Impact Statement
The growing challenge of drug-resistant tuberculosis underscores the need for innovative approaches to understand and disable Mtb resistance mechanisms. This study highlights the complexities of drug resistance in Ethiopian Mtb sub-lineage 4.2.2.2 isolates. By integrating phenotypic, transcriptomic, and genomic analyses, it reveals reduced expression of six non-canonical resistance-associated genes, offering fresh insights into the molecular adaptations accompanying drug resistance. Moreover, the identified discrepancies between phenotypic resistance profiles and genome-based predictions could contribute evidence towards the utility of these methods for diagnostic applications. These findings advance our understanding of Mtb resistance mechanisms and pathways associated with drug resistance.
Introduction
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), is a historically significant pathogen that continues to pose a global public health challenge. TB affects millions of lives and remains one of the most lethal microorganisms worldwide (Goletti et al., ). Over the past two centuries, Mtb has been responsible for the deaths of approximately one billion individuals, underscoring its impact on human health (Torres Ortiz et al. , ). The Mycobacterium tuberculosis complex (MTBC) comprises a group of mycobacteria species that share 99.9% genetic similarity at the nucleotide level and possess identical 16S rRNA sequences (Brosch et al., ). Within the MTBC, there are ten lineages adapted to human hosts, identified as Lineages 1 through 10. Africa is the only continent where all MTBC lineages can be found (Cerezo-Cortés et al., ; Guyeux et al., ). Ethiopia is recognized as a major hub encompassing most of these lineages; according to a study by Mekonnen et al. (), the proportions of isolates for L4, L3, L1, and L7 were 62.3%, 21.7%, 7.9%, and 3.4%, respectively. Among the sub-lineages, the most prevalent clustered isolates were L4.2.2. ETH/SIT149 (L4.2.2.2), L4.10/SIT53, L3.ETH1/SIT25, and L4.6/SIT37, contributing 14.4%, 9.7%, 7.2%, and 5.5% of the total isolates, respectively. In northwest Ethiopia (from 2020 to 2022), the dominant drug-resistant sub-lineage was L4.2.2.ETH, which accounted for 34.5% of cases, representing 50% of drug-resistant isolates (Mekonnen et al., ). These findings highlight L4.2.2.2 as a sub-lineage marked by high transmissibility and drug resistance.
Drug resistance in Mtb presents a significant challenge to human health. Mtb has evolved resistance to most anti-TB drugs. The emergence of drug-resistant strains of Mtb threatens the effectiveness of disease control programs worldwide (Miotto et al., ). Treatment success rates on a global scale range from 85% for drug-sensitive TB to 56% for MDR-TB, further dropping to 39% for extensively drug-resistant tuberculosis (XDR-TB) (Wan et al. ). Although well-established canonical mutations are known to confer high-level drug resistance in Mtb, recent research indicates that Mtb can develop non-canonical resistance-associated mutations, which provide survival advantages in the presence of specific drugs and may serve as intermediates in the progression to high-level resistance (Martini et al., ). There is an urgent need for new drugs to shorten the duration of TB treatment and address the growing prevalence of infections caused by drug-resistant strains. However, selecting suitable targets for drug development remains challenging due to our limited understanding of how Mtb responds when target function is interrupted, or adapts to accommodate drug resistance conferring mutations.
Given the limited genetic diversity among Mtb strains, variations in Mtb phenotypes primarily stem from differences in the regulation of biochemical networks by key transcriptional regulators. By analyzing the transcriptomic profiles associated with specific genotypes, we can gain valuable insights into the underlying biology. This knowledge may help to identify potential targets for the development of novel drugs (Gomez-Gonzalez et al., ). In this study, we examined the expression of genes using RNA sequencing in Mtb sub lineage 4.2.2.2 strains isolated from Ethiopia. We compared drug-resistant with drug-sensitive isolates using in vitro phenotypic drug susceptibility testing (DST) and in silico predictions from whole genome sequencing (WGS).
Materials and methods
Study M. tuberculosis strains
This study utilized Mtb isolates obtained from active Ethiopian pulmonary TB patients participating in the TBGEN-Africa tuberculosis study. Six clinical isolates of Mtb sub-lineage 4.2.2.2 were obtained from various regions of Ethiopia, including Addis Ababa, Hawassa, Wolayita Sodo, and Gambella to include three drug susceptible and three drug resistant 4.2.2.2 isolates, which exhibited at least isoniazid (INH) resistance. These six clinically characterized Mtb isolates were further investigated using WGS.
Growth of M. tuberculosis isolates in vitro
To generate the desired logarithmic-phase growth of bacilli, six clinical isolates of Mtb sub-lineage 4.2.2.2 were cultured to an optical density of 0.2 at 600 nm (OD600). These isolates were inoculated in a 1:4 ratio (32 ml total volume) using Middlebrook 7H9 broth, supplemented with 10% oleic acid, albumin, dextrose, catalase enrichment, 0.5% (v/v) glycerol, and 0.05% Tween 80, then incubated at 37°C for 11–15 days until they reached the mid-log phase of growth, with OD600 values ranging from 0.3 to 0.5.
Phenotypic DST
The phenotypic drug susceptibility patterns of the isolates were determined using the BACTEC MGIT™ 960 system with the SIRE kit. Frozen isolates were thawed and subcultured in MGIT tubes. The day the MGIT system indicated a positive signal was recorded as day 0, tubes were incubated for one more day before commencing the DST. For signals detected on days 1 or 2, no dilution was required. However, for signals observed on days 3 to 5, a 1:5 dilution with sterile saline was performed prior to DST inoculation. If growth was detected after day 5, the samples were vortexed, diluted 1:100 with sterile saline, and 0.5 ml of the diluted sample was inoculated into a MGIT tube. The experiment was performed in accordance with the manufacturer's established standard operating procedures (Siddiqi and Rüsch-Gerdes ; Tilahun et al., ).
DST for first- and second-line drug resistance was conducted following the WHO technical manual (Siddiqi and Rüsch-Gerdes, ). The drug concentrations used were as follows: streptomycin (STM) 1.0 μg ml−1, INH 0.1 μg ml−1, rifampicin (RIF) 1.0 μg ml−1, ethambutol (EMB) 5.0 μg ml−1, bedaquiline 1.0 μg ml−1, delamanid 0.06 μg ml−1, moxifloxacin 0.25 μg ml−1, levofloxacin 1.0 μg ml−1, linezolid 1.0 μg ml−1, ofloxacin 2.0 μg ml−1, and clofazimine 1.0 μg ml−1. The DST process was marked as complete by the instrument when the growth control reached a growth unit (GU) value of 400. If the GU value was 400 or higher and the drug-containing tube had a reading below 100, the result was reported as “susceptible.” Conversely, if the GU value reached 400 and the drug-containing tube reading exceeded 100, the result was reported as “resistant.” Results were deemed invalid if the growth control GU value reached 400 in less than 4 days or failed to reach 400 within 21 days, triggering error messages X400 and X200, respectively. Quality control was ensured by testing each batch of MGIT medium and SIRE Kit with the pan-susceptible laboratory strain M. tuberculosis H37Rv (Tilahun et al., ).
