. Leukemic IDH mutations cause changes in genome structure and gene activity, culminating in an arrest of normal myeloid differentiation []. Ivosidenib and enasidenib are recently developed IDH1 and IDH2 inhibitors, respectively, and both presented well-tolerated options in the setting of refractory or relapsed AML. Even in elderly and heavily pretreated patients, these 2 agents have shown response rates of 30–40% [, ]. Notably, Norsworthy et al. [] have reported that approximately 19% of the patients with IDH inhibitors were adjudicated as differentiation syndrome. They concluded that differentiation syndrome is a common and potentially fatal adverse reaction of IDH inhibitors and use of standardized diagnostic criteria may aid in earlier diagnosis and treatment. Another important epigenetic change in AML is alterations in histone methylation. This leads to the aberrant silencing of expression of multiple genes [] involved in tumor suppression and cell cycling, resulting in myeloid maturation arrest. The inhibition of lysine-specific demethylase-1 (LSD1) is a promising novel epigenetic approach for AML therapy. Schenk et al. [] demonstrated that pharmacologic LSD1 inhibitor, tranylcypromine, restore myeloid differentiation in AML cells. The precise mechanisms of the action were revealed by Ravasio et al. [] that integration of proteomic/epigenomic/mutational studies showed that LSD1 inhibitors alter the recruitment of LSD1-containing complexes to chromatin, inhibiting the interaction between LSD1 and the transcription factor GFI1. Association of LSD1 with GFI1 has been reported to be critical for maintaining the differentiation block of AML cells [, ]; therefore, the dissociation between these 2 factors by LSD1 inhibition plays a role in the induction of myeloid differentiation. Wass et al. [] recently demonstrated that combination of LSD1 inhibition by tranylcypromine with ATRA can induce differentiation of AML blasts and lead to clinical response in heavily pretreated patients with relapsed/refractory AML with acceptable toxicity. These suggest that the combination of epigenetic modifiers with conventional chemotherapy, or differentiation therapy, may change the use of standard chemotherapy.
microRNAs
Noncoding RNA molecules that regulate DNA transcription and translation include miRNAs, long noncoding RNAs, and circular RNAs []. miRNAs are a class of small, noncoding single-stranded RNAs comprising approximately 19–22 nucleotides. They typically inhibit mRNA translation by binding to the 3′-untranslated region of target mRNA []. miRNAs are involved in a variety of physiological processes including differentiation []. One of the most characterized miRNAs functioning in myeloid differentiation is miR-223, whose expression is regulated by myeloid transcription factor NFI-A and C/EBPα []. miR-223 overexpression in APL cell lines enhanced differentiation into granulocytes while inhibiting miR-223 reduced differentiation []. However, using a loss-of-function allele in mice, miR-223 was shown to negatively regulate progenitor proliferation and granulocyte differentiation and activation, suggesting that it acts as a fine-tuner of granulocyte production and the inflammatory response []. Numerous miRNAs other than miR-223 have been reported as being necessary for myeloid differentiation, including miR-34a [], miR-24 [], miR-27 [], miR-342 [], and miR-155 []. The aberrant expression and function of these miRNAs are important in leukemogenesis.
Vitamin Derivatives
Vitamin A and its analogs, in general, and ATRA, in particular, play a critical role in the differentiation of neutrophils []. ATRA exert their antileukemic effects by degrading tumor-specific protein PML-RAR alfa []. Similar to decitabine, as noted before, ATO and ATRA also induce proteasome-dependent degradation of NPM1 mutant leukemic protein, leading to cell growth inhibition and apoptosis in NPM1-mutated AML []. Additionally, Vitamin D derivatives such as 1,25-dihydroxyvitamin D3 (1,25D), the hormonal and active form of vitamin D, and its synthetic analogs regulate multiple cell events including cell proliferation, survival, differentiation, and immune responses [-]. Although the underlying mechanism of action remains unknown, vitamin K2 has also been reported to induce apoptosis and differentiation []. 1,25D induces monocyte/macrophage-like differentiation, and cell cycle arrest through activating various kinase pathways including mitogen-activated kinases, c-Jun NH2-terminal kinases, and p38 mitogen-activated kinases []. 1,25D also modulates the expression of several miRNAs []. A major limiting factor in the clinical application of 1,25D is the supraphysiologic dose required, which results in systemic hypercalcemia []. Vitamin D derivatives have been suggested to have anti-neoplastic properties, but the translation of epidemiological and laboratory findings to the clinic has so far not been successful [, , ]. However, it may possible to overcome this using combination therapies of 1,25D with HDAC inhibitors or HMAs [], thus, highlighting the importance of epigenetic modifiers in differentiation therapy.
