Introduction
Patients with diabetes have significant risk of mortality and developing both macrovascular (e.g., coronary and peripheral artery disease, and stroke) and microvascular (e.g., retinopathy, nephropathy, and neuropathy) complications due to unresolved metabolic, cellular, and inflammatory events []. Diabetes is the main cause of chronic kidney disease (CKD) (30–50% of all cases) [], which develops in approximately 40% of patients with diabetes []. The diagnosis of CKD depends on identifiable abnormalities in kidney structure or function present for more than 3 months, with associated symptomology []. An estimated glomerular filtration rate (eGFR) of less than 60 mL/min/1.73 m2 and/or a urine albumin:creatinine ratio (UACR) of ≥30 mg/g is diagnostic of CKD [] and diabetic kidney disease (DKD) []. Albuminuria is a predictor of cardiovascular outcomes and all-cause mortality in various populations [], including those with DKD [].
Although DKD is usually classified as a noninflammatory glomerular disease, the significant contribution of inflammation in its pathogenesis is recognized []. The three main driving forces in the initiation and progression of DKD are hemodynamic (systemic hypertension, glomerular hyperfiltration, glomerular hypertension and hypertrophy, and activation of the renin-angiotensin-aldosterone system [RAAS] and sympathetic nervous system), metabolic (hyperglycemia, formation of advanced glycation products, insulin resistance, and obesity), and inflammatory/fibrotic (increases in several proinflammatory and profibrotic cytokines, transforming growth factor β [TGF-β], tumor necrosis factor α, metalloproteinases, reactive oxygen species [ROS], angiotensin II, aldosterone, mineralocorticoid receptor [MR] activity, and the Rho family small guanosine triphosphatase, Ras-related C3 botulinum toxin substrate 1 [Rac1]) [, ].
Fibrosis is the final common pathway of kidney disease progression, regardless of etiology, and is initiated by epithelial trauma (due to toxins), inflammation (e.g., from acute interstitial nephritis), or direct epithelial injury, as seen with albuminuria in diabetes [, ]. Once kidney disease is established, irrespective of its cause, progression may continue even after the initial insult is resolved [, ]. Increasing evidence suggests a multidirectional interaction and upregulation between aldosterone, the MR, and Rac1, as driving forces in the onset of chronic interstitial nephritis and progression to fibrosis in CKD and DKD. In this review, we discuss the modes of action of aldosterone, the MR, and Rac1, and how their intricate relationship leads to kidney disease progression. We also discuss the clinical implications of MR inhibition as one approach to attenuate DKD progression.
The Aldosterone-MR-Rac1 Triangle
Aldosterone
Aldosterone is a mineralocorticoid belonging to the group of lipophilic steroid hormones that include sex hormones (i.e., testosterone, estrogen, and progesterone) and glucocorticoids []. It is synthesized primarily in the zona glomerulosa of the adrenal gland via aldosterone synthase in response to elevated K+ levels, volume depletion, and angiotensin II as part of the RAAS, regulating sodium and K+ homeostasis in the distal renal tubule and collecting duct. In addition to the kidneys, aldosterone can also act on other tissues such as the myocardium, blood vessels, adipose tissue, and macrophages [].
Expression of aldosterone is not limited to the adrenal gland or activation of the RAAS. In CKD and diabetes, aldosterone can be generated intrarenally, along with the local production of all other components of the RAAS, including angiotensinogen at the proximal tubules [], and renin release by the macula densa, as seen with hyperglycemia-induced mitochondrial succinate production acting on the GPR91 receptor []. Aldosterone is also ectopically produced by endothelial and smooth muscle cells, by neurons and glial cells, and by adipocytes []. In diabetes-associated obesity, aldosterone produced by adipocytes, also via aldosterone synthase, is linked to vascular dysfunction via MR-mediated actions [].
The classic, genomic, MR-mediated action of aldosterone on the distal tubules and collecting duct is to upregulate and increase the number of epithelial sodium channels located on the apical membrane []. These channels are also found on the endothelium glycocalyx, vascular smooth muscle, the choroid plexus, and immune cells, and respond to vasopressin, angiotensin II, Rac1, and insulin [].
Rapid, nongenomic, MR-mediated effects of aldosterone increase cytosolic calcium, produce ROS by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox) in mitochondria, and initiate inflammatory pathways via nuclear factor ĸB and serum- and glucocorticoid-inducible protein kinase 1 (SGK1), resulting in inflammation and fibrosis [, ]. In the kidney, expression of aldosterone has profibrotic effects and stimulates plasminogen activator inhibitor-1 via MRs in podocytes, macrophages, mesangial and tubular epithelial cells. It acts synergistically with TGF-β by promoting fibroblast formation and fibrosis [], as well as activating the nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome via ROS. This further promotes podocyte apoptosis and tubular injury [].
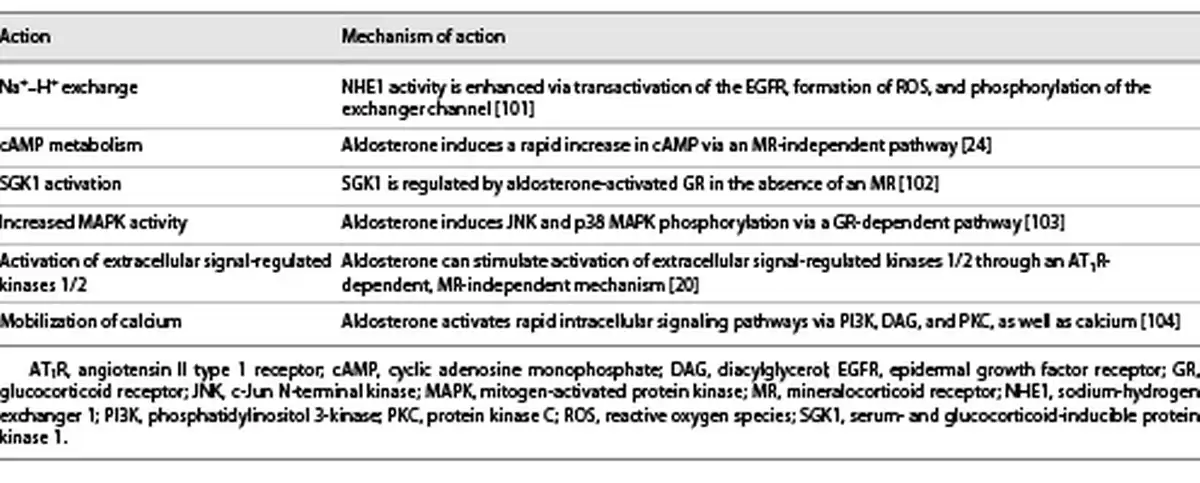
Through its MR-independent action [], aldosterone stimulates pathways involved in the development of DKD, such as cyclic adenosine monophosphate production [], and activates protein kinase C α/β [] and the mitogen-activated protein kinases (Table 1) []. Importantly, aldosterone can mobilize the extracellular signal-regulated kinases 1/2 through an angiotensin II type 1 receptor-dependent, MR-independent mechanism to further promote a mitogenic and profibrotic phenotype (Table 1) []. Recently, fibroblast growth factor 23 has been found to be elevated in CKD and appears to be directly stimulated by aldosterone, as well as involved in the activation of aldosterone and the RAAS. This leads to further fibroblast growth factor 23 elevation and a dysregulated phosphate metabolism with a risk of cardiac fibrosis and hypertrophy, as well as CKD progression [].
The MR
The MR is located intracellularly and found in many tissues, including endothelial cells, vascular smooth muscle cells, adipocytes, fibroblasts, cardiomyocytes, and macrophages [, ]. In the kidney, the MR localizes to the distal tubules, collecting duct, podocytes, fibroblasts, and mesangial cells []. Once aldosterone binds to the MR, the new complex attaches to hormone response elements in the DNA, resulting in target gene transcription [, ]. Although cortisol and other ligands also recognize the receptor with similar affinity, aldosterone is the principal steroid for MR activation [].
Upregulation of the MR is seen in multiple clinical conditions including hyperglycemia, obesity, insulin resistance, high salt intake, and CKD, as outlined in more detail in Table 2. This upregulation causes increased transcription of profibrotic genes (e.g., TGF-β1, plasminogen activator inhibitor-1, connective tissue growth factor, and extracellular matrix proteins [collagens, fibronectin]), all of which are linked to renal and cardiac fibrosis (Fig. 1) []. Moreover, high serum aldosterone levels are associated with a greater risk of renal failure in patients with and without diabetes []. Accumulating evidence suggests that MR activation is associated with podocyte injury via i) Rac1 [], ii) reducing autophagy necessary for podocyte maintenance [], and iii) increased Nox activity, leading to oxidative stress with upregulation of a cascade of proinflammatory cytokines and profibrotic proteins []. The subsequent development of albuminuria (even without hypertension), reduced renal blood flow, and acute kidney injury result in the progression of chronic interstitial inflammation and fibrosis in the kidney (Table 2) [, ]. Therefore, MR antagonists (MRAs) may well delay CKD progression, regardless of etiology [].
