Introduction
Transplant glomerulopathy (TG) is a morphological lesion characterized by reduplication and/or multilayering of the glomerular basement membrane (GBM), and it is strongly associated with poor graft survival [-]. The pathogenesis of TG clearly involves chronic, repetitive endothelial cell injury that can be caused by chronic antibody-mediated rejection, hepatitis C, thrombotic microangiopathy, cell-mediated immune injury, or various autoantibodies []. Several studies have demonstrated that podocyte injury and depletion may play a role in the progression of TG and contribute to proteinuria, reduced glomerular filtration rate, and allograft loss [-].
Focal segmental glomerulosclerosis (FSGS) represents a common histological pattern of podocyte injury and depletion. Subclasses of FSGS include primary, genetic, and secondary forms, the latter comprising maladaptive, viral, and drug-induced FSGS []. For the most part, the interplay among these drivers is unclear and likely complex. For instance, maladaptive FSGS involves both podocyte stress and genetic susceptibility []. TG coexisting with FSGS can be found in renal allograft biopsies, but the etiologies, clinical features, and prognosis of patients with these 2 overlapping lesions remain largely unknown. Therefore, we explored the clinicopathological characteristics of TG with FSGS and without FSGS and analyzed the association of FSGS with allograft loss in this cohort of patients with TG.
Materials and Methods
Patient Population
Cases of all kidney transplant recipients with biopsy-proven TG from the National Clinical Research Center of Kidney Diseases in Jinling Hospital between January 2013 and December 2019 were reviewed. The inclusion criteria were as follows: (1) GBM double contours by light microscopy (LM) (Banff chronic glomerulopathy [cg] score ≥1b) [] and (2) ≥10 glomeruli on periodic acid-Schiff staining. Patients who had TG concomitant with other kidney diseases, including IgA nephropathy, immune complex-mediated glomerulonephritis, C3 glomerulonephritis, Henoch-Schonlein purpura nephritis, BK virus-associated nephropathy, and diabetic nephropathy, were excluded. All cases of TG were from for-cause biopsies (elevated serum creatinine, proteinuria, and/or positive panel reactive antibodies [PRAs]). In patients with multiple biopsies, only the first biopsy showing TG was included for subsequent analysis. According to the presence of FSGS, the patients were divided into the TG with FSGS and without FSGS groups. This study was approved by the Ethics Committee of the Jinling Hospital (2017NZKYKS-002-01) and conformed to the tenets of the Declaration of Helsinki.
Clinical Information and Primary Outcome
Demographic and clinical information at the time of renal allograft biopsy and follow-up information were collected from patient medical records. Laboratory data were collected within 1 week of the allograft biopsy. The estimated glomerular filtration rate (eGFR) was calculated from the Chronic Kidney Disease Epidemiology Collaboration formula []. The primary outcome was death-censored allograft loss defined as eGFR declining to <15 mL/min/1.73 m2 or the initiation of dialysis or re-transplantation. Clinical manifestations were defined as follows: proteinuria: urinary protein excretion >0.4 g/24 h; nephrotic-range proteinuria: urinary protein excretion >3.5 g/24 h; microscopic hematuria: a red blood cell count in urinary sediment >12/µL; allograft dysfunction: eGFR <60 mL/min/1.73 m2; and anemia: hemoglobin <120 g/L in males and hemoglobin <110 g/L in females. Due to the detection method and reference value updates, the definitions of elevated urinary N-acetyl-β-D-glucosaminidase (urNAG) and retinol binding protein (urRBP) were different before and after 2017. Elevated urNAG was defined as urNAG >16.5 U/g/cr before 2017 and >9.7 U/g/cr after 2017. Elevated urRBP was defined as urRBP >0.5 mg/L before 2017 and >0.7 mg/L after 2017.