DNA extraction
For WGS of Mtb, high-quality DNA ranging from 0.5 to 5 µg with a concentration above 20 ng µl−1 was extracted using the Restricted fragment length polymorphism (RFLP) method. In brief, mycobacterial colonies grown on Lowenstein Jensen growth media were harvested into distilled water, followed by heat treatment and incubation with lysozyme, proteinase K, and NaCl/CTAB solutions. After DNA extraction with chloroform/isoamyl alcohol, the nucleic acids were precipitated with isopropanol and subsequently washed with ethanol. The DNA was dried, dissolved in molecular-grade water, and integrity assessed by agarose gel electrophoresis and Nanodrop spectrophotometer. DNA concentration was quantified using a Qubit fluorometer to ensure that the DNA met the necessary quality standards for sequencing (Dashti et al., ; Riaz et al., ).
WGS bioinformatic analyses
The quality of the raw sequence reads was first evaluated using FASTQC (v0.12.1) (Yang et al., ). Low-quality reads, short fragments, and adapter sequences were then removed with Trimmomatic (v0.39) (Daniyarov et al., ), The remaining reads were subsequently mapped to the H37Rv reference genome (NC 000962.3) using BWA (v0.7.18) (Li, ). Single nucleotide polymorphisms (SNPs) were identified using the bcftools (v1.8) (Phelan et al., ). Drug resistance profiles and lineages were predicted with TBProfiler (v6.2.0) (Verboven et al., ). SNP based phylogenetic trees were constructed using IQ-REE (v2.0.3) with maximum likelihood model by replicating 1000 bootstraps value, and annotated and visualized using interactive Tree of Life (iTOL) (v6.9) (Yu ).
Total RNA extraction
A 30-ml mid-log phase culture was transferred to 50 ml screw-cap tubes. The tubes were centrifuged at 2200 g at 4°C for 10 min to collect mycobacterial cells, discarding the supernatant. The resulting cell pellet was mixed with 1.2 ml TRIzol™ Reagent (Thermo Fisher Scientific, USA). The mixture was then transferred to a Lysing Matrix B tube (MP Biomedicals, Santa Ana, CA, USA) containing 0.1 mm silica beads. Bead beating was performed using a Fast Prep-24 5 G instrument (MP Biomedicals) at a speed setting of 6.5 for 45 s, followed by chloroform clean-up as previously described (Garton et al., ; Wildner et al., ).
Samples were purified and DNase-treated using RNeasy® Mini Columns (Qiagen) following the manufacturer’s protocol. The purified RNA samples were quantified using the Nanodrop Spectrophotometer and RNA quality was assessed using the Agilent 4150 TapeStation system. Quality of the RNA samples was ensured by including samples with an RNA integrity number (RIN) >7 and an absorbance A260/A280 ratio between 1.8 and 2.1.
Ribosomal RNA was removed using the Illumina Ribo-Zero Plus Microbiome rRNA Depletion Kit (Illumina, USA) and libraries prepared for sequencing using TruSeq Stranded Total RNA kit designed for Illumina sequencing systems. Sequencing was performed using the Illumina NextSeq2000 platform, utilizing a P3 100-cycle protocol.
RNAseq data analysis
FASTQC (v0.12.1) was used to visualize the quality of raw reads. Reads with low quality sequences, a Phred score less than 20 (Q < 20), and short fragments having <50 base-pairs were filtered out, and adapter sequences were trimmed using Fastp (v0.23.4) (Mohideen et al., ). Taxonomic identification and relative abundance of reads were assessed using Kraken software (Lu et al., ). Using the M. tuberculosis H37Rv (NC 000962.3) reference sequence obtained from the NCBI Database, clean reads were aligned and quantified using Hisat2 version 2.1.0 (Kim et al., ) and featureCounts version 2.0.6 (Liao et al., ), respectively. Subsequently, differential expression of genes was analyzed using DESeq2 package in R version 4.3.2 (Liu et al., ). Differentially expressed genes (DEGs) were identified, applying a cutoff threshold of log2 fold change > 1 comparing drug-resistant to drug-susceptible lineage 4.2.2.2 clinical isolates. P-values were adjusted for multiple comparisons using the Benjamini–Hochberg procedure with a significance cutoff P < 0.05 (Anders and Huber, ). Finally, Gene Ontology (GO) analysis, functional enrichment, and pathways analysis were conducted.
Ethical clearance
Written informed consent was obtained from each patient prior to sample collection, ensuring their confidentiality and anonymity throughout the process. This study was approved by the Institutional Review Board of the College of Health Sciences, Addis Ababa University (Approval No. 072/21/Sop). This research is part of the TBGEN study—An integrated approach to unraveling susceptibility to tuberculosis in Africa, and received ethical approval from the Armauer Hansen Research Institute Institutional Ethics Review Committee (Approval No. P031/18).