Hematopoietic Cytokines and Chemotherapeutic Agents
Many hematopoietic cytokines can trigger the lineage-specific differentiation of leukemia cells, which may have important implications in clinical settings []. However, despite numerous laboratory investigations into the differentiation-inducing activities of cytokines, relatively few studies have considered the applications of hematopoietic cytokines in the treatment of leukemia patients []. Cytokine therapy implies the stimulation of leukemia cells, but 2 major hematopoietic growth factors, G-CSF and granulocyte macrophage-CSF, are in clinical use to shorten the duration of chemotherapy-induced neutropenia and to prevent infections []. The use of these agents in leukemia was used for 2 major indications: the first, as a primary or secondary prophylaxis of infectious events after therapy in susceptible, older, populations and the second, as a priming strategy to potentiate response to chemotherapy. There is always a theoretical concern that G-CSF may stimulate and mobilize leukemic blasts, and thus, this intervention is approached with caution by most clinicians. Therefore, only few sporadic case reports [] have described the effectiveness of cytokine therapy for AML treatment. Its therapeutic role remains to be established in more controlled clinical trials.
Various low-dose chemotherapeutic agents have been tested using in vitro differentiation models, including Ara-C [], antifolates [], anthracyclines [], and camptothecin []. Among these, LDAC has been used for AML treatment since the 1980s []. The mechanisms for differentiation by LDAC remain unclear; however, several findings have been reported. Wang et al. [] reported that LDAC led to a marked increase of cyclin-dependent kinase inhibitor p21CIP1, which plays a pivotal role in human myelomonocytic leukemia U937 cells. This effect is profoundly impaired in the p21CIP1 antisense stably expressed U937 cells, suggesting that p21CIP1 is playing an important role in the LDAC-mediated U937 differentiation. Chen et al. [] recently found that LDAC can induce autophagy in AML cells and appeared to play an important role in differentiation and death. They revealed that low-dose (50 nM) cytarabine does not cause apoptosis but induces significant increase in differentiation marker CD11b in monocytic leukemia U937 cells. However, there are dose-dependent increases of apoptotic cells with >500 nM of cytarabine. LDAC induces autophagy, and downregulation of Akt-mTOR pathway is involved in these processes. The CAG regimen, a combination of LDAC, aclarubicin, and G-CSF, has been widely employed in patients with relapsed or refractory AML []. G-CSF stimulates the proliferation of myeloid cells and intensifies Ara-C metabolism. The efficacy of other agents, such as low-dose oral idarubicin treatment, has also been reported []. Recently, these low-dose chemotherapeutic treatments have drawn increasing attention, from the development of several promising combination therapies for LDAC. In the pivotal clinical trials evaluating venetoclax, a BCL2 inhibitor described in epigenetic modifiers section, either in combination with LDAC or with HMA, the rates of complete remission plus complete remission with incomplete hematological recovery were 54 and 67%, respectively, and the median overall survival was 10.4 and 17.5 months, respectively, comparing favorably with outcomes in clinical trials evaluating single-agent LDAC or HMA []. Glasdegib, a hedgehog pathway inhibitor, may be used in combination with LDAC for the same indication and improves survival compared with LDAC alone []. From randomized phase II, open-label, and multicenter study, it was reported that glasdegib plus LDAC has a favorable benefit-risk profile and may be a promising option for AML patients unsuitable for intensive chemotherapy [].
Conclusion
To date, numerous differentiation inducers have been reported, but development of their further efficacy is important to expand future applications for differentiation therapies. Attempts to improve the outcome of therapy for AML with HMA or LDAC are, given separately, sometimes in combination with a second agent such as a BCL2 inhibitor venetoclax and the hedgehog inhibitor glasdegib or with novel experimental agents such as IDH inhibitors [, , -, , , -]. Among the agents described in this review (Fig. 2; Table 1), in my opinion, epigenomic modifiers seem promising [-, -, , , ], and the combination of these drugs may make an epoch for AML therapy. Particular attention should also be paid to glycosylation modifiers as combinational agents for leukemia therapy because changes in glycosylation patterns are a unique characteristic of cancer [], indicating that these inhibitors will work efficiently for hematological malignancies. Further clarifying the mechanism of myeloid differentiation may lead to the development of efficient differentiation therapy for AML.