Allograft Pathology
LM, immunofluorescence (IF), and electron microscopy (EM) were routinely performed using standard techniques. Paraffin sections for LM were stained with hematoxylin and eosin, periodic acid-Schiff, periodic acid-silver methenamine, and Masson’s trichrome. Pathological diagnosis and scoring of histological lesions were performed by 2 experienced pathologists using the 2017 Banff Classification []. Discrepancies in scoring were resolved through discussion until a consensus was reached. Morphological variants of FSGS were evaluated according to the Columbia classification []. In addition, several common pathological changes, including global sclerosis, crescents, mesangiolysis, podocyte hyperplasia, pseudocrescents, pseudotubules, acute tubular injury, striped interstitial fibrosis, and nodular arteriolar hyalinosis, were also evaluated. IF was performed on cryosections (4 μm) using fluorescein isothiocyanate-conjugated polyclonal antibodies to IgG, IgA, IgM, C3, C1q, and C4d. Cases with C4d2 (10–50%) or C4d3 (>50% of peritubular capillaries) were considered positive. Immunohistochemical staining of CD3, CD4, CD8, CD20, CD68, and human leukocyte antigen (HLA)-DR was performed on paraffin sections. The expression of HLA-DR in tubule cells ≥10% of the specimen was thought to be positive. Ten peritubular capillaries were observed randomly on EM, and the mean value of peritubular capillary basement membrane multilayering was calculated for each specimen. Degrees of foot process effacement (FPE) and subendothelial rarefaction were assessed semiquantitatively and classified as negative (0%), focal (<50%), or diffuse (≥50% of the foot process or subendothelial area).
The glomerular tuft area and podocyte number were identified and calculated automatically by the analytic renal pathology system as previously reported []. A biopsy specimen with 5 or more glomeruli without global sclerosis, segmental sclerosis, and crescents was analyzed. The standard glomerular tuft area was determined to be 0.025 mm2 in this study. Podocytes were expressed as the number of podocytes per standard glomerular tuft area.
Statistical Analysis
Analysis was performed using the SPSS, version 25.0, statistical software package (SPSS Inc). Continuous variables were expressed as the mean ± the standard deviation or the median with the interquartile range (IQR), and comparisons between groups were performed using the t test or Mann-Whitney U test. Categorical variables were expressed as counts and percentages, and differences between groups were analyzed using the χ2 test or Fisher’s exact test. Allograft outcomes were assessed using Kaplan-Meier curves and the log-rank test. Associations between clinicopathological findings and allograft outcomes were calculated using the Cox regression model. Multivariate Cox regression was used to determine the independent predictors of allograft loss. Univariate and multivariate logistic regression models were used to identify pathological variables associated with proteinuria. Candidate variables with p < 0.20 on univariate analysis were included in multivariable models with the backward stepwise method. Two-sided p < 0.05 was considered statistically significant.
Results
Demographic and Clinical Characteristics
A total of 66 patients with biopsy-proven TG were enrolled and subdivided into 2 groups: TG with FSGS (40 patients) and TG without FSGS (26 patients). Forty-six of 66 (69.7%) were male. Eight (12.1%) patients underwent kidney transplantation in our hospital and 58 (87.9%) at other hospitals. Forty-two patients (63.6%) received kidneys from living related donors, and 24 (36.4%) from donors after cardiac death. Thirteen (19.7%) patients underwent native kidney biopsy, and 7 (10.6%) patients had donor kidney biopsy information. The clinical data of each patient with TG are presented in online supplementary Table 1 (for all online suppl. material, see http://www.karger.com/doi/10.1159/000519648). The median time from transplantation to allograft biopsy diagnosis of TG was 4.8 years (range 2.4 months to 15.3 years). The mean age was 40.3 ± 9.9 years at the time of biopsy. The median serum creatinine and proteinuria were 2.2 (IQR, 1.4–3.0) mg/dL and 1.4 (IQR, 0.7–2.9) g/24 h, respectively. Fifty-seven of 66 (86.4%) patients had proteinuria, and 11 (16.7%) patients presented with nephrotic-range proteinuria. Fifty-three (80.3%) patients were class I or class II PRA positive. Compared with TG patients without FSGS, TG patients with FSGS had higher levels of proteinuria (2.6 [1.2–3.6] vs. 0.8 [0.4–1.3] g/24 h, p < 0.001) and serum creatinine (2.5 [1.5–3.5] vs. 2.1 [1.3–2.5] mg/dL, p = 0.04), lower levels of serum albumin (34.2 ± 6.5 vs. 38.6 ± 4.8 g/L, p = 0.004), a greater proportion of elevated urNAG (p = 0.01) and urRBP (p = 0.002), and a higher incidence of microscopic hematuria (p = 0.03) and edema (p = 0.04), while there were no significant differences in other clinical parameters (Table 1).