Results
Discordance of phenotypic and whole genome-based drug resistance determination in M. tuberculosis clinical isolates
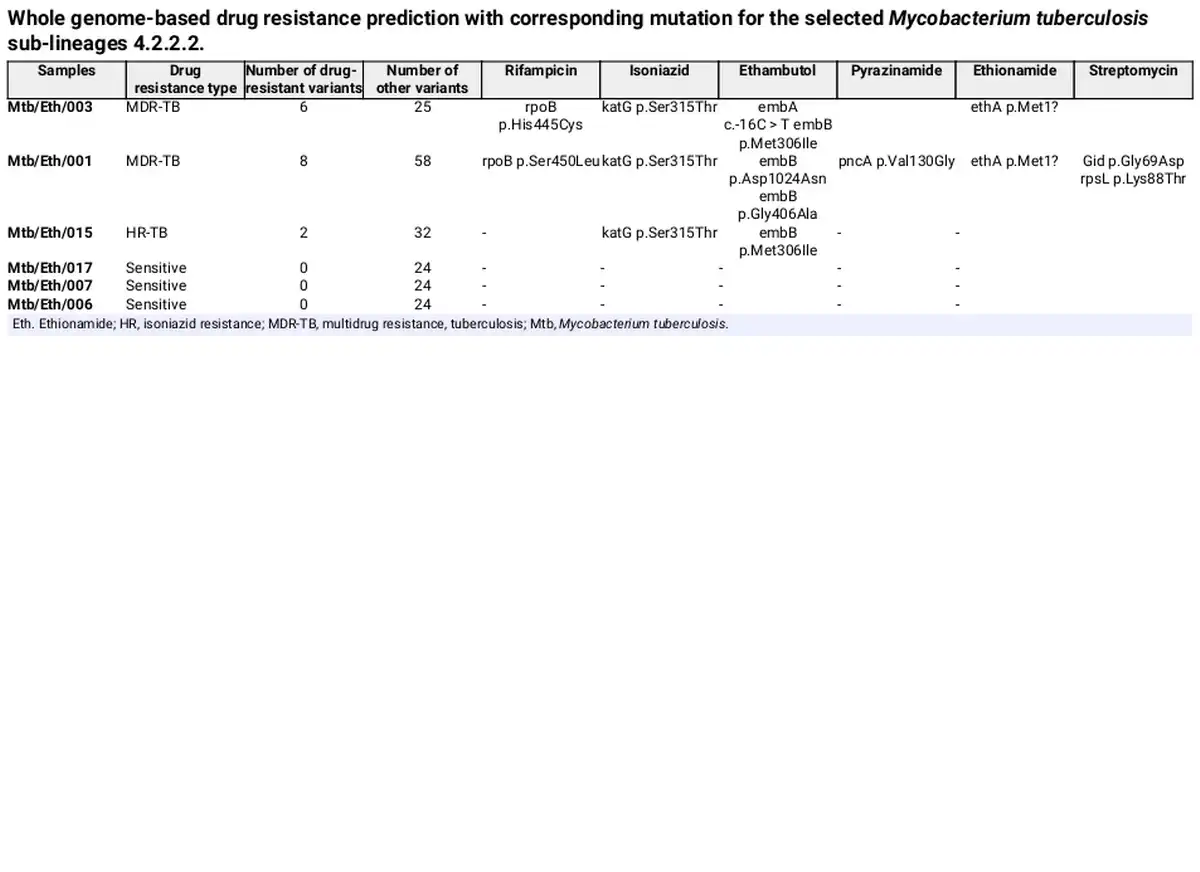
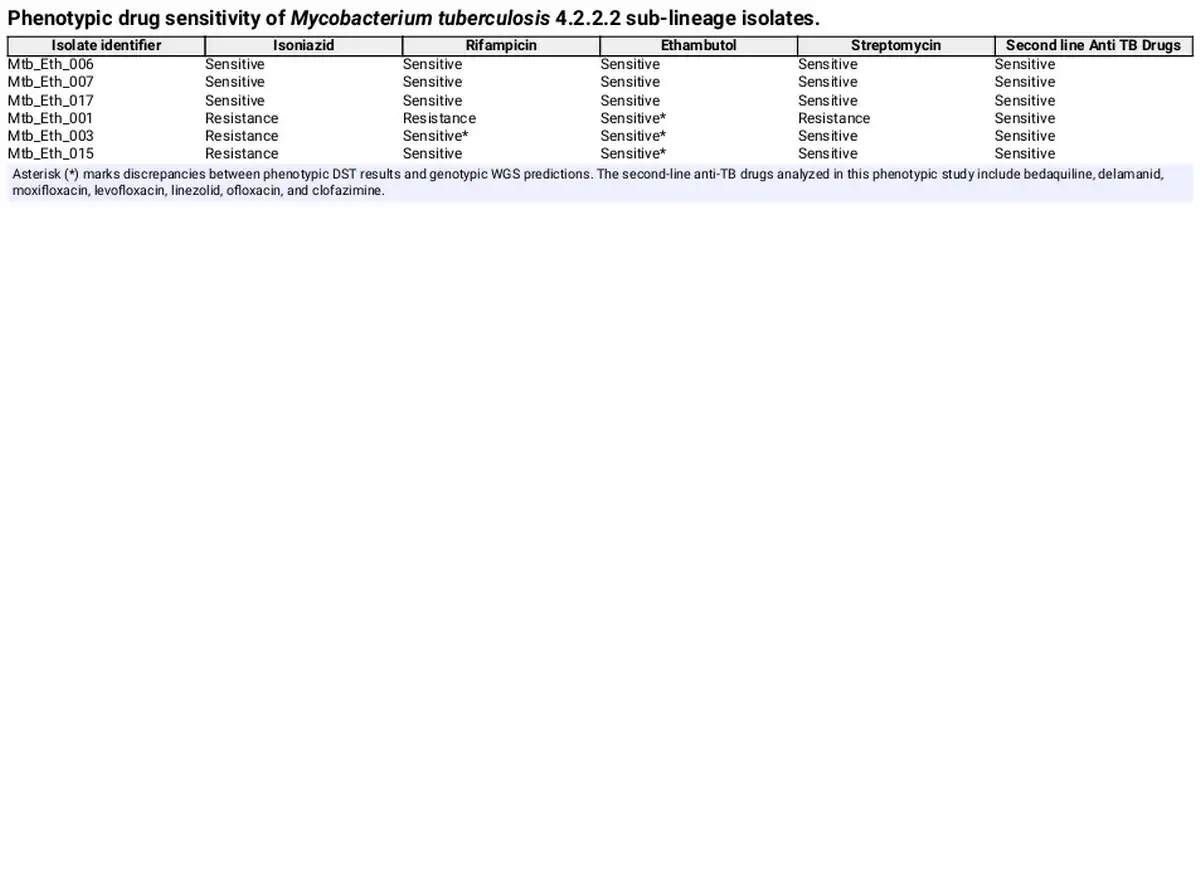
WGS was conducted on six Mtb sublineage 4.2.2.2 clinical isolates, including three drug-resistant and three drug-sensitive strains. From WGS analysis, two isolates (Mtb/Eth/001 and Mtb/Eth/003) were predicted as MDR (INH and RIF resistance), while one isolate (Mtb/Eth/015) was predicted to be resistant to INH and EMB (Fig. 1). All three resistant strains shared the same INH resistance conferring katG p.Ser315Thr mutation (Table 1). Additionally, the two isolates (Mtb/Eth/001 and Mtb/Eth/003) resistant to ethionamide carried an identical ethA p.Met1 mutation (loss of start codon). WGS analysis revealed distinct mutations associated with resistance to RIF in the two MDR-TB isolates (Table 1). Interestingly, phenotypic drug sensitivity testing indicated discrepancies, identifying only one isolate as RIF resistant and therefore MDR (isolate Mtb/Eth/001) as the WGS predicted RIF and STM resistance was not observed in phenotypic DST for isolate Mtb/Eth/003 (Table 2).