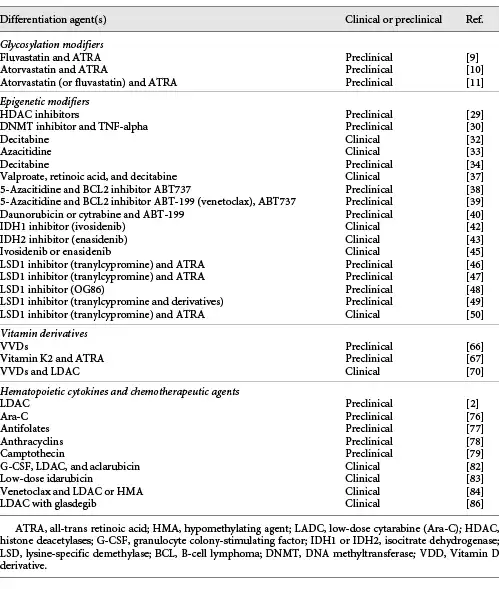
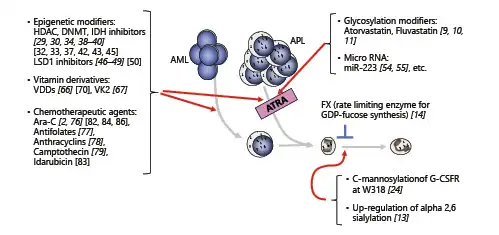
Fig. 2
Myeloid differentiation inducers are described in this review. The reference number of important papers demonstrating clinical significances is shown in roman, while the reference number of basic research papers is shown in italic. Red and blue lines are showing the inducible and suppressive effect on myeloid differentiation, respectively.
Acknowledgement
We thank Sarah Williams, PhD, from Edanz Group (https://en-author-services.edanzgroup.com/) for editing a draft of this manuscript.
Conflict of Interest Statement
The author declares no competing interests.
Funding Sources
This work was supported in part by a Grant-in-Aid for Scientific Research (No. 17K09019) from the Ministry of Education, Science and Culture, Japan, and Daiichi-Sankyo Research Support (Daiichi-Sankyo Inc.).
Author Contributions
S.T. designed the study, obtained the grants, analyzed the data, and wrote the manuscript.
References
- 1. Vey N. Low-intensity regimens versus standard-intensity induction strategies in acute myeloid leukemia. Ther Adv Hematol. 2020;11:2040620720913010.http://dx.doi.org/10.1177/2040620720913010.
- 2. Wang Z, Wang S, Fisher PB, Dent P, Grant S. Evidence of a functional role for the cyclin-dependent kinase inhibitor p21CIP1 in leukemic cell (U937) differentiation induced by low concentrations of 1-beta-D-arabinofuranosylcytosine. Differentiation. 2000 Aug;66(1):1–13.http://dx.doi.org/10.1046/j.1432-0436.2000.066001001.x.
- 3. Bohl SR, Bullinger L, Rücker FG. Epigenetic therapy: azacytidine and decitabine in acute myeloid leukemia. Expert Rev Hematol. 2018 May;11(5):361–71.http://dx.doi.org/10.1080/17474086.2018.1453802.
- 4. Cossu G, Kuo AL, Pessano S, Warren L, Cooper RA. Decreased synthesis of high-molecular-weight glycopeptides in human promyelocytic leukemic cells (HL-60) during phorbol ester-induced macrophage differentiation. Cancer Res. 1982 Feb;42(2):484–9..
- 5. Mizoguchi A, Takasaki S, Maeda S, Kobata A. Changes in asparagine-linked sugar chains of human promyelocytic leukemic cells (HL-60) during monocytoid differentiation and myeloid differentiation. Decrease of high-molecular-weight oligosaccharides in acidic fraction. J Biol Chem. 1984 Oct 10;259(19):11949–57..
- 6. Mizoguchi A, Maeda S, Shiraishi N, Yoshima H, Sugiyama T. Cell surface asparagine-linked sugar chains of human early myeloblastic leukemic cells (KG-1a). Cancer Res. 1986 Jan;46(1):29–37..
- 7. Corsini A, Maggi FM, Catapano AL. Pharmacology of competitive inhibitors of HMG-CoA reductase. Pharmacol Res. 1995 Jan;31(1):9–27.http://dx.doi.org/10.1016/1043-6618(95)80042-5.
- 8. Mo H, Elson CE. Studies of the isoprenoid-mediated inhibition of mevalonate synthesis applied to cancer chemotherapy and chemoprevention. Exp Biol Med. 2004 Jul;229(7):567–85.http://dx.doi.org/10.1177/153537020422900701.
- 9. Sassano A, Katsoulidis E, Antico G, Altman JK, Redig AJ, Minucci S, et al. Suppressive effects of statins on acute promyelocytic leukemia cells. Cancer Res. 2007 May 1;67(9):4524–32.http://dx.doi.org/10.1158/0008-5472.CAN-06-3686.