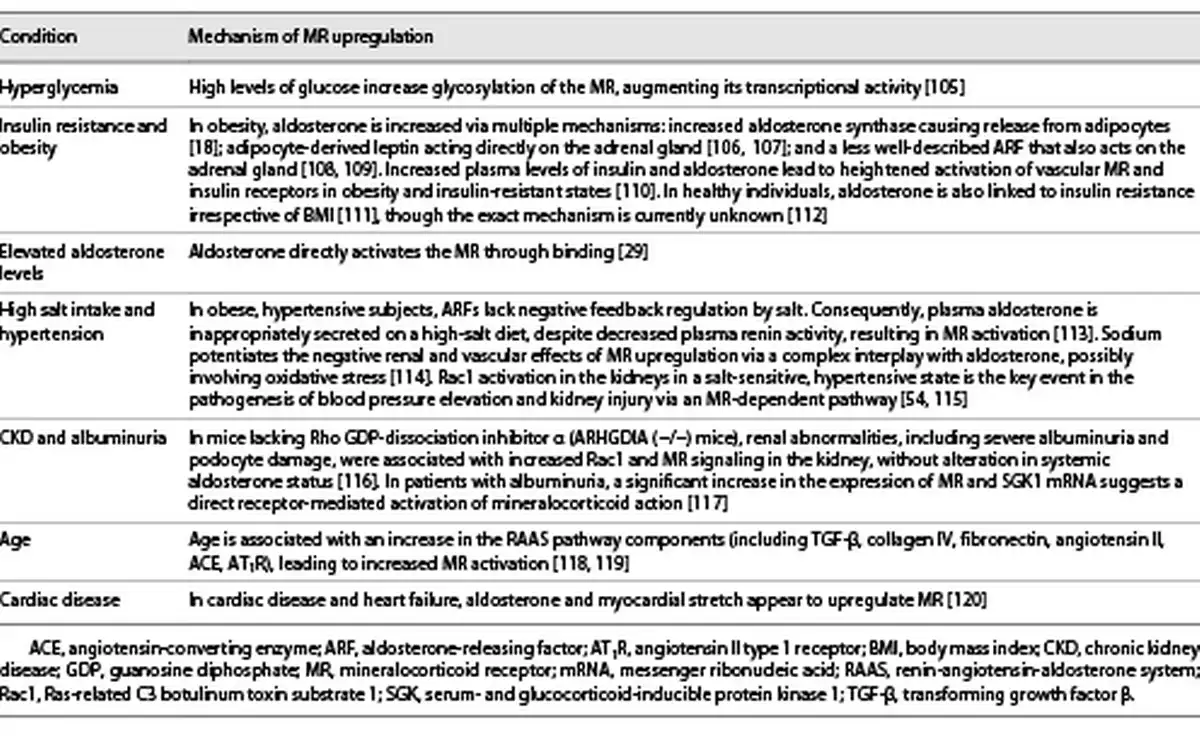
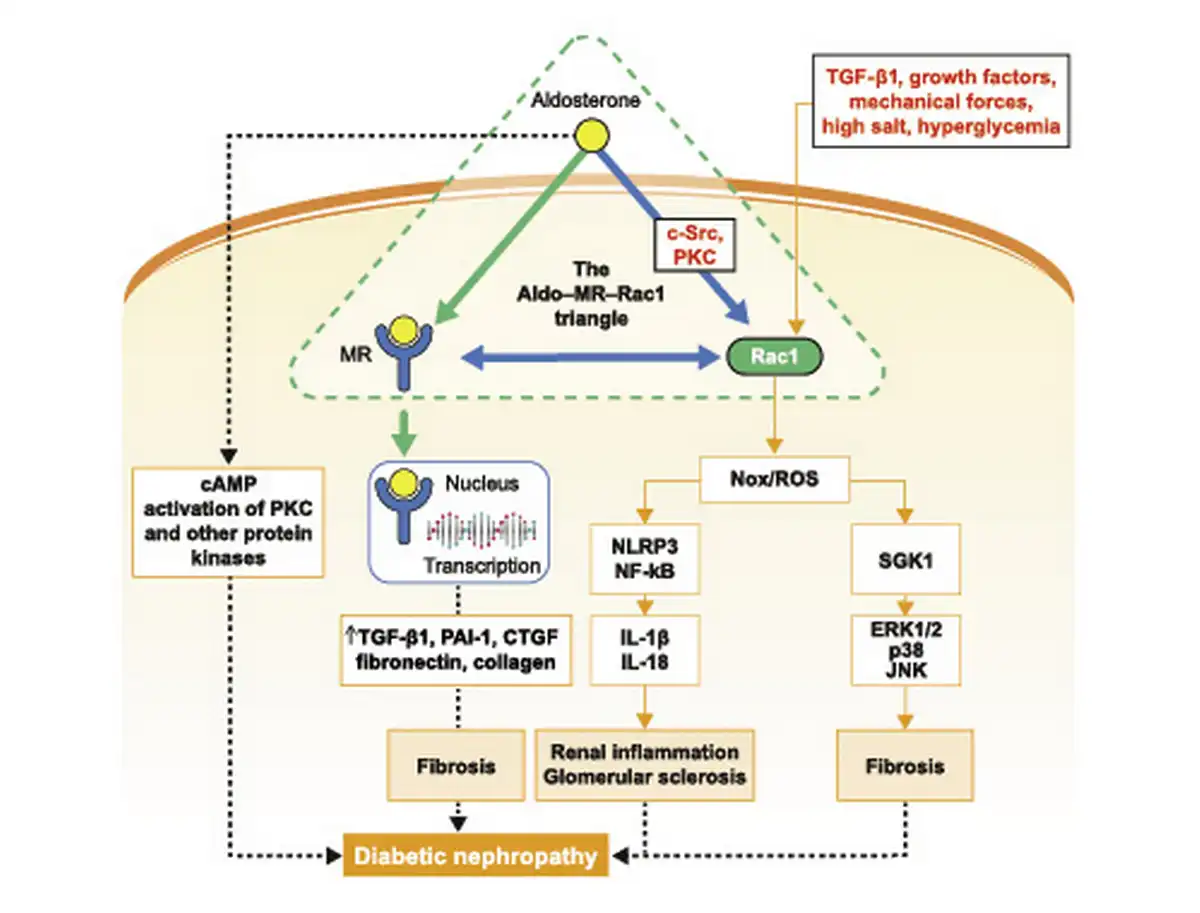
Fig. 1
A model signaling pathway highlighting Aldo, MR, and Rac1 crosstalk in the progression of DKD. Aldo, MR, and Rac1 function in a triangle, with bidirectional interconnection, and are the main forces behind the initiation and progression of chronic interstitial nephritis to fibrosis. Rac/Nox/ROS- and MR-dependent signaling pathways, downstream of the aldosterone-MR-Rac1 triad, stimulate ERK1/2, p38, JNK, PKC, NF-κB, and NLRP3 activity to promote production of inflammatory cytokines (e.g., IL-1β, IL-18), fibrotic factors (such as PAI-1 and CTGF), and extracellular matrix (fibronectin and collagen) accumulation, culminating in glomerular and tubular injury, interstitial fibrosis, and DKD progression. Aldo, aldosterone; cAMP, cyclic adenosine monophosphate; CTGF, connective tissue growth factor; DKD, diabetic kidney disease; ERK1/2, extracellular signal-regulated kinase 1/2; IL, interleukin; JNK, c-Jun N-terminal kinase; MR, mineralocorticoid receptor; NF-κB, nuclear factor κB; NLRP3, NOD-like receptor family pyrin domain-containing 3; Nox, nicotinamide adenine dinucleotide phosphate oxidase; PAI-1, plasminogen activator inhibitor-1; PKC, protein kinase C; Rac1, Ras-related C3 botulinum toxin substrate 1; ROS, reactive oxygen species; SGK1, serum- and glucocorticoid-inducible protein kinase 1; TGF-β1, transforming growth factor β1.
Rac1
The Rho family of small guanosine triphosphatases, which include RhoA, Rac1, and Cdc42, are key effectors of normal kidney function and important regulators of structure and function in mesangial cells, podocytes, and the tubular epithelium []. Rac1, an intracellular switch and second messenger, has important physiologic functions involving vesicular trafficking, including glucose-stimulated insulin secretion [] and the membrane translocation of glucose transporter GLUT4 []. GTP-activated Rac1 is an essential partner of the Nox1/2 holoenzyme system and mediates glucose-stimulated Nox-dependent production of ROS [-]. Increased oxidative stress is a major contributor to the development of organ fibrosis, including the kidney []. Increased expression of various components of NADPH oxidases is evident during diabetic renal injury, and inhibition of Nox4 attenuates DKD progression [, ]. Rac1 also coordinates tissue inflammatory and fibrotic responses by activating a complex network of multiple kinases and cellular signaling effectors [].