Figure 1
Phylogeny tree with drug susceptibility of Mycobacterium tuberculosis sub-lineage 4.2.2.2 isolates. NC 000962.3 is a Mtb H37Rv reference genome downloaded from NCBI. Shaded circles indicate a prediction of drug-resistance from the WGS analysis.
Transcriptomic profiles of resistant and sensitive M. tuberculosis
Following quality control and trimming procedures, ~12 to 16 million single-ended reads for each sample were aligned to the M. tuberculosis (H37Rv) reference genome, with an average alignment rate of 85.26%. Differential expression analysis revealed six genes that were differentially regulated in drug-resistant compared to the drug-sensitive isolates. All the six genes: Rv0096, Rv2780, Rv3136, Rv3136A, Rv3137, and Rv3230c were downregulated in the drug-resistant group. These DEGs were visualized using a clustered heat map and volcano plot as depicted in Figs 2 and 3. The DEGs were classified according to functional categories as detailed in Table 3.
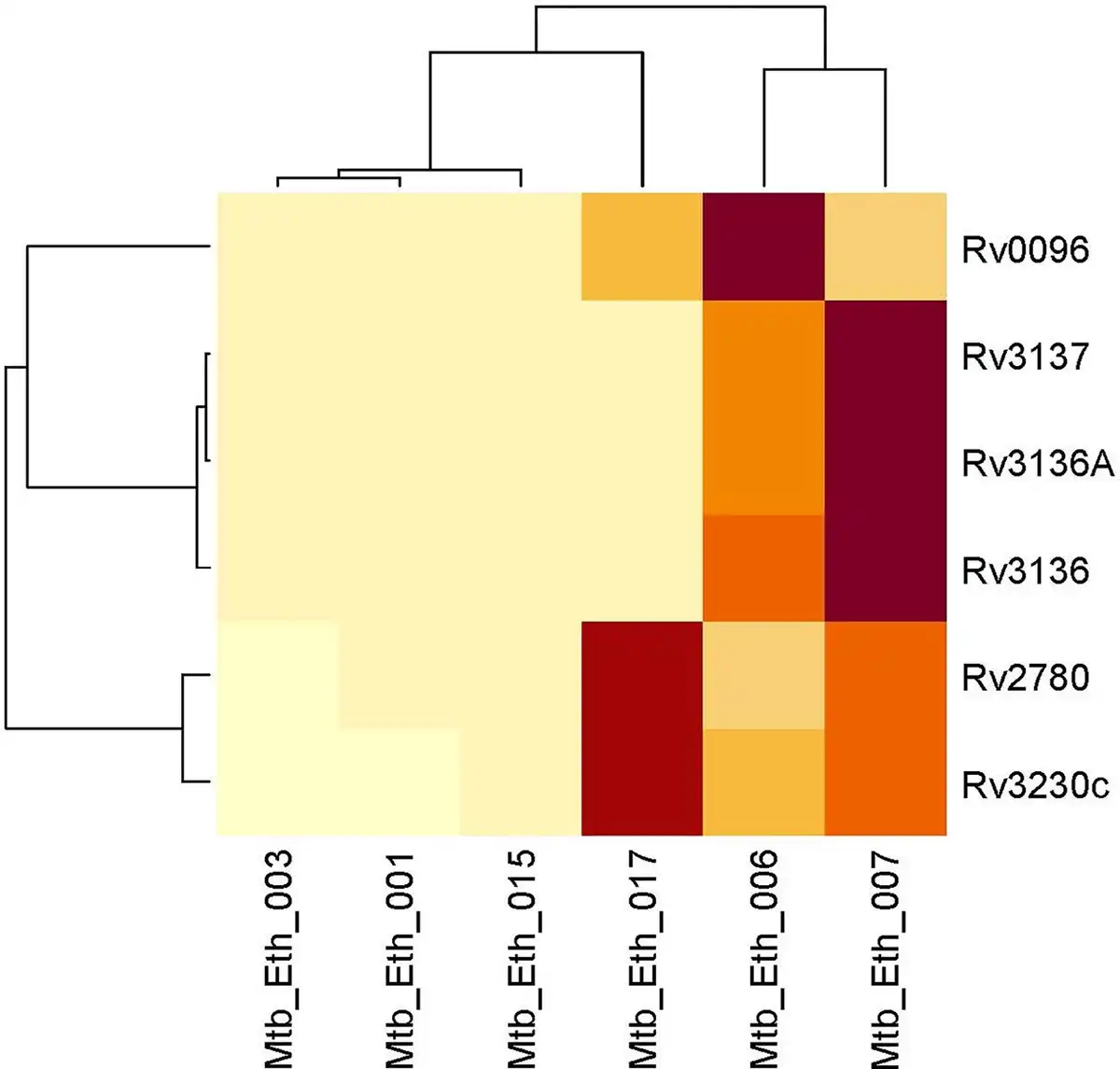
Figure 2
Clustered heatmap of significantly differentially expressed genes between drug resistant and drug-sensitive Mycobacterium tuberculosis lineage 4.2.2.2 isolates. The heat map displays the expression levels of genes across different isolates, with rows representing individual genes and columns representing clinical isolates. The color intensity indicates expression levels, with light yellow signifying lower expression and dark red indicating higher expression. The dendrograms on the top and left show hierarchical clustering, grouping genes and samples based on their expression similarities.