- 10. Sassano A, Lo Iacono M, Antico G, Jordan A, Uddin S, Calogero RA, et al. Regulation of leukemic cell differentiation and retinoid-induced gene expression by statins. Mol Cancer Ther. 2009 Mar;8(3):615–25.http://dx.doi.org/10.1158/1535-7163.MCT-08-1196.
- 11. Sassano A, Altman JK, Gordon LI, Platanias LC. Statin-dependent activation of protein kinase Cδ in acute promyelocytic leukemia cells and induction of leukemic cell differentiation. Leuk Lymphoma. 2012 Sep;53(9):1779–84.http://dx.doi.org/10.3109/10428194.2012.668287.
- 12. Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015 Sep;15(9):540–55.http://dx.doi.org/10.1038/nrc3982.
- 13. Le Marer N, Skacel PO. Up-regulation of alpha2,6 sialylation during myeloid maturation: a potential role in myeloid cell release from the bone marrow. J Cell Physiol. 1999 Jun;179(3):315–24.http://dx.doi.org/10.1002/(SICI)1097-4652(199906)179:3<315::AID-JCP9>3.0.CO;2-U.
- 14. Zhou L, Li LW, Yan Q, Petryniak B, Man Y, Su C, et al. Notch-dependent control of myelopoiesis is regulated by fucosylation. Blood. 2008 Jul 15;112(2):308–19.http://dx.doi.org/10.1182/blood-2007-11-115204.
- 15. Takahashi S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: biology and therapeutic implications. J Hematol Oncol. 2011;4:13.http://dx.doi.org/10.1186/1756-8722-4-13.
- 16. Duan C, Fukuda T, Isaji T, Qi F, Yang J, Wang Y, et al. Deficiency of core fucosylation activates cellular signaling dependent on FLT3 expression in a Ba/F3 cell system. FASEB J. 2020 Feb;34(2):3239–52.http://dx.doi.org/10.1096/fj.201902313RR.
- 17. Lee BH, Williams IR, Anastasiadou E, Boulton CL, Joseph SW, Amaral SM, et al. FLT3 internal tandem duplication mutations induce myeloproliferative or lymphoid disease in a transgenic mouse model. Oncogene. 2005 Nov 24;24(53):7882–92.http://dx.doi.org/10.1038/sj.onc.1208933.
- 18. Rillahan CD, Antonopoulos A, Lefort CT, Sonon R, Azadi P, Ley K, et al. Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nat Chem Biol. 2012 Jul;8(7):661–8.http://dx.doi.org/10.1038/nchembio.999.
- 19. Okeley NM, Alley SC, Anderson ME, Boursalian TE, Burke PJ, Emmerton KM, et al. Development of orally active inhibitors of protein and cellular fucosylation. Proc Natl Acad Sci U S A. 2013 Apr 2;110(14):5404–9.http://dx.doi.org/10.1073/pnas.1222263110.
- 20. Kizuka Y, Nakano M, Yamaguchi Y, Nakajima K, Oka R, Sato K, et al. An alkynyl-fucose halts hepatoma cell migration and invasion by inhibiting GDP-fucose-synthesizing enzyme FX, TSTA3. Cell Chem Biol. 2017 Dec 21;24(12):1467–78.e5.http://dx.doi.org/10.1016/j.chembiol.2017.08.023.
- 21. Takahashi S, Harigae H, Yokoyama H, Ishikawa I, Abe S, Imaizumi M, et al. Synergistic effect of arsenic trioxide and flt3 inhibition on cells with flt3 internal tandem duplication. Int J Hematol. 2006 Oct;84(3):256–61.http://dx.doi.org/10.1532/IJH97.06076.
- 22. Takahashi S. Combination therapy with arsenic trioxide for hematological malignancies. Anticancer Agents Med Chem. 2010 Jul 1;10(6):504–10.http://dx.doi.org/10.2174/1871520611009060504.
- 23. Nagai K, Hou L, Li L, Nguyen B, Seale T, Shirley C, et al. Combination of ATO with FLT3 TKIs eliminates FLT3/ITD+ leukemia cells through reduced expression of FLT3. Oncotarget. 2018 Aug 31;9(68):32885–99.http://dx.doi.org/10.18632/oncotarget.25972.
- 24. Otani K, Niwa Y, Suzuki T, Sato N, Sasazawa Y, Dohmae N, et al. Regulation of granulocyte colony-stimulating factor receptor-mediated granulocytic differentiation by C-mannosylation. Biochem Biophys Res Commun. 2018 Apr 6;498(3):466–72.http://dx.doi.org/10.1016/j.bbrc.2018.02.210.