Pathological Characteristics
Light Microscopy
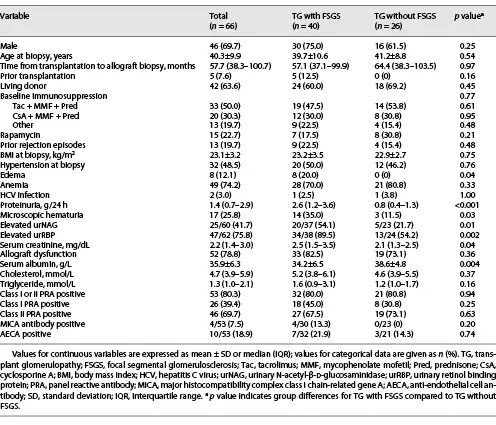
All patients with TG were characterized by GBM duplication (Fig. 1a) (cg score: cg1 on 14 cases, cg2 on 16 cases, and cg3 on 36 cases). Glomerulitis (100%), peritubular capillaritis (92.4%), tubular atrophy (97.0%), interstitial fibrosis (92.4%), and arteriolar hyalinosis (89.4%) were found in most patients. Nodular arteriolar hyalinosis and striped interstitial fibrosis were observed in 19 (28.8%) and 5 (7.6%) cases. Global sclerosis, mesangiolysis, and crescents were present in 59 (89.4%), 28 (42.4%), and 5 (7.6%) cases, respectively. Forty (60.6%) cases had segmental sclerosis (Fig. 1b), and the majority were the not otherwise specified (NOS) variant (70%), followed by the tip variant (15%), with the collapsing, cellular, and perihilar variants each accounting for 5%. Podocyte hyperplasia was found around the sclerotic capillary loops in 21 of 40 (52.5%) patients (Fig. 1c), and pseudocrescents were present in 2 (5%) patients. TG patients with FSGS had a higher cg score (cg3, 72.5% vs. 26.9%, p = 0.001, Table 2), larger glomerular tuft area (p = 0.02), lower number of podocytes (p = 0.04), higher incidences of podocyte hyperplasia (p = 0.001), pseudotubule formation (p = 0.01), and acute tubular injury (p < 0.001) than patients without FSGS (Table 3). There were no statistically significant differences in other histological features.
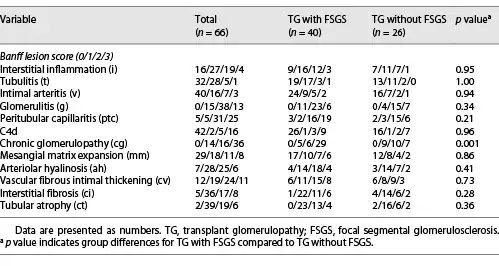
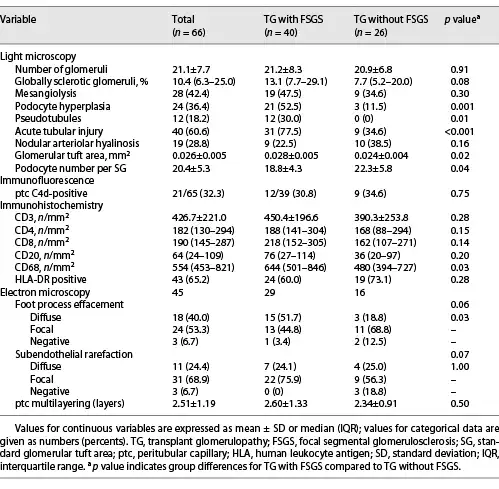
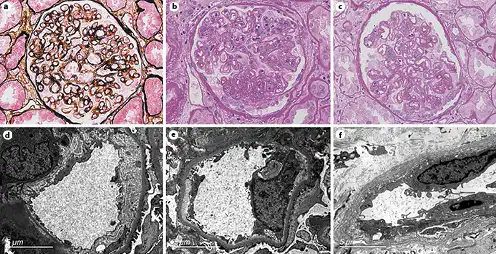
Fig. 1
Pathological features of TG patients with and without FSGS. a GBM double contours (PASM; original magnification, ×400). b GBM double contours and segmental sclerosis (PAS; original magnification, ×400). c Podocyte hyperplasia around the sclerotic capillary loops (PAS; original magnification, ×400). d Subendothelial widening and rarefaction (EM; original magnification, ×6,800). e Podocyte foot process effacement (70%–80%) (EM; original magnification, ×6,800). f Peritubular capillary basement membrane multilayering (EM; original magnification, ×6,800). TG, transplant glomerulopathy; FSGS, focal segmental glomerulosclerosis; GBM, glomerular basement membrane; PASM, periodic acid-silver methenamine; PAS, periodic acid-Schiff; EM, electron microscopy.