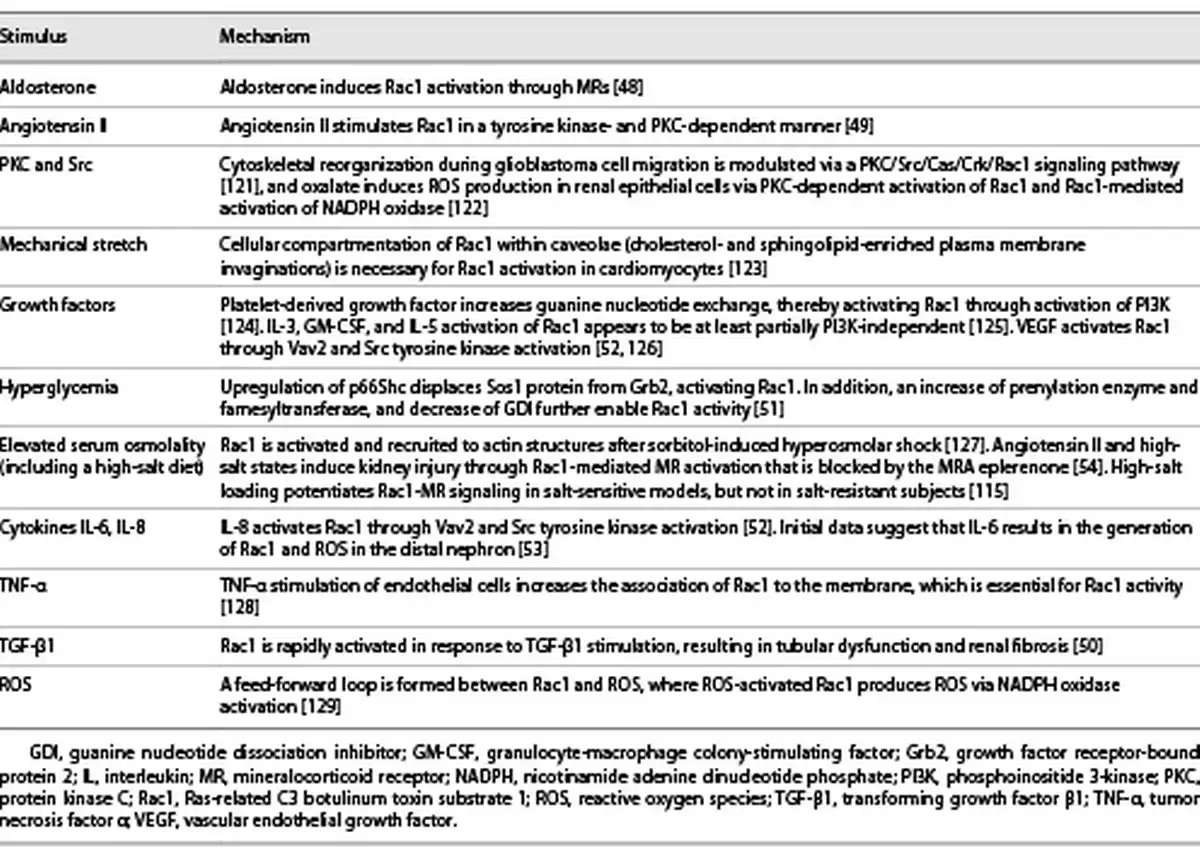
Rac1 can be activated by GTP loading or upregulated by several mechanisms, including aldosterone [], angiotensin II [], TGF-β [], high glucose [], and interleukin (IL)-6/8 stimulation [, ] (Table 3). High glucose levels in diabetic nephropathy mobilize the Rho pathway in mesangial cells, and Rac1 appears crucial for insulin signaling in muscle glucose uptake, implicating Rac1 in human insulin resistance and dysglycemia []. Pharmacological inhibition of Rac1 attenuates endothelial dysfunction in experimental models of diabetes and reduced platelet hyperaggregation in patients with diabetes []. Furthermore, glucose-induced Rac1 stimulation contributed to renal MR activation in cultured mesangial cells, resulting in their apoptosis [] and eventual nephropathy in an animal model of obesity-related type 2 diabetes (T2D) []. Rac1 is also rapidly activated in response to TGF-β, leading to profibrotic action of this cytokine (Table 3) []. In diabetic kidney injury, TGF-β1 activation and engagement of the Rac1 pathway appear to be critical elements in mesangial/podocyte dysfunction and eventual glomerular scarring []. Experimentally, Rac1 upregulation via transient receptor potential cation channels can cause direct podocyte injury and detachment without coaction of aldosterone or MR, leading to glomerulosclerosis [], posing a risk for interstitial inflammation and fibrosis in the kidney []. As urinary Rac1 was found to be elevated in focal segmental glomerular sclerosis and DKD, it may also represent a new biomarker (and target) for therapy [, ].
Aldosterone, MR Activation, and Rac1-Induced Inflammation and Fibrosis
The chronic inflammation induced by aldosterone, the MR, and Rac1, and its progression to glomerular and interstitial fibrosis, is a complex process. Aldosterone and/or the MR start the inflammatory cycle with the generation of ROS by NADPH in the mitochondria, potentiated by Rac1 (Fig. 1; Tables 1, 3) []. Aldosterone also increases expression and activation of SGK1, which can lead to renal fibrosis []. A similar positive correlation between SGK1 and Rac1 in the renal cortex links SGK1 with Rac1-MR and progression of DKD and hypertension (shown in Fig. 1) [].
Both Rac1 and aldosterone promote fibrosis through the action of inflammasomes. These are multiprotein complexes that activate intracellular caspase-1 with the release of cytokines (e.g., interleukin [IL]-1β, IL-18), regulating inflammation, cytokine maturity, and cell death []. These complexes are formed, in part, from pattern recognition receptors, such as the nucleotide-binding oligomerization domain-like receptor family that sense intracellular pathogens, damage-associated molecular patterns, or toxic signals, including mitochondrial ROS. A well-described inflammasome is NLRP3, the expression of which is increased in the glomeruli of patients with DKD, correlating with the degree of albuminuria []. Within podocytes, aldosterone, via an MR-mediated mechanism, and Rac1 activate the NLRP3 inflammasome resulting in podocyte injury and glomerular sclerosis [] (Fig. 1).
Aldosterone and MR activation promotes polarization of macrophages toward a proinflammatory M1 phenotype, a process inhibited by MR blockade []. In macrophages, aldosterone activates inflammasomes, evoking inflammatory responses and renal interstitial fibrosis by accelerating macrophage infiltration in the tubulointerstitial region []. Macrophages are also major producers of renal TGF-β, which plays a central role in inflammation and renal fibrosis []. TGF-β-driven renal fibrosis appears to be mediated via Rac1, which acts as a major redox-dependent non-SMAD (i.e., noncanonical) control element [, ].
MRAs in CKD and DKD
The MR is a key element of the aldosterone-MR-Rac1 triangle, and studies have highlighted the benefits of MRAs in preventing or reducing aldosterone- or Rac1-related effects. From a clinical perspective, systematic reviews indicate that MRAs may offer cardiorenal protection in a variety of populations [-]. A recent Cochrane review evaluated the effect of MRAs, alone or in combination with angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) []. Based on 44 studies involving 5,745 adults with CKD with proteinuria (23 studies included participants with DKD), the authors concluded that aldosterone antagonists, in addition to an ACE inhibitor or ARB (or both), may reduce proteinuria, eGFR, and systolic blood pressure []. However, there was an increased risk of hyperkalemia and incidence of gynecomastia, and the data were insufficient to conclude on the effect on outcomes, such as progression to kidney failure, major cardiovascular events, and death [].
Steroidal MRAs
The first identified and approved MRA was spironolactone, a nonselective steroidal MRA used to treat primary aldosteronism, hypertension, edema due to heart failure, and liver cirrhosis, preceding the discovery of the MR by 27 years []. Spironolactone is also a relatively nonselective progesterone receptor agonist and testosterone antagonist, associated with gynecomastia, dysmenorrhea, impotence, and hyperkalemia []. The addition of spironolactone to ACE inhibitor treatment in a small study cohort of 21 patients with DKD reduced albuminuria by 33%, albumin clearance by 50%, and reduced blood pressure significantly []. The first clinical trial that evaluated the aldosterone blockade in combination with an ARB-based regiment in 136 patients with DKD found that treatment with spironolactone significantly reduced systolic and diastolic blood pressure, and urinary albumin excretion after 18 months. Asymptomatic hyperkalemia occurred in 3.6% of patients []. The Randomized Aldactone Evaluation Study demonstrated significantly improved outcomes with spironolactone treatment in patients with severe heart failure, but also occurrence of severe hyperkalemia. This prompted a follow-up study that reported a correlation between spironolactone treatment and increased hyperkalemia-associated morbidity and mortality, warranting caution when treating with spironolactone []. However, a judicious selection of patients with mild to moderate CKD with a serum K+ <5.5 mmol/L and no history of hyperkalemia (n = 117) resulted in low rates of serious hyperkalemia (<1%) and worsening renal function (<3%) when treating with low-dose spironolactone [].