- 25. Freund P, Kerenyi MA, Hager M, Wagner T, Wingelhofer B, Pham HTT, et al. O-GlcNAcylation of STAT5 controls tyrosine phosphorylation and oncogenic transcription in STAT5-dependent malignancies. Leukemia. 2017 Oct;31(10):2132–42.http://dx.doi.org/10.1038/leu.2017.4.
- 26. Takahashi S. PU.1, a versatile transcription factor and a suppressor of myeloid leukemia. In: Myeloid leukemia: basic mechanisms of leukemogenesis. 2011. p. 239–62.
- 27. Alvarez-Errico D, Vento-Tormo R, Sieweke M, Ballestar E. Epigenetic control of myeloid cell differentiation, identity and function. Nat Rev Immunol. 2015 Jan;15(1):7–17.
- 28. Takahashi S. Epigenetic aberrations in myeloid malignancies (Review). Int J Mol Med. 2013 Sep;32(3):532–8.http://dx.doi.org/10.3892/ijmm.2013.1417.
- 29. Kosugi H, Towatari M, Hatano S, Kitamura K, Kiyoi H, Kinoshita T, et al. Histone deacetylase inhibitors are the potent inducer/enhancer of differentiation in acute myeloid leukemia: a new approach to anti-leukemia therapy. Leukemia. 1999 Sep;13(9):1316–24.http://dx.doi.org/10.1038/sj.leu.2401508.
- 30. Laurenzana A, Petruccelli LA, Pettersson F, Figueroa ME, Melnick A, Baldwin AS, et al. Inhibition of DNA methyltransferase activates tumor necrosis factor alpha-induced monocytic differentiation in acute myeloid leukemia cells. Cancer Res. 2009 Jan 1;69(1):55–64.http://dx.doi.org/10.1158/0008-5472.CAN-08-0245.
- 31. Bewersdorf JP, Shallis R, Stahl M, Zeidan AM. Epigenetic therapy combinations in acute myeloid leukemia: what are the options?Ther Adv Hematol. 2019;10:2040620718816698.http://dx.doi.org/10.1177/2040620718816698.
- 32. Kantarjian HM, Thomas XG, Dmoszynska A, Wierzbowska A, Mazur G, Mayer J, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol. 2012 Jul 20;30(21):2670–7.http://dx.doi.org/10.1200/JCO.2011.38.9429.
- 33. Dombret H, Seymour JF, Butrym A, Wierzbowska A, Selleslag D, Jang JH, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood. 2015 Jul 16;126(3):291–9.http://dx.doi.org/10.1182/blood-2015-01-621664.
- 34. Blagitko-Dorfs N, Schlosser P, Greve G, Pfeifer D, Meier R, Baude A, et al. Combination treatment of acute myeloid leukemia cells with DNMT and HDAC inhibitors: predominant synergistic gene downregulation associated with gene body demethylation. Leukemia. 2019 Apr;33(4):945–56.http://dx.doi.org/10.1038/s41375-018-0293-8.
- 35. Gu X, Ebrahem Q, Mahfouz RZ, Hasipek M, Enane F, Radivoyevitch T, et al. Leukemogenic nucleophosmin mutation disrupts the transcription factor hub that regulates granulomonocytic fates. J Clin Invest. 2018 Oct 1;128(10):4260–79.http://dx.doi.org/10.1172/JCI97117.
- 36. Takahashi S. Current findings for recurring mutations in acute myeloid leukemia. J Hematol Oncol. 2011;4:36.http://dx.doi.org/10.1186/1756-8722-4-36.
- 37. Lubbert M, Grishina O, Schmoor C, Schlenk RF, Jost E, Crysandt M, et al. Valproate and retinoic acid in combination with decitabine in elderly nonfit patients with acute myeloid leukemia: results of a multicenter, randomized, 2 x 2, phase II trial. J Clin Oncol. 2020 Jan 20;38(3):257–70.
- 38. Tsao T, Shi Y, Kornblau S, Lu H, Konoplev S, Antony A, et al. Concomitant inhibition of DNA methyltransferase and BCL-2 protein function synergistically induce mitochondrial apoptosis in acute myelogenous leukemia cells. Ann Hematol. 2012 Dec;91(12):1861–70.http://dx.doi.org/10.1007/s00277-012-1537-8.
- 39. Bogenberger JM, Delman D, Hansen N, Valdez R, Fauble V, Mesa RA, et al. Ex vivo activity of BCL-2 family inhibitors ABT-199 and ABT-737 combined with 5-azacytidine in myeloid malignancies. Leuk Lymphoma. 2015 Jan;56(1):226–9.http://dx.doi.org/10.3109/10428194.2014.910657.