Immunofluorescence and Immunohistochemistry
Peritubular capillary C4d positive staining was present in 21 of 65 (32.3%) patients (C4d2 in 5 cases and C4d3 in 16 cases), and the 2 groups had similar positive rates of C4d. Forty-one (62.1%) and 17 (25.8%) patients had IgM and complement C3 granular deposition (+) in the mesangium and glomerular capillary loops, respectively. Thirty (45.5%) patients had trace glomerular IgA stains, and 9 (13.6%) had trace to + stains of IgG and C1q. Immunohistochemical staining showed that TG patients with FSGS had more infiltration of CD68+ cells in renal interstitium than that of patients without FSGS (p = 0.03, Table 3). The infiltration of other cells in the 2 groups was similar.
Electron Microscopy
EM was available in 52 patients, and 7 had no glomeruli within their specimens. Forty-two of 45 (93.3%) cases showed subendothelial widening and rarefaction (Fig. 1d) and a loss of endothelial fenestrations. Podocyte FPE was observed in 42 (93.3%) patients (Fig. 1e); 18 (40%) presented with diffuse FPE (12 of 18 had FPE covering 50%–60% of the glomerular capillary surface), and 24 (53.3%) with focal FPE. Peritubular capillary basement membrane multilayering was found in all patients (Fig. 1f), and the mean number of layers was 2.51 ± 1.19. As shown in Table 3, the incidence of diffuse FPE was higher in TG patients with FSGS than in patients without FSGS (p = 0.03), and there were no statistically significant differences in other ultrastructural changes.
FSGS Associated with Allograft Loss in TG
Overall, clinical follow-up was available for 59 of 66 (89.4%) patients. The median follow-up time was 16.1 (IQR, 9.9–26.2) months after biopsy, and 32 (54.2%) patients lost their grafts. A greater proportion of patients with FSGS developed death-censored allograft loss than those without FSGS (65.7% vs. 37.5%, p = 0.03). Kaplan-Meier analysis revealed that renal graft survival rates were significantly lower in TG patients with -FSGS than in patients without FSGS (p = 0.001, Fig. 2). In the univariate Cox regression model, higher proteinuria, lower eGFR, elevated urRBP, presence of FSGS, and higher cg score were found to be associated with allograft loss in TG (all p < 0.05). After adjusting for significant covariates, proteinuria (hazard ratio (HR) = 1.18, 95% confidence interval (CI): 1.02–1.37, p = 0.02), eGFR (HR = 0.94, 95% CI: 0.91–0.97, p < 0.001), FSGS (HR = 3.42, 95% CI: 1.30–8.98, p = 0.01), and PRA (HR = 3.99, 95% CI: 1.14–13.99, p = 0.03) were independent predictors of allograft loss (Table 4).
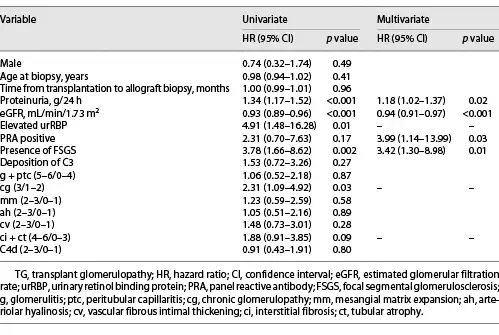
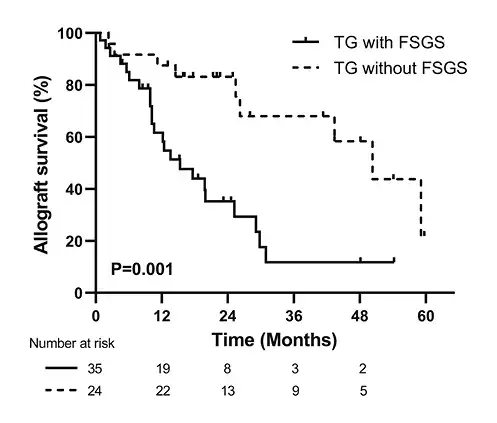
Fig. 2
Kaplan-Meier renal graft survival analysis comparing TG with and without FSGS groups. TG, transplant glomerulopathy; FSGS, focal segmental glomerulosclerosis.