In 2002, eplerenone, a second-generation selective steroidal MRA, was approved and found to be more selective in its MRA effect, devoid of androgen antagonism and progesterone agonism, but still with a risk of hyperkalemia []. Eplerenone reduced albuminuria when used in combination with an ACE inhibitor (enalapril or ramipril) in the two studies available to date that specifically focused on patients with DKD [, ]. Doses of 50–100 mg eplerenone added to enalapril resulted in significant reduction in albuminuria from baseline (UACR reduction of 41% in the 50 mg group and 48.4% in the 100 mg group) []. A single-blind, randomized clinical trial of 75 hypertensive patients with T2D and microalbuminuria on either monotherapy or a combination therapy of eplerenone and ramipril reported a significant reduction of UACR after 24 weeks for the monotherapy groups, and an even greater reduction for the combination therapy group. Incidence of hyperkalemia occurred in 4% of both monotherapy groups and in 8% of the eplerenone and ramipril combination therapy group []. A safety study of eplerenone in relation to the adverse event, hyperkalemia, in 31 kidney-transplant recipients with impaired renal function on cyclosporine A showed that eplerenone could be safely given to these patients if K+ levels were closely monitored []. To date, no phase 3 clinical trials have been performed with steroidal MRAs in the DKD indication.
Nonsteroidal MRAs
In the last decade, first-in-class nonsteroidal MRAs were developed, although only one is currently approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA). Finerenone, a selective, nonsteroidal dihydropyridine MRA that differs significantly from aldosterone [-] is indicated for the treatment of CKD associated with T2D [], and to reduce the risk of sustained eGFR decline, end-stage kidney disease, cardiovascular death, nonfatal myocardial infarction, and hospitalization for heart failure in this population []. The first clinical studies (ARTS-DN and ARTS-DN Japan) investigating the effects of finerenone in patients with albuminuric DKD reported that finerenone treatment reduced UACR up to 38% compared with placebo, with low discontinuation rates owing to hyperkalemia (K+ >5.6 mmol/L) (1.8% of patients in the ARTS-DN) [, ]. Renal outcomes of finerenone in patients with T2D and albuminuric CKD in combination with ACE inhibitor or ARB therapy were investigated in the FIDELIO-DKD study (n = 5,734). After a mean follow-up period of 2.6 years, finerenone reduced the risk of the primary composite endpoint (time to first occurrence of kidney failure, sustained reduction in eGFR of ≥40%, or death from renal causes) by 18% (p = 0.001), the risk of the secondary endpoint (major adverse cardiovascular events) by 14%, and albuminuria at 4 months by 31% from baseline compared with placebo []. Adverse events of finerenone versus placebo included hyperkalemia (18.3% vs. 9.0%), hypotension (4.8% vs. 3.4%), and hyponatremia (1.4% vs. 0.71%) []. Cardiovascular outcomes of patients with DKD on RAAS inhibitors were investigated in the FIGARO-DKD study (n = 7,352) []. After a mean follow-up of 3.4 years, the risk of the primary composite endpoint (time to first occurrence of death from cardiovascular causes, nonfatal myocardial infarction, nonfatal stroke, or hospitalization for heart failure) was lower in the finerenone group compared with placebo (HR, 0.87; p = 0.03) []. Moreover, finerenone was associated with fewer patients developing end-stage kidney disease compared with placebo (0.9% vs. 1.3%), a greater UACR reduction from baseline to month 4, and a lower rate of the composite outcome of a sustained decrease in eGFR of ≥57% from baseline, kidney failure, or death from renal causes (2.9% vs. 3.8%) []. Studies examining the role of finerenone in DKD indicate its therapeutic value in delaying the progression of DKD; moreover, it appears to have a better safety profile than other MRAs, with a low rate of drug discontinuation owing to hyperkalemia [].
Esaxerenone, another nonsteroidal MRA, is approved in Japan for treating hypertension []. A phase 2 clinical trial (n = 365) of esaxerenone reported that doses of 1.25, 2.5, and 5 mg/d esaxerenone significantly reduced UACR after 12 weeks of treatment (38%, 50%, and 56%, respectively) in patients with T2D and microalbuminuria []. The safety assessment of esaxerenone marked that serum K+ ≥6.0 mEq/L was similar in the 0.625 (3%), 1.25 (3%), and 2.5 (3%) mg/d treatment groups compared with placebo (1%), but was higher in the 5 mg/d (10%) group []. Moreover, the ESAX-DN phase 3 study (n = 449) investigated the effects of esaxerenone on UACR levels in patients with T2D and a UACR of 45 to <300 mg/g creatinine treated with RAAS inhibitors. Results showed that treatment of esaxerenone for up to 52 weeks was associated with a >30% reduction in UACR in ∼70% of patients; in 22% of patients, UACR returned to <30 mg/g creatinine compared with placebo. Furthermore, a smaller number of patients progressed to UACR ≥300 mg/g creatinine in the esaxerenone group (1%) compared with the placebo group (7%) []. Increased serum K+ levels leading to discontinuation of treatment during the phase 3 trial occurred in 4% of the patients [].
KBP-5074 is a new, currently in development, highly selective nonsteroidal MRA that is being evaluated for its blood pressure-lowering efficacy, safety, and pharmacokinetics in patients with CKD [, ]. The BLOCK-CKD phase 2b study reported a 7.0–12.0 mm Hg (p = 0.0026–0.0399) reduction in systolic blood pressure in patients (n = 162) with uncontrolled hypertension and stage 3b or 4 CKD. Hyperkalemia incidence (K+ ≥5.6 to <6 mmol/L) was similar among the treatment groups and the placebo group, while no hyperkalemia of serum K+ ≥6.0 mmol/L was observed []. A phase 3 randomized double-blind placebo controlled multicenter study is currently ongoing (https://clinicaltrials.gov/ct2/show/NCT04968184).
Several other MRAs are also being investigated. A phase 2 study of apararenone (n = 293) at concentrations of 2.5, 5, and 10 mg reported baseline UACR reduction of 62.9%, 50.8%, and 46.5%, respectively (all p < 0.001 vs. placebo), at week 24 in patients with stage 2 DKD. Even though serum K+ levels tended to increase, this was not clinically significant []. The MIRACLE study (https://clinicaltrials.gov/ct2/show/NCT04595370) is currently investigating the use of AZD9977, a nonsteroidal, selective MR modulator, in combination with dapagliflozin compared with dapagliflozin alone in patients with CKD and heart failure.
Clinical Implications for Use of MRAs in DKD
The anti-inflammatory and antifibrosis actions of MRAs address an important mechanism of DKD not specifically modulated by glucose-lowering medications such as sodium-glucose co-transporter 2 (SGLT2) inhibitors or glucagon-like peptide-1 receptor agonists. The residual mean loss of eGFR in DKD on maximally tolerated doses of ACE inhibitors (or ARBs) and SGLT2 inhibitors remains high (least-squares mean change in eGFR slope of −3.19 mL/min/1.73 m2/year; standard error, 0.15) []. A potential synergistic effect of combination therapy with SGLT2 inhibitors and MRAs in slowing the progression of kidney disease is suggested, owing to the mechanism of action of the two different classes of drugs []. The FIDELITY pooled analysis of the FIDELIO-DKD and FIGARO-DKD trials, where 6.7% of patients received an SGLT2 inhibitor and 7.2% a glucagon-like peptide-1 receptor agonist at baseline, showed that the cardiorenal benefits from finerenone remained independent of SGLT2 inhibitor use []. Triple therapy with an ACE inhibitor, an SGLT2 inhibitor, and an MRA, especially in patients with residual albuminuria of ≥300 mg/g, might have the potential to slow DKD progression, reduce albuminuria, and lower cardiovascular events. Further studies could help determine the patient population who would benefit most from this combination and delineate the additional reduction of yearly eGFR decline (as well as UACR reduction) when finerenone is added to an SGLT2 inhibitor. A study that will evaluate the combination of these agents is ongoing (https://clinicaltrials.gov/ct2/show/NCT05254002).
Conclusion
Recent advances highlight the aldosterone-MR-Rac1 triangle as a potential focus for novel therapies. The overactivity of each component of the triangle in diabetes, CKD, and cardiovascular disease, and its multidirectional capacity potentiate the risks toward a proinflammatory and profibrotic milieu, and disease progression. Although aldosterone has MR-independent actions that are involved in the development of DKD, and Rac1 can be upregulated independently of MR activation, the MR is still a key element of this triangle, with clinical data supporting the use of MRAs in delaying the progression of DKD. With FDA and EMA approval of finerenone, the treatment of DKD has entered an exciting new era in MRA therapy addressing the inflammation and fibrosis component in slowing DKD progression, as well as reducing cardiovascular events.
Acknowledgment
Medical writing support, under the guidance of the authors, was provided by Constantinos Bezos, MCh, 3 Stories High, UK.