- 40. Niu X, Zhao J, Ma J, Xie C, Edwards H, Wang G, et al. Binding of released bim to Mcl-1 is a mechanism of intrinsic resistance to ABT-199 which can be overcome by combination with daunorubicin or cytarabine in AML cells. Clin Cancer Res. 2016 Sep 1;22(17):4440–51.http://dx.doi.org/10.1158/1078-0432.CCR-15-3057.
- 41. Teh TC, Nguyen NY, Moujalled DM, Segal D, Pomilio G, Rijal S, et al. Enhancing venetoclax activity in acute myeloid leukemia by co-targeting MCL1. Leukemia. 2018 Feb;32(2):303–12.http://dx.doi.org/10.1038/leu.2017.243.
- 42. Becker JS, Fathi AT. Targeting isocitrate dehydrogenase mutations (IDH) in AML: wielding the double-edged sword of differentiation. Curr Cancer Drug Targets. 2020 Apr 24;20(7):490–500.
- 43. DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018 Jun 21;378(25):2386–98.http://dx.doi.org/10.1056/NEJMoa1716984.
- 44. Stein EM, DiNardo CD, Fathi AT, Pollyea DA, Stone RM, Altman JK, et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood. 2019 Feb 14;133(7):676–87.http://dx.doi.org/10.1182/blood-2018-08-869008.
- 45. Norsworthy KJ, Mulkey F, Scott EC, Ward AF, Przepiorka D, Charlab R, et al. Differentiation syndrome with ivosidenib and enasidenib treatment in patients with relapsed or refractory IDH-mutated AML: a U.S. Food and Drug Administration systematic analysis. Clin Cancer Res. 2020 Aug 15;26(16):4280–8.
- 46. Schenk T, Chen WC, Göllner S, Howell L, Jin L, Hebestreit K, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. 2012 Mar 11;18(4):605–11.http://dx.doi.org/10.1038/nm.2661.
- 47. Ravasio R, Ceccacci E, Nicosia L, Hosseini A, Rossi PL, Barozzi I, et al. Targeting the scaffolding role of LSD1 (KDM1A) poises acute myeloid leukemia cells for retinoic acid-induced differentiation. Sci Adv. 2020 Apr;6(15):eaax2746.http://dx.doi.org/10.1126/sciadv.aax2746.
- 48. Maiques-Diaz A, Spencer GJ, Lynch JT, Ciceri F, Williams EL, Amaral FMR, et al. Enhancer activation by pharmacologic displacement of LSD1 from GFI1 induces differentiation in acute myeloid leukemia. Cell Rep. 2018 Mar 27;22(13):3641–59.http://dx.doi.org/10.1016/j.celrep.2018.03.012.
- 49. Barth J, Abou-El-Ardat K, Dalic D, Kurrle N, Maier AM, Mohr S, et al. LSD1 inhibition by tranylcypromine derivatives interferes with GFI1-mediated repression of PU.1 target genes and induces differentiation in AML. Leukemia. 2019 Jun;33(6):1411–26.http://dx.doi.org/10.1038/s41375-018-0375-7.
- 50.
- 51. Liu Y, Cheng Z, Pang Y, Cui L, Qian T, Quan L, et al. Role of microRNAs, circRNAs and long noncoding RNAs in acute myeloid leukemia. J Hematol Oncol. 2019 May 24;12(1):51.http://dx.doi.org/10.1186/s13045-019-0734-5.
- 52. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009 Jan 23;136(2):215–33.http://dx.doi.org/10.1016/j.cell.2009.01.002.
- 53. Li M, Cui X, Guan H. MicroRNAs: pivotal regulators in acute myeloid leukemia. Ann Hematol. 2020 Mar;99(3):399–412.http://dx.doi.org/10.1007/s00277-019-03887-5.
- 54. Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell. 2005 Dec 2;123(5):819–31.http://dx.doi.org/10.1016/j.cell.2005.09.023.
- 55. Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008 Feb 28;451(7182):1125–9.http://dx.doi.org/10.1038/nature06607.
- 56. Pulikkan JA, Peramangalam PS, Dengler V, Ho PA, Preudhomme C, Meshinchi S, et al. C/EBPα regulated microRNA-34a targets E2F3 during granulopoiesis and is down-regulated in AML with CEBPA mutations. Blood. 2010 Dec 16;116(25):5638–49.http://dx.doi.org/10.1182/blood-2010-04-281600.