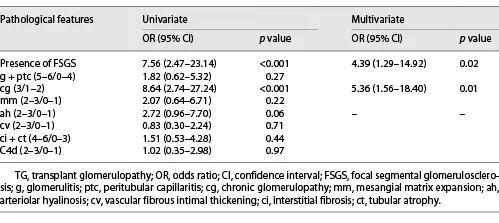
Histological Lesions Associated with Proteinuria
Table 5 shows the associations between histological lesions and proteinuria. The Banff cg score (p < 0.001) and presence of FSGS (p < 0.001) were significantly associated with proteinuria by univariate analysis. In the multivariate logistic regression model, cg (odds ratio = 5.36, 95% CI: 1.56–18.40, p = 0.01) and FSGS (odds ratio = 4.39, 95% CI: 1.29–14.92, p = 0.02) were still independent risk factors for proteinuria (>1 g/24 h) in TG.
Discussion
In renal allografts, TG is a common glomerular lesion rather than a specific entity. This is a morphological study including TG of all potential etiologies. We explored the clinicopathological characteristics and allograft outcomes of TG with FSGS and without FSGS and found that the presence of FSGS was significantly associated with allograft loss in patients with TG.
The clinical manifestations in the entire group of TG patients were similar to those in prior studies (median proteinuria and serum creatinine levels, 1.6–1.9 g/24 h and 2.0–2.3 mg/dL, respectively) [-]. However, previous studies on TG have paid little attention to concomitant FSGS. Our study showed that TG patients with FSGS had higher levels of proteinuria and serum creatinine and a greater proportion of elevated tubular injury markers than those without FSGS. In a previous study of chronic allograft nephropathy (that was eliminated from the Banff classification at the 8th Banff Conference in 2005 and replaced by “interstitial fibrosis and tubular atrophy without evidence of any specific etiology”), Cosio et al. [] found that FSGS was more common in patients with high-grade proteinuria, elevated serum cholesterol, and developed later post-transplantation than patients without FSGS. The levels of proteinuria and serum creatinine and the time from transplantation to biopsy diagnosis of FSGS in our study were similar to those in Cosio’s study. However, compared with recurrent FSGS which usually occurs within a few days or months post-transplantation and presents with nephrotic-range proteinuria [-], TG patients with FSGS were diagnosed much later after kidney transplantation and had relatively less proteinuria.
In a prior study, Agustian et al. [] found that the mean podocyte number per glomerulus was significantly lower in nephrectomy specimens with advanced TG (cg3) than in zero-hour biopsies and regular transplant biopsies without TG. In our study, TG patients with FSGS had more severe GBM duplication (cg3) than patients without FSGS. These results highly suggested that advanced TG may promote the development of FSGS. Prior studies have shown the association between cg and proteinuria [, ]. In our study, not only cg, but also FSGS was an independent risk factor for proteinuria. In other words, the presence of FSGS could aggravate proteinuria in patients with TG. Podocyte injury plays a key role in the development of proteinuria in native kidney diseases. In renal allograft biopsy, changes in podocyte number or density associated with proteinuria have also been reported. Pippin et al. [] found that decreased podocyte number and decreased expression of slit diaphragm proteins (nephrin and synaptopodin) were associated with proteinuria and glomerulosclerosis in a rat model of chronic allograft nephropathy. Yang et al. [] demonstrated that TG patients had 10- to 20-fold increased urine podocin/creatinine ratios and decreased glomerular podocyte densities, and this was strongly correlated with proteinuria, reduced eGFR, and glomerulosclerosis. Although TG is initiated with endothelial cell injury, podocyte injury and resulting FSGS may enhance proteinuria and further impair graft function.