Conflict of Interest Statement
C. Mende has received payments or honoraria for lectures, presentations, speaker bureaus, manuscript writing, or educational events from Bayer, Boehringer, Lilly, AstraZeneca, and Janssen. R. Samarakoon has received grants or contracts from Capital District Medical Research Institute (to institution AMC); honoraria from DOD-Kidney Cancer Research Program-Grant Review Panel; been a member of the Dean’s Advisory Board, Iona College, NY, USA (unpaid); and been on the Editorial Board of Frontiers in Pharmacology-Renal (unpaid). P. Higgins has been funded by the NIH and DOD and received honoraria from both the NIH and DOD Cancer Research Programs for Chairing grant review panels. He is a member of the Board of Directors of the Albany Research Institute.
Funding Sources
Medical writing support and article processing charges were funded by Bayer in accordance with Good Publication Practice (GPP 2022) guidelines (DeTora LM, et al., Ann Intern Med., August 30, 2022. Epub ahead of print. doi:10.7326/M22-1460).
Author Contributions
Christian W. Mende: conceptualization, literature search, figure concept, and writing (original draft, review, and editing). Rohan Samarakoon: literature search, figure drawing, and writing (review and editing). Paul J. Higgins: literature search and writing (review and editing).
References
- 1. Babel RA, Dandekar MP. A review on cellular and molecular mechanisms linked to the development of diabetes complications. <X00_Journal>Curr Diabetes Rev</X00_Journal>. 2021;17(4):457–73.
- 2. Vaidya S, Aeddula N. <X00_Journal>Chronic renal failure</X00_Journal>. Treasure Island (FL): StatPearls Publishing; 2021. [Updated 2021 Jul 16]. StatPearls.
- 3. Lin X, Xu Y, Pan X, Xu J, Ding Y, Sun X, et al. Global, regional, and national burden and trend of diabetes in 195 countries and territories: an analysis from 1990 to 2025. <X00_Journal>Sci Rep</X00_Journal>. 2020;10(1):14790.
- 4. Kidney Disease: Improving Global Outcomes (KDIGO) Blood Pressure Work Group. KDIGO 2021 Clinical practice guideline for the management of blood pressure in chronic kidney disease. <X00_Journal>Kidney Int</X00_Journal>. 2021;99(3S):S1-87.
- 5. García-Carro C, Vergara A, Bermejo S, Azancot MA, Sánchez-Fructuoso AI, Sánchez de la Nieta MD, et al. How to assess diabetic kidney disease progression? From albuminuria to GFR. <X00_Journal>J Clin Med</X00_Journal>. 2021;10(11):2505.
- 6. Aiumtrakul N, Phichedwanichskul K, Saravutthikul S, Ottasat K, Visuthitepkul K, Jaruthiti T, et al. Urine albumin dipstick independently predicts cardiovascular and renal outcomes among rural Thai population: a 14-year retrospective cohort study. <X00_Journal>BMC Nephrol</X00_Journal>. 2021;22(1):18.
- 7. Jung SW, Moon JY. The role of inflammation in diabetic kidney disease. <X00_Journal>Korean J Intern Med</X00_Journal>. 2021;36(4):753–66.
- 8. Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: challenges, progress, and possibilities. <X00_Journal>Clin J Am Soc Nephrol</X00_Journal>. 2017;12(12):2032–45.
- 9. Zoccali C, Vanholder R, Massy ZA, Ortiz A, Sarafidis P, Dekker FW, et al. The systemic nature of CKD. <X00_Journal>Nat Rev Nephrol</X00_Journal>. 2017;13(6):344–58.
- 10. Humphreys BD. Mechanisms of renal fibrosis. <X00_Journal>Annu Rev Physiol</X00_Journal>. 2018;80(1):309–26.
- 11. Levey AS, Coresh J. Chronic kidney disease. <X00_Journal>Lancet</X00_Journal>. 2012;379(9811):165–80.
- 12. Turner JM, Bauer C, Abramowitz MK, Melamed ML, Hostetter TH. Treatment of chronic kidney disease. <X00_Journal>Kidney Int</X00_Journal>. 2012;81(4):351–62.
- 13. LaPointe MS. Genomic and non-genomic aldosterone signaling. <X00_Journal>Anat Physiol Biochem Int J</X00_Journal>. 2017;1(5):113–8.
- 14. Rentoukas EI, Lazaros GA, Zirogiannis PN. Aldosterone in heart and kidney diseases. <X00_Journal>Hellenic J Cardiol</X00_Journal>. 2005;46(6):408–19.
- 15. Siragy HM, Carey RM. Role of the intrarenal renin-angiotensin-aldosterone system in chronic kidney disease. <X00_Journal>Am J Nephrol</X00_Journal>. 2010;31(6):541–50.
- 16. Peti-Peterdi J. High glucose and renin release: the role of succinate and GPR91. <X00_Journal>Kidney Int</X00_Journal>. 2010;78(12):1214–7.
- 17. Dinh Cat AN, Friederich-Persson M, White A, Touyz RM. Adipocytes, aldosterone and obesity-related hypertension. <X00_Journal>J Mol Endocrinol</X00_Journal>. 2016;57(1):F7-21.
- 18. Briones AM, Nguyen Dinh Cat A, Callera GE, Yogi A, Burger D, He Y, et al. Adipocytes produce aldosterone through calcineurin-dependent signaling pathways: implications in diabetes mellitus-associated obesity and vascular dysfunction. <X00_Journal>Hypertension</X00_Journal>. 2012;59(5):1069–78.
- 19. Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PM, Kohan DE. Collecting duct principal cell transport processes and their regulation. <X00_Journal>Clin J Am Soc Nephrol</X00_Journal>. 2015;10(1):135–46.
- 20. Brown NJ. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. <X00_Journal>Nat Rev Nephrol</X00_Journal>. 2013;9(8):459–69.
- 21. Ritz E, Tomaschitz A. Aldosterone and the kidney: a rapidly moving frontier (an update). <X00_Journal>Nephrol Dial Transplant</X00_Journal>. 2014;29(11):2012–9.
- 22. Luther JM, Fogo AB. The role of mineralocorticoid receptor activation in kidney inflammation and fibrosis. <X00_Journal>Kidney Int Suppl</X00_Journal>. 2022;12(1):63–8.
- 23. Bai M, Chen Y, Zhao M, Zhang Y, He JCJ, Huang S, et al. NLRP3 inflammasome activation contributes to aldosterone-induced podocyte injury. <X00_Journal>Am J Physiol Renal Physiol</X00_Journal>. 2017;312(4):F556-64.
- 24. Sheader EA, Wargent ET, Ashton N, Balment RJ. Rapid stimulation of cyclic AMP production by aldosterone in rat inner medullary collecting ducts. <X00_Journal>J Endocrinol</X00_Journal>. 2002;175(2):343–7.
- 25. Eiam-Ong S, Chaipipat M, Manotham K, Eiam-Ong S. Rapid action of aldosterone on protein levels of sodium-hydrogen exchangers and protein kinase C beta isoforms in rat kidney. <X00_Journal>Int J Endocrinol</X00_Journal>. 2017;2017:2975853.
- 26. Wang YQ, Fan CC, Chen BP, Shi J. Resistin-like molecule beta (RELM-β) regulates proliferation of human diabetic nephropathy mesangial cells via mitogen-activated protein kinases (MAPK) signaling pathway. <X00_Journal>Med Sci Monit</X00_Journal>. 2017;23:3897–903.
- 27. Epstein M. Considerations for the future: current and future treatment paradigms with mineralocorticoid receptor antagonists-unmet needs and underserved patient cohorts. <X00_Journal>Kidney Int Suppl</X00_Journal>. 2022;12(1):69–75.
- 28. Messaoudi S, Azibani F, Delcayre C, Jaisser F. Aldosterone, mineralocorticoid receptor, and heart failure. <X00_Journal>Mol Cell Endocrinol</X00_Journal>. 2012;350(2):266–72.
- 29. Jaisser F, Farman N. Emerging roles of the mineralocorticoid receptor in pathology: toward new paradigms in clinical pharmacology. <X00_Journal>Pharmacol Rev</X00_Journal>. 2016;68(1):49–75.
- 30. Bertocchio JP, Warnock DG, Jaisser F. Mineralocorticoid receptor activation and blockade: an emerging paradigm in chronic kidney disease. <X00_Journal>Kidney Int</X00_Journal>. 2011;79(10):1051–60.
- 31. Pascual-Le Tallec L, Lombès M. The mineralocorticoid receptor: a journey exploring its diversity and specificity of action. <X00_Journal>Mol Endocrinol</X00_Journal>. 2005;19(9):2211–21.
- 32. Tesch GH, Young MJ. Mineralocorticoid receptor signaling as a therapeutic target for renal and cardiac fibrosis. <X00_Journal>Front Pharmacol</X00_Journal>. 2017;8:313.
- 33. Verma A, Vaidya A, Subudhi S, Waikar SS. Aldosterone in chronic kidney disease and renal outcomes. <X00_Journal>Eur Heart J</X00_Journal>. 2022;43(38):3781–91.