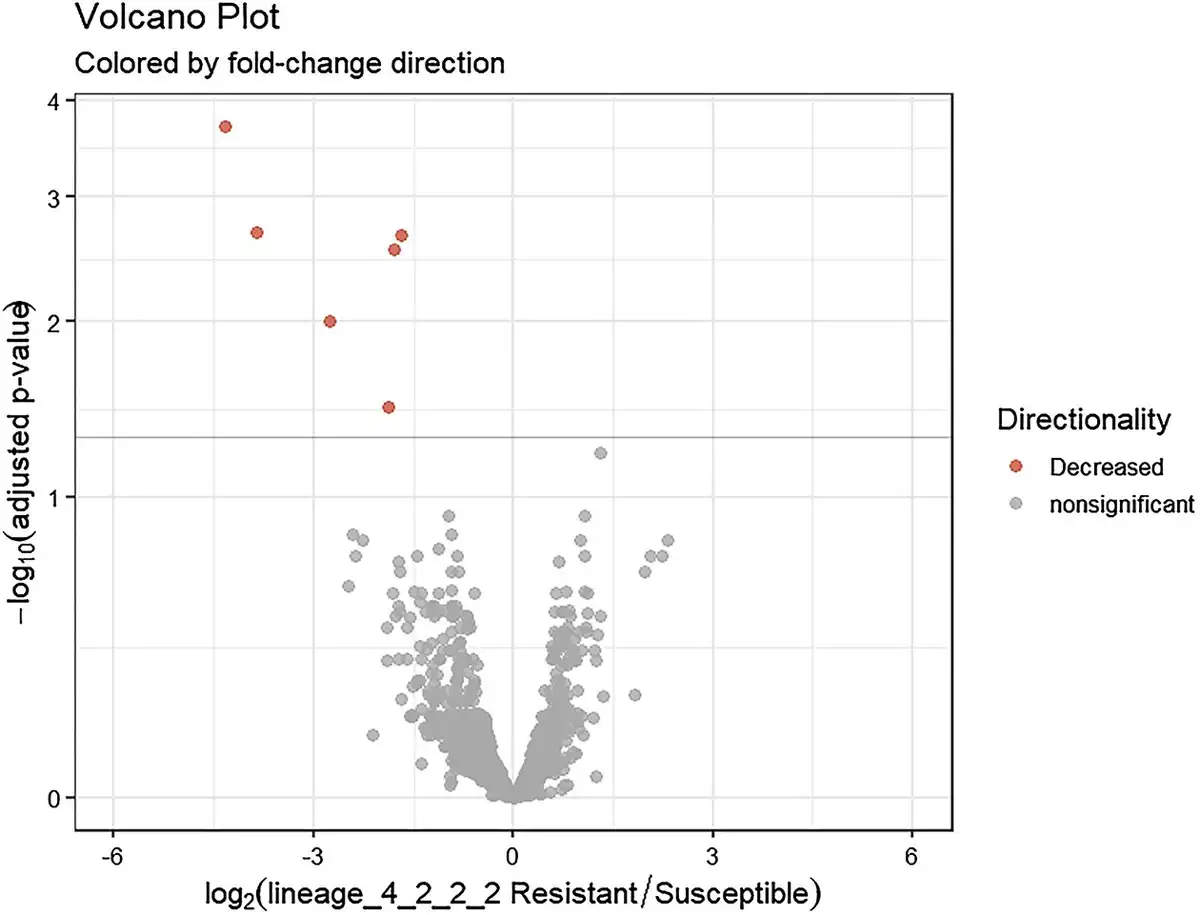
Figure 3
Volcano plot illustrating differentially expressed genes between drug-resistant and drug-sensitive Mtb sub-lineage 4.2.2.2 isolates. The x-axis represents the log2 fold change and the y-axis shows the −log10 P-values. Negative values indicate downregulation in drug-resistant relative to drug sensitive isolates. Red coloring denotes significantly downregulated genes with >1 log2 fold change and <0.05 adjusted P-value.
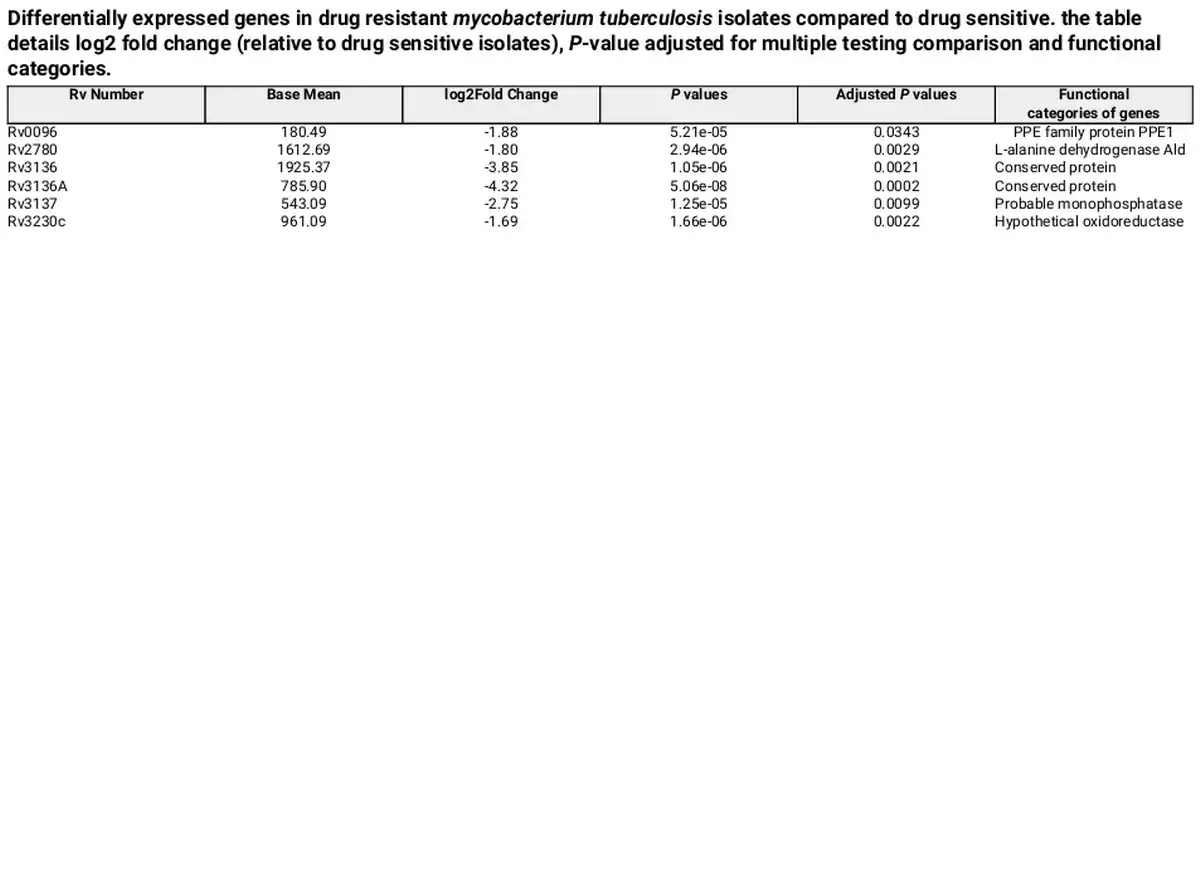
Table 3. Differentially expressed genes in drug resistant mycobacterium tuberculosis isolates compared to drug sensitive. the table details log2 fold change (relative to drug sensitive isolates), P-value adjusted for multiple testing comparison and functional categories.