However, the mechanisms underlying podocyte injury and how it further leads to FSGS in TG remain unclear. Agustian et al. [] indicated that disruption of antiapoptotic HGF/Met signaling from endothelial cells to podocytes is a possible mechanism for podocyte loss and FSGS in patients with TG. Yang et al. [] examined podocyte density and related glomerular parameters in serial renal allograft biopsies and found that a reduction in podocyte number per glomerulus was similar in patients with early TG and late TG, but the glomerular volume was significantly increased in late TG. It is suggested that nonimmune mechanisms may be involved in podocyte injury and detachment in late TG. Glomerular enlargement exceeds the capacity of podocytes to adapt, leading to podocyte stress, injury, and ultimately detachment with resulting FSGS [, ]. Our study showed that the glomerular tuft area was enlarged, podocytes were decreased, and the incidence of diffuse FPE (roughly two-thirds had FPE 50%–60%) was increased in TG patients with FSGS, supporting the hypothesis that maladaptive FSGS plays a part in TG. However, whether immune mechanisms are involved in this remains unclear. Haas [] suggested that antibody-mediated active immune mechanisms are largely responsible for podocyte injury and depletion in early TG. Bhargava et al. [] recently demonstrated that N-glycosylated IgG in patients with TG upregulates calcium/calmodulin kinase IV, which reduces the expression of nephrin by inhibiting GSK3β, modulates podocyte migration, and remodels the actin cytoskeleton, thus leading to podocyte injury. Since the renal graft is affected by many complicated factors after transplantation, the specific etiology and mechanism of FSGS caused by podocyte injury in TG need further study.
Renal graft survival in patients with TG was poor, manifested by a median graft survival time (3.25 years) less than that in patients without TG (18.82 years) []. Aubert et al. [] identified 5 archetypes of TG with distinct clinical, functional, immunological, and histological features, using an unsupervised learning method, and they found that the 5-year graft survival rates in different TG archetypes ranged from 88% to 22%, and the worst archetype was characterized by the highest proteinuria level, the highest mean cg score, and high chronic lesions. This suggests that the prognosis of patients with TG is highly heterogeneous. Our study showed that TG patients with FSGS had worse allograft survival than those without FSGS. The presence of FSGS was significantly associated with allograft loss in TG, independent of other clinicopathological features except for proteinuria, eGFR, and PRA. Therefore, individualized treatment strategies and close follow-up monitoring should be adopted for patients with FSGS to improve allograft outcomes. Previous studies [, , ] have indicated that proteinuria and impaired renal graft function at biopsy were independent risk factors for allograft loss in TG. Relative histopathological risk factors including interstitial fibrosis [, ], chronic score [, ], cg [], and glomerular C3 deposition [] have also been reported but are not seen in our study, probably due to the limited sample size and to the differences in follow-up time.
The limitations of this study must be recognized. First, this was a retrospective study, and selection bias of patients who underwent renal allograft biopsy could not be avoided. Second, donor-specific antibody and pretransplant HLA mismatch data were not available, and various treatment modalities were used in the patients of this study, preventing us from analyzing the group differences in these features and their impact on graft survival. Finally, the majority of native kidney biopsies and donor kidney biopsies of these patients were not available.
In summary, the clinicopathological findings of TG patients with FSGS were more severe than those of TG patients without FSGS. The presence of FSGS was independently associated with allograft loss in patients with TG. Substantial emphasis should be placed on the development of FSGS in TG, and the specific mechanisms deserve further study.
Acknowledgments
We thank all the participants of this study.
Statement of Ethics
This single-center retrospective study was carried out with the approval of the Ethics Committee of the Jinling Hospital (2017NZKYKS-002-01) and conducted ethically in accordance with the World Medical Association Declaration of Helsinki.
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
Funding Sources
This research was funded by the National Key Research and Development Program of China (2016YFC0901202), the National Natural Science Foundation of China (82070793), and the Project of Invigorating Health Care through Science, Technology and Education of Jiangsu Province Medical Key Talent (ZDRCA2016098).
Author Contributions
C.Z. and Y.Z. conceived and designed the study; C.Z., Y.Z., Y.F., F.X., S.L., D.L., and P.L. performed acquisition, analysis, and interpretation of the data; Y.X., X.Z., F.Y., and J.C. participated in analysis and interpretation of the data; Y.Z. drafted the manuscript; C.Z. revised the manuscript. All the authors approved the final version of the manuscript.
Data Availability Statement
The data that support the findings of this study are not publicly available due to their containing information that could compromise the privacy of research participants but are available from the corresponding author upon reasonable request.
References
- 1. Husain S, Sis B. Advances in the understanding of transplant glomerulopathy. Am J Kidney Dis. 2013 Aug;62(2):352–63. http://dx.doi.org/10.1053/j.ajkd.2012.10.026.