- 34. Nagase M, Fujita T. Role of Rac1-mineralocorticoid-receptor signalling in renal and cardiac disease. <X00_Journal>Nat Rev Nephrol</X00_Journal>. 2013;9(2):86–98.
- 35. Tang C, Livingston MJ, Liu Z, Dong Z. Autophagy in kidney homeostasis and disease. <X00_Journal>Nat Rev Nephrol</X00_Journal>. 2020;16(9):489–508.
- 36. Bauersachs J, Jaisser F, Toto R. Mineralocorticoid receptor activation and mineralocorticoid receptor antagonist treatment in cardiac and renal diseases. <X00_Journal>Hypertension</X00_Journal>. 2015;65(2):257–63.
- 37. Steichen C, Hervé C, Hauet T, Bourmeyster N. Rho GTPases in kidney physiology and diseases. <X00_Journal>Small GTPases</X00_Journal>. 2022;13(1):141–61.
- 38. Asahara S, Shibutani Y, Teruyama K, Inoue HY, Kawada Y, Etoh H, et al. Ras-related C3 botulinum toxin substrate 1 (RAC1) regulates glucose-stimulated insulin secretion via modulation of F-actin. <X00_Journal>Diabetologia</X00_Journal>. 2013;56(5):1088–97.
- 39. Ueda S, Kitazawa S, Ishida K, Nishikawa Y, Matsui M, Matsumoto H, et al. Crucial role of the small GTPase Rac1 in insulin-stimulated translocation of glucose transporter 4 to the mouse skeletal muscle sarcolemma. <X00_Journal>FASEB J</X00_Journal>. 2010;24(7):2254–61.
- 40. Abo A, Pick E, Hall A, Totty N, Teahan CG, Segal AW. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. <X00_Journal>Nature</X00_Journal>. 1991;353(6345):668–70.
- 41. Abo A, Webb MR, Grogan A, Segal AW. Activation of NADPH oxidase involves the dissociation of p21rac from its inhibitory GDP/GTP exchange protein (rhoGDI) followed by its translocation to the plasma membrane. <X00_Journal>Biochem J</X00_Journal>. 1994;298 Pt 3(3Pt 3):585–91.
- 42. Pi J, Bai Y, Zhang Q, Wong V, Floering LM, Daniel K, et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. <X00_Journal>Diabetes</X00_Journal>. 2007;56(7):1783–91.
- 43. Kowluru A. Friendly, and not so friendly, roles of Rac1 in islet β-cell function: lessons learnt from pharmacological and molecular biological approaches. <X00_Journal>Biochem Pharmacol</X00_Journal>. 2011;81(8):965–75.
- 44. Samarakoon R, Overstreet JM, Higgins PJ. TGF-β signaling in tissue fibrosis: redox controls, target genes and therapeutic opportunities. <X00_Journal>Cell Signal</X00_Journal>. 2013;25(1):264–8.
- 45. Sedeek M, Gutsol A, Montezano AC, Burger D, Nguyen Dinh Cat A, Kennedy CRJ, et al. Renoprotective effects of a novel Nox1/4 inhibitor in a mouse model of Type 2 diabetes. <X00_Journal>Clin Sci</X00_Journal>. 2013;124(3):191–202.
- 46. Jha JC, Gray SP, Barit D, Okabe J, El-Osta A, Namikoshi T, et al. Genetic targeting or pharmacologic inhibition of NADPH oxidase Nox4 provides renoprotection in long-term diabetic nephropathy. <X00_Journal>J Am Soc Nephrol</X00_Journal>. 2014;25(6):1237–54.
- 47. Marei H, Malliri A. Rac1 in human diseases: the therapeutic potential of targeting Rac1 signaling regulatory mechanisms. <X00_Journal>Small GTPases</X00_Journal>. 2017;8(3):139–63.
- 48. Iwashima F, Yoshimoto T, Minami I, Sakurada M, Hirono Y, Hirata Y. Aldosterone induces superoxide generation via Rac1 activation in endothelial cells. <X00_Journal>Endocrinology</X00_Journal>. 2008;149(3):1009–14.
- 49. Schmitz U, Thömmes K, Beier I, Wagner W, Sachinidis A, Düsing R, et al. Angiotensin II-induced stimulation of p21-activated kinase and c-Jun NH2-terminal kinase is mediated by Rac1 and Nck. <X00_Journal>J Biol Chem</X00_Journal>. 2001;276(25):22003–10.
- 50. Patel S, Tang J, Overstreet JM, Anorga S, Lian F, Arnouk A, et al. Rac-GTPase promotes fibrotic TGF-β1 signaling and chronic kidney disease via EGFR, p53, and Hippo/YAP/TAZ pathways. <X00_Journal>FASEB J</X00_Journal>. 2019;33(9):9797–810.
- 51. Sahajpal N, Kowluru A, Kowluru RA. The regulatory role of Rac1, a small molecular weight GTPase, in the development of diabetic retinopathy. <X00_Journal>J Clin Med</X00_Journal>. 2019;8(7):965.
- 52. Ju L, Zhou Z, Jiang B, Lou Y, Guo X. Autocrine VEGF and IL-8 promote migration via Src/Vav2/Rac1/PAK1 signaling in human umbilical vein endothelial cells. <X00_Journal>Cell Physiol Biochem</X00_Journal>. 2017;41(4):1346–59.
- 53. Paul O, Moseley A, Song C, Al-Khalili OO, Gomez-Sanchez C, Eaton DC, et al. IL-6 increases ROS via Rac1, leading to mineralocorticoid receptor and ENaC activation. <X00_Journal>FASEB J</X00_Journal>. 2020;34(S1):1.
- 54. Kawarazaki W, Nagase M, Yoshida S, Takeuchi M, Ishizawa K, Ayuzawa N, et al. Angiotensin II- and salt-induced kidney injury through Rac1-mediated mineralocorticoid receptor activation. <X00_Journal>J Am Soc Nephrol</X00_Journal>. 2012;23(6):997–1007.
- 55. Tung CW, Hsu YC, Shih YH, Chang PJ, Lin CL. Glomerular mesangial cell and podocyte injuries in diabetic nephropathy. <X00_Journal>Nephrology</X00_Journal>. 2018;23(Suppl 4):32–7.
- 56. Yoshida S, Ishizawa K, Ayuzawa N, Ueda K, Takeuchi M, Kawarazaki W, et al. Local mineralocorticoid receptor activation and the role of Rac1 in obesity-related diabetic kidney disease. <X00_Journal>Nephron Exp Nephrol</X00_Journal>. 2014;126(1):16–24.
- 57. Hall G, Spurney RF. Losing their footing: Rac1 signaling causes podocyte detachment and FSGS. <X00_Journal>Kidney Int</X00_Journal>. 2017;92(2):283–5.
- 58. Ayuzawa N, Fujita T. The mineralocorticoid receptor in salt-sensitive hypertension and renal injury. <X00_Journal>J Am Soc Nephrol</X00_Journal>. 2021;32(2):279–89.
- 59. Saleem MA, Welsh GI. Podocyte RhoGTPases: new therapeutic targets for nephrotic syndrome? <X00_Journal>F1000Res</X00_Journal>. 2019;8:F1000 Faculty Rev–1847.
- 60. Dagon Y, Raghu H, Ge T, Walsh L, Reilly JF. Urinary Rac1, a novel predictive biomarker, is elevated in FSGS and diabetic nephropathy patients and reduced by TRPC5 inhibition with GFB-887 in a rat FSGS model. <X00_Journal>Kidney Week</X00_Journal>. 2020.
- 61. Hirohama D, Nishimoto M, Ayuzawa N, Kawarazaki W, Fujii W, Oba S, et al. Activation of Rac1-mineralocorticoid receptor pathway contributes to renal injury in salt-loaded db/db mice. <X00_Journal>Hypertension</X00_Journal>. 2021;78(1):82–93.
- 62. Komada T, Muruve DA. The role of inflammasomes in kidney disease. <X00_Journal>Nat Rev Nephrol</X00_Journal>. 2019;15(8):501–20.
- 63. Shahzad K, Bock F, Dong W, Wang H, Kopf S, Kohli S, et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. <X00_Journal>Kidney Int</X00_Journal>. 2015;87(1):74–84.
- 64. Zhang Q, Conley SM, Li G, Yuan X, Li PL. Rac1 GTPase inhibition blocked podocyte injury and glomerular sclerosis during hyperhomocysteinemia via suppression of nucleotide-binding oligomerization domain-like receptor containing pyrin domain 3 inflammasome activation. <X00_Journal>Kidney Blood Press Res</X00_Journal>. 2019;44(4):513–32.
- 65. Martín-Fernández B, Rubio-Navarro A, Cortegano I, Ballesteros S, Alía M, Cannata-Ortiz P, et al. Aldosterone induces renal fibrosis and inflammatory M1-macrophage subtype via mineralocorticoid receptor in rats. <X00_Journal>PLoS One</X00_Journal>. 2016;11(1):e0145946.