Discussion
TB poses a significant global health challenge, and the emergence of drug-resistant Mtb strains further complicates control efforts. Unlike some bacteria, Mtb does not gain drug resistance through horizontal gene transfer but by the accumulation of drug-resistance conferring chromosomal mutations. A deeper understanding of drug resistance mechanisms is crucial for developing drugs that can overcome existing resistance or target alternative biochemical pathways. This knowledge will not only help prevent the emergence of drug resistance but also identify cellular components that impact the action of anti-TB drugs.
A missense mutation in rpoB p.Ser450Leu aligns with phenotypic drug resistance in MDR isolate Mtb/Eth/001, whereas the rpoB p.His445Cys missense mutation did not confer phenotypic resistance to rifampicin in isolate Mtb/Eth/003 in our hands. The rpoB p.Ser450Leu variant has been documented in the WHO Catalogue of Mutations in the M. tuberculosis complex and their association with drug resistance, demonstrating a sensitivity of 64.4% and a specificity of 99.3% for detecting rifampicin-resistant phenotypes. However, rpoB p.His445Cys variant has a sensitivity of 0.4% and a specificity of 100% for detecting rifampicin-resistant phenotypes (World Health Organization, ). The substitution of the polar amino acid serine with the non-polar leucine at position 450 may disrupt rifampicin binding to rpoB, leading to drug resistance (Go and Miyazawa, ). No additional variants or SNP differences were detected in the rpoB gene between the two clinical isolates, both of which were classified as MDR-TB based on WGS analysis. The katG p.Ser315Thr missense mutation was associated with phenotypic drug resistance in all drug resistant isolates and is also reported in WHO Catalogue with a sensitivity of 77.8% and a specificity of 99.1% for detecting INH-resistant bacilli (World Health Organization, ).
Our phenotypic drug sensitivity results indicate susceptibility to EMB (Mtb/Eth/001 and Mtb/Eth/003), whereas WGS-based drug sensitivity analysis identified resistance conferring mutations, with different mutations occurring at various positions for different samples. In line with our finding, routine phenotypic analysis failed to detect EMB resistance in 91.4% of resistant isolates, highlighting the challenges of EMB phenotypic testing. The inability of culture-based methods to reliably identify true EMB resistance negatively impacts TB control programs. However, a significant proportion of phenotypically EMB-resistant isolates (~30%) still lack identifiable mutations in embB, underscoring the need for a comprehensive understanding of EMB resistance mechanisms in clinical isolates (Johnson et al., ). Differences between molecular and phenotypic ethambutol resistance results are likely due to limitations in conventional susceptibility testing methods (Plinke et al., ). Our study found higher drug resistance predicted through WGS compared to phenotypic drug sensitivity testing. This discrepancy may stem from the difficulties in generating accurate and reproducible drug sensitivity data from pathogenic clinical isolates in the laboratory. Given these discrepancies, an improved and an integrative approach combining phenotypic and genotypic methods is essential for accurately characterizing drug resistance and optimizing TB treatment strategies.
By analyzing gene expression levels and genetic regions associated with differential expression, we identified six DEGs in our Mtb sub-lineage 4.2.2.2 drug-resistant isolates that may be linked to the acquisition of or adaptation to drug resistance. Rv0096, Rv2780, Rv3136, Rv3136A, Rv3137 and Rv3230c were downregulated in the drug-resistant isolates compared to drug sensitive Mtb sub-lineage 4.2.2.2. These genes are not direct targets of the current anti-TB drugs. Additionally, no common SNPs were observed in the six DEGs in the drug-resistant group compared to the drug sensitive group.
The Rv0096 (PPE1) gene encodes a putative member of the PPE (proline-glutamate and proline-proline-glutamate) family and is part of the rv0096–rv0101 operon, which has been shown to produce a virulence-associated lipopeptide (Sao Emani and Reiling, ). This operon encodes enzymes responsible for synthesizing an isonitrile lipopeptide, potentially involved in biofilm formation and copper acquisition under copper-limited conditions. Recent studies suggest that PPE1 is a functional component of this cluster, highlighting its co-essentiality with the other genes in the operon (Jinich et al., ). Consistent with our findings, downregulation of Rv0096 has been reported in rifampicin-resistant Mtb H37Rv strains carrying the H526Y mutation in the rpoB gene, but not in rifampicin-susceptible H37Rv wild-type strains (de Knegt et al., ). A loss-of-function mutation in Rv0096 was also found to confer resistance to D-cycloserine in MDR Mtb (Sao Emani and Reiling, ).
Rv2780, also downregulated in drug-resistant isolates, encodes L-alanine dehydrogenase (ald), an enzyme catalyzing the oxidative deamination of L-alanine to pyruvate, which is subsequently utilized in peptidoglycan synthesis (Sao Emani and Reiling, ). Rv2780 is also shown to catalyze the reductive amination of glyoxylate to glycine and is upregulated under conditions such as hypoxia, nutrient starvation, and when alanine serves as the sole nitrogen source, suggesting multiple physiological roles, particularly in carbon and nitrogen metabolism (Sao Emani and Reiling, ). Desjardins et al. () showed that deletion of ald increased Mtb resistance to D-cycloserine and reintroducing the gene partially restored susceptibility to the drug. L-alanine treatment effectively inhibited bacterial survival, comparable to RIF, and a combination of L-alanine with RIF further enhanced bacterial clearance, suggesting L-alanine as a promising adjunct to TB therapy. The small-molecule and Rv2780 inhibitor (S)-N-(5-(3-fluorobenzyl)-1H-1,2,4-triazol-3-yl) tetrahydrofuran-2-carboxamide (GWP-042) significantly reduced the survival of both Mtb H37Rv and multidrug-resistant (MDR-TB) strains.