- 57. Zaidi SK, Dowdy CR, van Wijnen AJ, Lian JB, Raza A, Stein JL, et al. Altered Runx1 subnuclear targeting enhances myeloid cell proliferation and blocks differentiation by activating a miR-24/MKP-7/MAPK network. Cancer Res. 2009 Nov 1;69(21):8249–55.http://dx.doi.org/10.1158/0008-5472.CAN-09-1567.
- 58. Feng J, Iwama A, Satake M, Kohu K. MicroRNA-27 enhances differentiation of myeloblasts into granulocytes by post-transcriptionally downregulating Runx1. Br J Haematol. 2009 May;145(3):412–23.http://dx.doi.org/10.1111/j.1365-2141.2009.07632.x.
- 59. De Marchis ML, Ballarino M, Salvatori B, Puzzolo MC, Bozzoni I, Fatica A. A new molecular network comprising PU.1, interferon regulatory factor proteins and miR-342 stimulates ATRA-mediated granulocytic differentiation of acute promyelocytic leukemia cells. Leukemia. 2009 May;23(5):856–62.http://dx.doi.org/10.1038/leu.2008.372.
- 60. Palma CA, Al Sheikha D, Lim TK, Bryant A, Vu TT, Jayaswal V, et al. MicroRNA-155 as an inducer of apoptosis and cell differentiation in acute myeloid leukaemia. Mol Cancer. 2014 Apr 5;13:79.http://dx.doi.org/10.1186/1476-4598-13-79.
- 61. Mehta K. Retinoic acid: a player that rules the game of life and death in neutrophils. Indian J Exp Biol. 2002 Aug;40(8):874–81..
- 62. de The H, Le Bras M, Lallemand-Breitenbach V. The cell biology of disease: acute promyelocytic leukemia, arsenic, and PML bodies. J Cell Biol. 2012 Jul 9;198(1):11–21.
- 63. Martelli MP, Gionfriddo I, Mezzasoma F, Milano F, Pierangeli S, Mulas F, et al. Arsenic trioxide and all-trans retinoic acid target NPM1 mutant oncoprotein levels and induce apoptosis in NPM1-mutated AML cells. Blood. 2015 May 28;125(22):3455–65.http://dx.doi.org/10.1182/blood-2014-11-611459.
- 64. Feldman D, Krishnan AV, Swami S, Giovannucci E, Feldman BJ. The role of vitamin D in reducing cancer risk and progression. Nat Rev Cancer. 2014 May;14(5):342–57.http://dx.doi.org/10.1038/nrc3691.
- 65. Studzinski GP, Harrison JS, Wang X, Sarkar S, Kalia V, Danilenko M. Vitamin D control of hematopoietic cell differentiation and leukemia. J Cell Biochem. 2015 Aug;116(8):1500–12.http://dx.doi.org/10.1002/jcb.25104.
- 66. Nachliely M, Sharony E, Kutner A, Danilenko M. Novel analogs of 1,25-dihydroxyvitamin D2 combined with a plant polyphenol as highly efficient inducers of differentiation in human acute myeloid leukemia cells. J Steroid Biochem Mol Biol. 2016 Nov;164:59–65.http://dx.doi.org/10.1016/j.jsbmb.2015.09.014.
- 67. Yaguchi M, Miyazawa K, Katagiri T, Nishimaki J, Kizaki M, Tohyama K, et al. Vitamin K2 and its derivatives induce apoptosis in leukemia cells and enhance the effect of all-trans retinoic acid. Leukemia. 1997 Jun;11(6):779–87.http://dx.doi.org/10.1038/sj.leu.2400667.
- 68. Gocek E, Studzinski GP. The potential of vitamin D-regulated intracellular signaling pathways as targets for myeloid leukemia therapy. J Clin Med. 2015 Mar 25;4(4):504–34.http://dx.doi.org/10.3390/jcm4040504.
- 69. Cao H, Xu Y, de Necochea-Campion R, Baylink DJ, Payne KJ, Tang X, et al. Application of vitamin D and vitamin D analogs in acute myelogenous leukemia. Exp Hematol. 2017 Jun;50:1–12.http://dx.doi.org/10.1016/j.exphem.2017.01.007.
- 70. Yamada K, Mizusawa M, Harima A, Kajiwara K, Hamaki T, Hoshi K, et al. Induction of remission of relapsed acute myeloid leukemia after unrelated donor cord blood transplantation by concomitant low-dose cytarabine and calcitriol in adults. Eur J Haematol. 2006 Oct;77(4):345–8.http://dx.doi.org/10.1111/j.0902-4441.2006.t01-1-EJH2570.x.