- 66. Kadoya H, Satoh M, Sasaki T, Taniguchi S, Takahashi M, Kashihara N. Excess aldosterone is a critical danger signal for inflammasome activation in the development of renal fibrosis in mice. <X00_Journal>FASEB J</X00_Journal>. 2015;29(9):3899–910.
- 67. Wang X, Chen J, Xu J, Xie J, Harris DCH, Zheng G. The role of macrophages in kidney fibrosis. <X00_Journal>Front Physiol</X00_Journal>. 2021;12:705838.
- 68. Berbenetz NM, Mrkobrada M. Mineralocorticoid receptor antagonists for heart failure: systematic review and meta-analysis. <X00_Journal>BMC Cardiovasc Disord</X00_Journal>. 2016;16(1):246.
- 69. Abdelhakim AM, Abd-ElGawad M. Impact of mineralocorticoid receptor antagonist in renal transplant patients: a systematic review and meta-analysis of randomized controlled trials. <X00_Journal>J Nephrol</X00_Journal>. 2020;33(3):529–38.
- 70. Tromp J, Ouwerkerk W, van Veldhuisen DJ, Hillege HL, Richards AM, van der Meer P, et al. A systematic review and network meta-analysis of pharmacological treatment of heart failure with reduced ejection fraction. <X00_Journal>JACC Heart Fail</X00_Journal>. 2022;10(2):73–84.
- 71. Chung EY, Ruospo M, Natale P, Bolignano D, Navaneethan SD, Palmer SC, et al. Aldosterone antagonists in addition to renin angiotensin system antagonists for preventing the progression of chronic kidney disease. <X00_Journal>Cochrane Database Syst Rev</X00_Journal>. 2020;10(10):CD007004.
- 72. Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, et al. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. <X00_Journal>Science</X00_Journal>. 1987;237(4812):268–75.
- 73. Gomez-Sanchez EP. Third-generation mineralocorticoid receptor antagonists: why do we need a fourth? <X00_Journal>J Cardiovasc Pharmacol</X00_Journal>. 2016;67(1):26–38.
- 74. Rossing K, Schjoedt KJ, Smidt UM, Boomsma F, Parving HH. Beneficial effects of adding spironolactone to recommended antihypertensive treatment in diabetic nephropathy: a randomized, double-masked, cross-over study. <X00_Journal>Diabetes Care</X00_Journal>. 2005;28(9):2106–12.
- 75. Esteghamati A, Noshad S, Jarrah S, Mousavizadeh M, Khoee SH, Nakhjavani M. Long-term effects of addition of mineralocorticoid receptor antagonist to angiotensin II receptor blocker in patients with diabetic nephropathy: a randomized clinical trial. <X00_Journal>Nephrol Dial Transpl</X00_Journal>. 2013;28(11):2823–33.
- 76. Juurlink DN, Mamdani MM, Lee DS, Kopp A, Austin PC, Laupacis A, et al. Rates of hyperkalemia after publication of the randomized aldactone evaluation study. <X00_Journal>N Engl J Med</X00_Journal>. 2004;351(6):543–51.
- 77. Edwards NC, Steeds RP, Chue CD, Stewart PM, Ferro CJ, Townend JN. The safety and tolerability of spironolactone in patients with mild to moderate chronic kidney disease. <X00_Journal>Br J Clin Pharmacol</X00_Journal>. 2012;73(3):447–54.
- 78. Epstein M, Williams GH, Weinberger M, Lewin A, Krause S, Mukherjee R, et al. Selective aldosterone blockade with eplerenone reduces albuminuria in patients with type 2 diabetes. <X00_Journal>Clin J Am Soc Nephrol</X00_Journal>. 2006;1(5):940–51.
- 79. El Mokadem M, Abd El Hady Y, Aziz A. A prospective single-blind randomized trial of ramipril, eplerenone and their combination in type 2 diabetic nephropathy. <X00_Journal>Cardiorenal Med</X00_Journal>. 2020;10(6):392–401.
- 80. Bertocchio JP, Barbe C, Lavaud S, Toupance O, Nazeyrollas P, Jaisser F, et al. Safety of eplerenone for kidney-transplant recipients with impaired renal function and receiving cyclosporine A. <X00_Journal>PLoS One</X00_Journal>. 2016;11(4):e0153635.
- 81. Hou J, Xiong W, Cao L, Wen X, Li A. Spironolactone add-on for preventing or slowing the progression of diabetic nephropathy: a meta-analysis. <X00_Journal>Clin Ther</X00_Journal>. 2015;37(9):2086–103.e10.
- 82. Kolkhof P, Nowack C, Eitner F. Nonsteroidal antagonists of the mineralocorticoid receptor. <X00_Journal>Curr Opin Nephrol Hypertens</X00_Journal>. 2015;24(5):417–24.
- 83. Rossing P. Mineralocorticoid receptor antagonists for diabetic kidney disease. <X00_Journal>Clin J Am Soc Nephrol</X00_Journal>. 2020;15(12):1696–8.
- 84. Bayer AG. <X00_Journal>KERENDIA (finerenone) summary of product characteristics</X00_Journal>; 2022.
- 85. Bayer Healthcare Pharmaceuticals Inc. <X00_Journal>KERENDIA (finerenone) prescribing information</X00_Journal>; 2021.
- 86. Bakris GL, Agarwal R, Chan JC, Cooper ME, Gansevoort RT, Haller H, et al. Effect of finerenone on albuminuria in patients with diabetic nephropathy: a randomized clinical trial. <X00_Journal>JAMA</X00_Journal>. 2015 Sep 1;314(9):884–94.
- 87. Katayama S, Yamada D, Nakayama M, Yamada T, Myoishi M, Kato M, et al. A randomized controlled study of finerenone versus placebo in Japanese patients with type 2 diabetes mellitus and diabetic nephropathy. <X00_Journal>J Diabetes Complications</X00_Journal>. 2017;31(4):758–65.
- 88. Bakris GL, Agarwal R, Anker SD, Pitt B, Ruilope LM, Rossing P, et al. Effect of finerenone on chronic kidney disease outcomes in type 2 diabetes. <X00_Journal>N Engl J Med</X00_Journal>. 2020;383(23):2219–29.
- 89. Pitt B, Filippatos G, Agarwal R, Anker SD, Bakris GL, Rossing P, et al. Cardiovascular events with finerenone in kidney disease and type 2 diabetes. <X00_Journal>N Engl J Med</X00_Journal>. 2021;385(24):2252–63.
- 90. Bomback AS, Kshirsagar AV, Amamoo MA, Klemmer PJ. Change in proteinuria after adding aldosterone blockers to ACE inhibitors or angiotensin receptor blockers in CKD: a systematic review. <X00_Journal>Am J Kidney Dis</X00_Journal>. 2008;51(2):199–211.
- 91. Vodošek Hojs N, Bevc S, Ekart R, Piko N, Petreski T, Hojs R. Mineralocorticoid receptor antagonists in diabetic kidney disease. <X00_Journal>Pharmaceuticals</X00_Journal>. 2021;14(6):561.
- 92. Ito S, Shikata K, Nangaku M, Okuda Y, Sawanobori T. Efficacy and safety of esaxerenone (CS-3150) for the treatment of type 2 diabetes with microalbuminuria: a randomized, double-blind, placebo-controlled, phase II trial. <X00_Journal>Clin J Am Soc Nephrol</X00_Journal>. 2019;14(8):1161–72.
- 93. Ito S, Kashihara N, Shikata K, Nangaku M, Wada T, Okuda Y, et al. Esaxerenone (CS-3150) in patients with type 2 diabetes and microalbuminuria (ESAX-DN): phase 3 randomized controlled clinical trial. <X00_Journal>Clin J Am Soc Nephrol</X00_Journal>. 2020;15(12):1715–27.
- 94. Sato A, Hayashi K, Naruse M, Saruta T. Effectiveness of aldosterone blockade in patients with diabetic nephropathy. <X00_Journal>Hypertension</X00_Journal>. 2003;41(1):64–8.
- 95. Schjoedt KJ, Andersen S, Rossing P, Tarnow L, Parving HH. Aldosterone escape during blockade of the renin-angiotensin-aldosterone system in diabetic nephropathy is associated with enhanced decline in glomerular filtration rate. <X00_Journal>Diabetologia</X00_Journal>. 2004;47(11):1936–9.
- 96. Bakris G, Pergola PE, Delgado B, Genov D, Doliashvili T, Vo N, et al. Effect of KBP-5074 on blood pressure in advanced chronic kidney disease: results of the BLOCK-CKD study. <X00_Journal>Hypertension</X00_Journal>. 2021;78(1):74–81.
- 97. Wada T, Inagaki M, Yoshinari T, Terata R, Totsuka N, Gotou M, et al. Apararenone in patients with diabetic nephropathy: results of a randomized, double-blind, placebo-controlled phase 2 dose-response study and open-label extension study. <X00_Journal>Clin Exp Nephrol</X00_Journal>. 2021;25(2):120–30.