This study also revealed that the cluster of genes Rv3136, Rv3136A, and Rv3137 were repressed in drug-resistant relative to drug-sensitive sub-lineage 4.2.2.2 isolates. Rv3136 encodes the PPE51 protein, which has been implicated in Mtb’s ability to uptake disaccharides. Nonsynonymous mutations in Rv3136 were resistant to high concentrations of thio-disaccharides, known for their bactericidal effect on Mtb, restoring with a functional gene reinstated their sensitivity to the wild type level (Korycka-Machała et al., ). Our study also observed a downregulation of Rv3135 (ppe50), with a log fold change of 2, though the difference was not statistically significant (Supplementary Table 1). The ppe50-ppe51 operon is critical for INH and RIF tolerance phenotypes in RNase J (Rv2752c) mutant Mtb strains. Deleting this operon induced drug tolerance and overexpression restored drug sensitivity, highlighting its role in drug susceptibility (Martini et al., ). Additionally, a study identified a crucial role of ppe50-ppe51 mutants in enhancing survival and contributing to the development of resistance (Bellerose, ). Our study also demonstrated reduced expression of the Rv3136A gene, encoding a conserved 110-amino-acid protein of unknown function. Rv3137, a member of the inositol monophosphatase (IMPase) gene family was also downregulated. This gene functions as the Mtb histidinol phosphate phosphatase (HolPase), specifically catalyzing the dephosphorylation of histidinol phosphate (Jha et al., ). Mtb has developed strategies to acquire amino acids from host cells, while also maintaining some de novo biosynthesis pathways. In response, the host mounts an immune defense by upregulating histidine-catabolizing enzymes through interferon gamma (IFN-γ)-mediated signaling, aiming to deprive the bacillus of intracellular free histidine. Mtb circumvents this immune response by synthesizing histidine de novo, and histidine auxotrophs are unable to proliferate (Dwivedy et al., ). Studies have demonstrated an association between INH resistance and reduced expression of Rv3137 in INH-resistant groups compared to susceptible groups (Mehaffy et al., ) (Martini et al., ). Rv3137 is also downregulated during exponential phase in the highly successful Mtb MtZ strain from Aragon, Spain (Comín et al. ). The observed downregulation of Rv3137 in highly successful Mtb strains as well as in drug-resistant isolates aligns with our findings and perhaps highlights the role of this gene in pathogenesis and drug resistance.
Rv3230c, also repressed in drug-resistant isolates, encodes an oxidoreductase that forms a functional complex with the integral membrane stearoyl-CoA desaturase DesA3 to convert saturated stearic acid into unsaturated oleic acid, utilizing molecular oxygen and NADPH in the process. Oleic acid is critical for maintaining membrane composition and physiological functions by regulating membrane fluidity through the incorporation of unsaturated fatty acids into membrane lipids. Oleic acid is thought to serve as a precursor in mycolic acid synthesis, contributing to the introduction of double bonds in specific mycolic acid structures. This suggests that DesA3 and Rv3230c are likely essential for lipid metabolism, playing key roles in the formation of both the cell membrane and cell wall in Mtb (Rehberg et al., ). A genomic study conducted on the clonal expansion of XDR-TB of the rare Proto-Beijing genotype identified a 1 285 bp deletion within the desA3 and oxidoreductase Rv3230c genes (Srilohasin et al., ). Furthermore, a WGS study conducted in Hanoi revealed a significant association between a 1-bp deletion in Rv3230c and clustered strains carrying the katG-S315T mutation, a key marker of INH resistance among the population (Hang et al., ).
A gene network analysis revealed that alternative sigma factors may regulate the downregulated genes. Specifically, sigma M (SigM) suppresses the expression of Rv3136 and Rv3137 while also modulating the regulation of Rv0096 (Raman et al., ). As a member of the extracytoplasmic function subfamily of alternative sigma factors, Mtb SigM is expressed at low levels in vitro and does not appear to play a role in stress response regulation. Instead, SigM positively regulates genes involved in the synthesis of surface or secreted molecules. Its role in repressing virulence-associated surface lipids while upregulating Esx family secreted proteins and nonribosomal peptide synthetase genes suggests that SigM may be involved in long-term adaptation to specific host environments during infection (Raman et al., ). The regulation of three of six genes by SigM suggests that it may play a role in the adaptation to drug resistance in lineage 4.2.2.2. Additionally, Rv3230c is negatively regulated by the alternative sigma factor SigD, encoded by Rv3414c (Raman et al., ). Further research should prioritize exploring the mechanisms underlying resistance to anti-TB drugs, including pathways differentially regulated in drug resistant isolates that may enable bacilli to adjust to the impact that drug-conferring mutations may have on Mtb metabolism.
Although this study offers insights into drug resistance mechanisms in Mtb, it has limitations. One notable limitation of the research lies in the small sample size, which can be attributed to the difficulties in obtaining suitable samples that adequately represent different drug resistance profiles in preferred Mtb sub-lineages.
Overall, our analysis of Mtb sub-lineage 4.2.2.2 clinical isolates comparing different drug resistance profiles to drug sensitive Mtb, revealed six common DEGs (Rv0096, Rv2780, Rv3136, Rv3136A, Rv3137, and Rv3230c) associated with drug resistance. The regulation of these genes was not directly linked to primary drug targets, nor impacted by common chromosomal SNPs. These findings highlight the potential of integrating phenotypic, genomic, and transcriptomic data to enhance our understanding of drug resistance mechanisms and identify promising drug targets.
Acknowledgments
We sincerely thank the Armauer Hansen Research Institute (AHRI) for hosting this study and granting access to its facilities. We are also grateful to Addis Ababa University for their support. Our deepest appreciation goes to the dedicated team members of the TBGEN project for their invaluable contributions. H3Africa is managed by the Science for Africa Foundation (SFA Foundation) in partnership with the Wellcome Trust, NIH, and AfSHG. The views expressed in this work are those of the authors and do not necessarily reflect those of the SFA Foundation or its partners.
References
- Anders S, Huber W. Differential expression of RNA-seq data at the gene level–the DESeq package. European Mol Biol Lab (EMBL), Heidelberg Germany . 2012;10:f1000research.
- Bellerose MM. Genetic identification of novel mycobacterium tuberculosis susceptibility and survival mechanisms during antibiotic treatment. A dissertation submitted to UMass Chan Medical School. 2020, 10.13028/kz6e-z069.
- Brosch R, Gordon SV, Marmiesse M, et al a new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci USA. 2002;99:3684–9. 10.1073/pnas.052548299
- Cerezo-Cortés MI, Rodríguez-Castillo JG, Mata-Espinosa DA, et al Close related drug-resistance Beijing isolates of Mycobacterium tuberculosis reveal a different transcriptomic signature in a murine disease progression model. Int J Mol Sci. 2022;23:5157. 10.3390/ijms23095157
- Comín J, Campos E, Gonzalo-Asensio J et al. Transcriptomic profile of the most successful Mycobacterium tuberculosis strain in Aragon, the MtZ strain, during exponential and stationary growth phases. Microbiol Spectr. 2023;11:e04685–04622. 10.1128/spectrum.04685-22
- Daniyarov A, Molkenov A, Rakhimova S, et al Whole genome sequence data of Mycobacterium tuberculosis XDR strain, isolated from patient in Kazakhstan. Data Brief. 2020;33:106416. 10.1016/j.dib.2020.106416
- Dashti AA, Jadaon MM, Abdulsamad AM et al. Heat treatment of bacteria: a simple method of DNA extraction for molecular techniques. Kuwait Med J. 2009;41:117–22.