- 2. Remport A, Ivanyi B, Mathe Z, Tinckam K, Mucsi I, Molnar MZ. Better understanding of transplant glomerulopathy secondary to chronic antibody-mediated rejection. Nephrol Dial Transplant. 2015 Nov;30(11):1825–33. http://dx.doi.org/10.1093/ndt/gfu371.
- 3. Filippone EJ, McCue PA, Farber JL. Transplant glomerulopathy. Mod Pathol. 2018 Feb;31(2):235–52. http://dx.doi.org/10.1038/modpathol.2017.123.
- 4. Agustian PA, Schiffer M, Gwinner W, Schäfer I, Theophile K, Modde F, et al. Diminished met signaling in podocytes contributes to the development of podocytopenia in transplant glomerulopathy. Am J Pathol. 2011 May;178(5):2007–19. http://dx.doi.org/10.1016/j.ajpath.2011.01.042.
- 5. Yang Y, Hodgin JB, Afshinnia F, Wang SQ, Wickman L, Chowdhury M, et al. The two kidney to one kidney transition and transplant glomerulopathy: a podocyte perspective. J Am Soc Nephrol. 2015 Jun;26(6):1450–65. http://dx.doi.org/10.1681/ASN.2014030287.
- 6. Bhargava R, Maeda K, Tsokos MG, Pavlakis M, Stillman IE, Tsokos GC. N-glycosylated IgG in patients with kidney transplants increases calcium/calmodulin kinase IV in podocytes and causes injury. Am J Transplant. 2021 Jan;21(1):148–60. http://dx.doi.org/10.1111/ajt.16140.
- 7. De Vriese AS, Sethi S, Nath KA, Glassock RJ, Fervenza FC. Differentiating primary, genetic, and secondary FSGS in adults: a clinicopathologic approach. J Am Soc Nephrol. 2018 Mar;29(3):759–74. http://dx.doi.org/10.1681/ASN.2017090958.
- 8. Rosenberg AZ, Kopp JB. Focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2017 Mar 7;12(3):502–17. http://dx.doi.org/10.2215/CJN.05960616.
- 9. Haas M, Loupy A, Lefaucheur C, Roufosse C, Glotz D, Seron D, et al. The Banff 2017 kidney meeting report: revised diagnostic criteria for chronic active T cell-mediated rejection, antibody-mediated rejection, and prospects for integrative endpoints for next-generation clinical trials. Am J Transplant. 2018 Feb;18(2):293–307. http://dx.doi.org/10.1111/ajt.14625.
- 10. Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009 May 5;150(9):604–12. http://dx.doi.org/10.7326/0003-4819-150-9-200905050-00006.
- 11. D’Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis. 2004 Feb;43(2):368–82. http://dx.doi.org/10.1053/j.ajkd.2003.10.024.
- 12. Zeng C, Nan Y, Xu F, Lei Q, Li F, Chen T, et al. Identification of glomerular lesions and intrinsic glomerular cell types in kidney diseases via deep learning. J Pathol. 2020 Sep;252(1):53–64. http://dx.doi.org/10.1002/path.5491.
- 13.
- 14. Li X, Chen J, Cheng D, Wang R, Wang W, Zhang M, et al. Proteinuria, estimated glomerular filtration rate and urinary retinol-binding protein as clinical predictors of long-term allograft outcomes in transplant glomerulopathy. Kidney Blood Press Res. 2018;43(6):1842–51. http://dx.doi.org/10.1159/000495816.
- 15. Panzer SE, Joachim E, Parajuli S, Zhong W, Astor BC, Djamali A. Glomerular C3 deposition is an independent risk factor for allograft failure in kidney transplant recipients with transplant glomerulopathy. Kidney Int Rep. 2019;4(4):582–93. http://dx.doi.org/10.1016/j.ekir.2019.01.018.
- 16. Cosio FG, Frankel WL, Pelletier RP, Pesavento TE, Henry ML, Ferguson RM. Focal segmental glomerulosclerosis in renal allografts with chronic nephropathy: implications for graft survival. Am J Kidney Dis. 1999 Oct;34(4):731–8. http://dx.doi.org/10.1016/S0272-6386(99)70400-2.