- 98. Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. <X00_Journal>N Engl J Med</X00_Journal>. 2019;380(24):2295–306.
- 99. Kolkhof P, Hartmann E, Freyberger A, Pavkovic M, Mathar I, Sandner P, et al. Effects of finerenone combined with empagliflozin in a model of hypertension-induced end-organ damage. <X00_Journal>Am J Nephrol</X00_Journal>. 2021;52(8):642–52.
- 100. Agarwal R, Filippatos G, Pitt B, Anker SD, Rossing P, Joseph A, et al. Cardiovascular and kidney outcomes with finerenone in patients with type 2 diabetes and chronic kidney disease: the FIDELITY pooled analysis. <X00_Journal>Eur Heart J</X00_Journal>. 2022;43(6):474–84.
- 101. De Giusti VC, Nolly MB, Yeves AM, Caldiz CI, Villa-Abrille MC, Chiappe de Cingolani GE, et al. Aldosterone stimulates the cardiac Na(+)/H(+) exchanger via transactivation of the epidermal growth factor receptor. <X00_Journal>Hypertension</X00_Journal>. 2011;58(5):912–9.
- 102. Chen SY, Bhargava A, Mastroberardino L, Meijer OC, Wang J, Buse P, et al. Epithelial sodium channel regulated by aldosterone-induced protein sgk. <X00_Journal>Proc Natl Acad Sci U S A</X00_Journal>. 1999;96(5):2514–9.
- 103. Chen F, Liu J, Wang Y, Wu T, Shan W, Zhu Y, et al. Aldosterone induces clonal β-cell failure through glucocorticoid receptor. <X00_Journal>Sci Rep</X00_Journal>. 2015;5:13215.
- 104. Christ M, Meyer C, Sippel K, Wehling M. Rapid aldosterone signaling in vascular smooth muscle cells: involvement of phospholipase C, diacylglycerol and protein kinase C alpha. <X00_Journal>Biochem Biophys Res Commun</X00_Journal>. 1995;213(1):123–9.
- 105. Kuppusamy M, Gomez-Sanchez E, Gomez-Sanchez C. Abstract 134: activation of the mineralocorticoid receptor by O-glycosylation through hyperglycemia. <X00_Journal>Hypertension</X00_Journal>. 2015;66(Suppl_1).
- 106. Huby AC, Antonova G, Groenendyk J, Gomez-Sanchez CE, Bollag WB, Filosa JA, et al. Adipocyte-derived hormone leptin is a direct regulator of aldosterone secretion, which promotes endothelial dysfunction and cardiac fibrosis. <X00_Journal>Circulation</X00_Journal>. 2015;132(22):2134–45.
- 107. Faulkner JL, Bruder-Nascimento T, Belin de Chantemèle EJ. The regulation of aldosterone secretion by leptin: implications in obesity-related cardiovascular disease. <X00_Journal>Curr Opin Nephrol Hypertens</X00_Journal>. 2018;27(2):63–9.
- 108. Ehrhart-Bornstein M, Lamounier-Zepter V, Schraven A, Langenbach J, Willenberg HS, Barthel A, et al. Human adipocytes secrete mineralocorticoid-releasing factors. <X00_Journal>Proc Natl Acad Sci U S A</X00_Journal>. 2003;100(24):14211–6.
- 109. Kargi AY, Iacobellis G. Adipose tissue and adrenal glands: novel pathophysiological mechanisms and clinical applications. <X00_Journal>Int J Endocrinol</X00_Journal>. 2014;2014:614074.
- 110. Koenen M, Hill MA, Cohen P, Sowers JR. Obesity, adipose tissue and vascular dysfunction. <X00_Journal>Circ Res</X00_Journal>. 2021;128(7):951–68.
- 111. Garg R, Hurwitz S, Williams GH, Hopkins PN, Adler GK. Aldosterone production and insulin resistance in healthy adults. <X00_Journal>J Clin Endocrinol Metab</X00_Journal>. 2010;95(4):1986–90.
- 112. Vecchiola A, Lagos CF, Carvajal CA, Baudrand R, Fardella CE. Aldosterone production and signaling dysregulation in obesity. <X00_Journal>Curr Hypertens Rep</X00_Journal>. 2016;18(3):20.
- 113. Kawarazaki W, Fujita T. The role of aldosterone in obesity-related hypertension. <X00_Journal>Am J Hypertens</X00_Journal>. 2016;29(4):415–23.
- 114. Jaques DA, Wuerzner G, Ponte B. Sodium intake as a cardiovascular risk factor: a narrative review. <X00_Journal>Nutrients</X00_Journal>. 2021;13(9):3177.
- 115. Shibata S, Mu S, Kawarazaki H, Muraoka K, Ishizawa K, Yoshida S, et al. Rac1 GTPase in rodent kidneys is essential for salt-sensitive hypertension via a mineralocorticoid receptor-dependent pathway. <X00_Journal>J Clin Invest</X00_Journal>. 2011;121(8):3233–43.
- 116. Shibata S, Nagase M, Yoshida S, Kawarazaki W, Kurihara H, Tanaka H, et al. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. <X00_Journal>Nat Med</X00_Journal>. 2008;14(12):1370–6.
- 117. Quinkler M, Zehnder D, Eardley KS, Lepenies J, Howie AJ, Hughes SV, et al. Increased expression of mineralocorticoid effector mechanisms in kidney biopsies of patients with heavy proteinuria. <X00_Journal>Circulation</X00_Journal>. 2005;112(10):1435–43.
- 118. Yoon HE, Kim EN, Kim MY, Lim JH, Jang IA, Ban TH, et al. Age-associated changes in the vascular renin-angiotensin system in mice. <X00_Journal>Oxid Med Cel Longev</X00_Journal>. 2016;2016:6731093.
- 119. Kim SK, McCurley AT, DuPont JJ, Aronovitz M, Moss ME, Stillman IE, et al. Smooth muscle cell-mineralocorticoid receptor as a mediator of cardiovascular stiffness with aging. <X00_Journal>Hypertension</X00_Journal>. 2018;71(4):609–21.
- 120. Bădilă E. The expanding class of mineralocorticoid receptor modulators: new ligands for kidney, cardiac, vascular, systemic and behavioral selective actions. <X00_Journal>Acta Endocrinol</X00_Journal>. 2020;16(4):487–96.
- 121. Nomura N, Nomura M, Sugiyama K, Hamada JI. Src regulates phorbol 12-myristate 13-acetate-activated PKC-induced migration via Cas/Crk/Rac1 signaling pathway in glioblastoma cells. <X00_Journal>Int J Mol Med</X00_Journal>. 2007;20(4):511–9.
- 122. Thamilselvan V, Menon M, Thamilselvan S. Selective Rac1 inhibition protects renal tubular epithelial cells from oxalate-induced NADPH oxidase-mediated oxidative cell injury. <X00_Journal>Urol Res</X00_Journal>. 2012;40(4):415–23.
- 123. Kawamura S, Miyamoto S, Brown JH. Initiation and transduction of stretch-induced RhoA and Rac1 activation through caveolae: cytoskeletal regulation of ERK translocation. <X00_Journal>J Biol Chem</X00_Journal>. 2003;278(33):31111–7.
- 124. Hawkins PT, Eguinoa A, Qiu R-G, Stokoe D, Cooke FT, Walters R, et al. PDGF stimulates an increase in GTP–Rac via activation of phosphoinositide 3-kinase. <X00_Journal>Curr Biol</X00_Journal>. 1995;5(4):393–403.
- 125. Grill B, Schrader JW. Activation of Rac-1, Rac-2, and Cdc42 by hemopoietic growth factors or cross-linking of the B-lymphocyte receptor for antigen. <X00_Journal>Blood</X00_Journal>. 2002;100(9):3183–92.
- 126. Garrett TA, Van Buul JD, Burridge K. VEGF-induced Rac1 activation in endothelial cells is regulated by the guanine nucleotide exchange factor Vav2. <X00_Journal>Exp Cell Res</X00_Journal>. 2007;313(15):3285–97.
- 127. Uhlik MT, Abell AN, Johnson NL, Sun W, Cuevas BD, Lobel-Rice KE, et al. Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. <X00_Journal>Nat Cell Biol</X00_Journal>. 2003;5(12):1104–10.
- 128. Papaharalambus C, Sajjad W, Syed A, Zhang C, Bergo MO, Alexander RW, et al. Tumor necrosis factor alpha stimulation of Rac1 activity. Role of isoprenylcysteine carboxylmethyltransferase. <X00_Journal>J Biol Chem</X00_Journal>. 2005;280(19):18790–6.
- 129. Nagase M, Ayuzawa N, Kawarazaki W, Ishizawa K, Ueda K, Yoshida S, et al. Oxidative stress causes mineralocorticoid receptor activation in rat cardiomyocytes: role of small GTPase Rac1. <X00_Journal>Hypertension</X00_Journal>. 2012;59(2):500–6.