- 71. Ferrara F, Fazi P, Venditti A, Pagano L, Amadori S, Mandelli F. Heterogeneity in the therapeutic approach to relapsed elderly patients with acute myeloid leukaemia: a survey from the Gruppo Italiano Malattie Ematologiche dell’ Adulto (GIMEMA) Acute Leukaemia Working Party. Hematol Oncol. 2008 Jun;26(2):104–7.http://dx.doi.org/10.1002/hon.846.
- 72. Karlic H, Varga F. Impact of vitamin D metabolism on clinical epigenetics. Clin Epigenetics. 2011 Apr;2(1):55–61.http://dx.doi.org/10.1007/s13148-011-0021-y.
- 73. Leung KN, Mak NK, Fung MC. Cytokines in the differentiation therapy of leukemia: from laboratory investigations to clinical applications. Crit Rev Clin Lab Sci. 2005;42(5–6):473–514.http://dx.doi.org/10.1080/10408360500295154.
- 74. Frankfurt O, Tallman MS. Growth factors in leukemia. J Natl Compr Canc Netw. 2007 Feb;5(2):203–15.http://dx.doi.org/10.6004/jnccn.2007.0020.
- 75. Nimubona S, Grulois I, Bernard M, Drénou B, Godard M, Fauchet R, et al. Complete remission in hypoplastic acute myeloid leukemia induced by G-CSF without chemotherapy: report on three cases. Leukemia. 2002 Sep;16(9):1871–3.http://dx.doi.org/10.1038/sj.leu.2402592.
- 76. Griffin J, Munroe D, Major P, Kufe D. Induction of differentiation of human myeloid leukemia cells by inhibitors of DNA synthesis. Exp Hematol. 1982 Oct;10(9):774–81..
- 77. Sokoloski JA, Blair OC, Carbone R, Sartorelli AC. Induction of the differentiation of synchronized HL-60 leukemia cells by tiazofurin. Exp Cell Res. 1989 May;182(1):234–41.http://dx.doi.org/10.1016/0014-4827(89)90294-2.
- 78. Schwartz EL, Sartorelli AC. Structure-activity relationships for the induction of differentiation of HL-60 human acute promyelocytic leukemia cells by anthracyclines. Cancer Res. 1982 Jul;42(7):2651–5..
- 79. Pantazis P. The water-insoluble camptothecin analogues: promising drugs for the effective treatment of haematological malignancies. Leuk Res. 1995 Nov;19(11):775–88.http://dx.doi.org/10.1016/0145-2126(95)00048-8.
- 80. Degos L. Differentiating agents in the treatment of leukemia and myelodysplastic syndromes. Leuk Res. 1990;14(8):731–3.http://dx.doi.org/10.1016/0145-2126(90)90103-g.
- 81. Chen L, Guo P, Zhang Y, Li X, Jia P, Tong J, et al. Autophagy is an important event for low-dose cytarabine treatment in acute myeloid leukemia cells. Leuk Res. 2017 Sep;60:44–52.http://dx.doi.org/10.1016/j.leukres.2017.06.007.
- 82. Yamada K, Furusawa S, Saito K, Waga K, Koike T, Arimura H, et al. Concurrent use of granulocyte colony-stimulating factor with low-dose cytosine arabinoside and aclarubicin for previously treated acute myelogenous leukemia: a pilot study. Leukemia. 1995 Jan;9(1):10–4..
- 83. Musso M, Porretto F, Crescimanno A, Bondì F, Polizzi V, Scalone R, et al. Low-dose long-term oral idarubicin in maintenance treatment of elderly acute myeloid leukemia. Haematologica. 1997 Sep–Oct;82(5 Suppl):4–8.
- 84. Richard-Carpentier G, DiNardo CD. Venetoclax for the treatment of newly diagnosed acute myeloid leukemia in patients who are ineligible for intensive chemotherapy. Ther Adv Hematol. 2019;10:2040620719882822.http://dx.doi.org/10.1177/2040620719882822.
- 85. Richard-Carpentier G, DiNardo CD. Single-agent and combination biologics in acute myeloid leukemia. Hematology Am Soc Hematol Educ Program. 2019 Dec 6;2019(1):548–56.http://dx.doi.org/10.1182/hematology.2019000059.
- 86. Cortes JE, Heidel FH, Hellmann A, Fiedler W, Smith BD, Robak T, et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia. 2019 Feb;33(2):379–89.http://dx.doi.org/10.1038/s41375-018-0312-9.
- 87. Costa AF, Campos D, Reis CA, Gomes C. Targeting glycosylation: a new road for cancer drug discovery. Trends Cancer. 2020 Sep;6(9):757–66.