- 17. Moroni G, Gallelli B, Quaglini S, Banfi G, Montagnino G, Messa P. Long-term outcome of renal transplantation in adults with focal segmental glomerulosclerosis. Transpl Int. 2010 Feb;23(2):208–16. http://dx.doi.org/10.1111/j.1432-2277.2009.00977.x.
- 18. Garrouste C, Canaud G, Büchler M, Rivalan J, Colosio C, Martinez F, et al. Rituximab for recurrence of primary focal segmental glomerulosclerosis after kidney transplantation: clinical outcomes. Transplantation. 2017 Mar;101(3):649–56. http://dx.doi.org/10.1097/TP.0000000000001160.
- 19. Allard L, Kwon T, Krid S, Bacchetta J, Garnier A, Novo R, et al. Treatment by immunoadsorption for recurrent focal segmental glomerulosclerosis after paediatric kidney transplantation: a multicentre French cohort. Nephrol Dial Transplant. 2018 Jun 1;33(6):954–63. http://dx.doi.org/10.1093/ndt/gfx214.
- 20. Mansur JB, Sandes-Freitas TV, Kirsztajn GM, Cristelli MP, Mata GF, de Paula MI, et al. Clinical features and outcomes of kidney transplant recipients with focal segmental glomerulosclerosis recurrence. Nephrology (Carlton). 2019 Nov;24(11):1179–88. http://dx.doi.org/10.1111/nep.13589.
- 21. Toki D, Inui M, Ishida H, Okumi M, Shimizu T, Shirakawa H, et al. Interstitial fibrosis is the critical determinant of impaired renal function in transplant glomerulopathy. Nephrology (Carlton). 2016 Jul;21(Suppl 1):20–5. http://dx.doi.org/10.1111/nep.12765.
- 22. Li X, Chen J, Cheng D, Wang W, Xie K, Zhang M, et al. Histopathologic features that predict transplant glomerulopathy progression in a Chinese cohort. Am J Nephrol. 2019;49(6):425–34. http://dx.doi.org/10.1159/000500043.
- 23. Pippin J, Kumar V, Stein A, Jablonski P, Shankland SJ, Davis CL. The contribution of podocytes to chronic allograft nephropathy. Nephron Exp Nephrol. 2009;111(1):e1–10. http://dx.doi.org/10.1159/000178762.
- 24. Haas M. Transplant glomerulopathy: the view from the other side of the basement membrane. J Am Soc Nephrol. 2015 Jun;26(6):1235–7. http://dx.doi.org/10.1681/ASN.2014090945.
- 25. Nishizono R, Kikuchi M, Wang SQ, Chowdhury M, Nair V, Hartman J, et al. FSGS as an adaptive response to growth-induced podocyte stress. J Am Soc Nephrol. 2017 Oct;28(10):2931–45. http://dx.doi.org/10.1681/ASN.2017020174.
- 26. Kovacs G, Devercelli G, Zelei T, Hirji I, Voko Z, Keown PA. Association between transplant glomerulopathy and graft outcomes following kidney transplantation: a meta-analysis. PLoS One. 2020;15(4):e0231646.
- 27. Aubert O, Higgins S, Bouatou Y, Yoo D, Raynaud M, Viglietti D, et al. Archetype analysis identifies distinct profiles in renal transplant recipients with transplant glomerulopathy associated with allograft survival. J Am Soc Nephrol. 2019 Apr;30(4):625–39. http://dx.doi.org/10.1681/ASN.2018070777.
- 28. Patri P, Seshan SV, Matignon M, Desvaux D, Lee JR, Lee J, et al. Development and validation of a prognostic index for allograft outcome in kidney recipients with transplant glomerulopathy. Kidney Int. 2016 Feb;89(2):450–8. http://dx.doi.org/10.1038/ki.2015.288.
- 29. Zhang Q, Rudolph B, Choi M, Bachmann F, Schmidt D, Duerr M, et al. The relationship between proteinuria and allograft survival in patients with transplant glomerulopathy: a Retrospective Single-Center Cohort Study. Transpl Int. 2021 Nov 17;34(2):259–71. http://dx.doi.org/10.1111/tri.13787.
- 30. John R, Konvalinka A, Tobar A, Kim SJ, Reich HN, Herzenberg AM. Determinants of long-term graft outcome in transplant glomerulopathy. Transplantation. 2010 Oct 15;90(7):757–64. http://dx.doi.org/10.1097/TP.0b013e3181efcffd.