- de Knegt GJ, Bruning O, ten Kate MT, et al Rifampicin-induced transcriptome response in rifampicin-resistant Mycobacterium tuberculosis. Tuberculosis. 2013;93:96–101. 10.1016/j.tube.2012.10.013
- Desjardins CA, Cohen KA, Munsamy V, et al Genomic and functional analyses of Mycobacterium tuberculosis strains implicate ald in D-cycloserine resistance. Nat Genet. 2016;48:544–51. 10.1038/ng.3548
- Dwivedy A, Ashraf A, Jha B, et al De novo histidine biosynthesis protects Mycobacterium tuberculosis from host IFN-γ mediated histidine starvation. Commun Biol. 2021;4:410. 10.1038/s42003-021-01926-4
- Garton NJ, Waddell S, Sherratt AL, et al Cytological and transcript analyses reveal fat and lazy persister-like bacilli in tuberculous sputum. PLoS Med. 2008;5:e75. 10.1371/journal.pmed.0050075
- Go M, Miyazawa S. Relationship between mutability, polarity and exteriority of amino acid residues in protein evolution. Int J Pept Protein Res. 1980;15:211–24. 10.1111/j.1399-3011.1980.tb02570.x
- Goletti D, Meintjes G, Andrade BB, et al Insights from the 2024 WHO Global Tuberculosis Report–More comprehensive action, innovation, and investments required for achieving WHO End TB goals. Int J Infect Dis. 2025;150:107325. 10.1016/j.ijid.2024.107325
- Gomez-Gonzalez PJ, Andreu N, Phelan JE, et al An integrated whole genome analysis of Mycobacterium tuberculosis reveals insights into relationship between its genome, transcriptome and methylome. Sci Rep. 2019;9:5204. 10.1038/s41598-019-41692-2
- Guyeux C, Senelle G, Le Meur A, et al Newly identified Mycobacterium africanum lineage 10, Central Africa. Emerg Infect Dis. 2024;30:560. 10.3201/eid3003.231466
- Hang NTL, Hijikata M, Maeda S, et al Whole genome sequencing, analyses of drug resistance-conferring mutations, and correlation with transmission of mycobacterium tuberculosis carrying katG-S315T in Hanoi, Vietnam. Sci Rep. 2019;9:15354. 10.1038/s41598-019-51812-7
- Jha B, Kumar D, Sharma A, et al Identification and structural characterization of a histidinol phosphate phosphatase from Mycobacterium tuberculosis. J Biol Chem. 2018;293:10102–18. 10.1074/jbc.RA118.002299
- Jinich A, Nazia SZ, Tellez AV, et al Genome-wide co-essentiality analysis in Mycobacterium tuberculosis reveals an itaconate defense enzyme module. bioRxiv. 2022. 10.1101/2022.09.27.509804
- Johnson R, Streicher EM, Louw GE, et al Drug resistance in mycobacterium tuberculosis. Curr Issues Mol Biol. 2006;8:97–112.
- Kim D, Paggi JM, Park C, et al Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907–15. 10.1038/s41587-019-0201-4
- Korycka-Machała M, Pawełczyk J, Borówka P, et al PPE51 is involved in the uptake of disaccharides by mycobacterium tuberculosis. Cells. 2020;9:603. 10.3390/cells9030603
- Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. [q-bio.gn]. 2013, 10.48550/ar.Xiv/.1303.3997
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30. 10.1093/bioinformatics/btt656
- Liu S, Wang Z, Zhu R, et al Three differential expression analysis methods for RNA sequencing: limma, EdgeR, DESeq2. JoVE (J Visual Exp). 2021;175:e62528.
- Lu J, Breitwieser FP, Thielen P et al. Bracken: estimating species abundance in metagenomics data. PeerJ Computer Sci. 2017;3:e104. 10.7717/peerj-cs.104
- Martini MC, Hicks ND, Xiao J, et al Loss of RNase J leads to multi-drug tolerance and accumulation of highly structured mRNA fragments in Mycobacterium tuberculosis. PLoS Pathog. 2022;18:e1010705. 10.1371/journal.ppat.1010705
- Mehaffy C, Islam MN, Fitzgerald B, et al Biochemical characterization of isoniazid-resistant mycobacterium tuberculosis: can the analysis of clonal strains reveal novel targetable pathways?. Mol Cell Proteomics. 2018;17:1685–701.
- Mekonnen D, Derbie A, Chanie A, et al Molecular epidemiology of M. tuberculosis in Ethiopia: a systematic review and meta-analysis. Tuberculosis. 2019;118:101858. 10.1016/j.tube.2019.101858
- Mekonnen D, Munshea A, Nibret E, et al Mycobacterium tuberculosis Sub-lineage 4.2. 2/SIT149 as dominant drug-resistant clade in northwest Ethiopia 2020–2022: in-silico whole-genome sequence analysis. IDR. 2023;16:6859–70. 10.2147/IDR.S429001
- Miotto P, Sorrentino R, De Giorgi S, et al Transcriptional regulation and drug resistance in Mycobacterium tuberculosis. Front Cell Infect Microbiol. 2022;12:990312. 10.3389/fcimb.2022.990312
- Mohideen AM, Johansen SD, Babiak I. High-throughput identification of adapters in single-read sequencing data. Biomolecules. 2020;10:878. 10.3390/biom10060878
- Phelan JE, Lim DR, Mitarai S, et al Mycobacterium tuberculosis whole genome sequencing provides insights into the Manila strain and drug-resistance mutations in the Philippines. Sci Rep. 2019;9:9305. 10.1038/s41598-019-45566-5
- Plinke C, Cox HS, Kalon S, et al Tuberculosis ethambutol resistance: concordance between phenotypic and genotypic test results. Tuberculosis. 2009;89:448–52. 10.1016/j.tube.2009.09.001
- Raman S, Hazra R, Dascher CC et al. Transcription regulation by the mycobacterium tuberculosis alternative sigma factor SigD and its role in virulence. J Bacteriol. 2004;186:6605–16. 10.1128/JB.186.19.6605-6616.2004
- Raman S, Puyang X, Cheng T-Y, et al Mycobacterium tuberculosis SigM positively regulates Esx secreted protein and nonribosomal peptide synthetase genes and down regulates virulence-associated surface lipid synthesis. J Bacteriol. 2006;188:8460–8. 10.1128/JB.01212-06
- Rehberg N, Omeje E, Ebada SS, et al 3-O-methyl-alkylgallates inhibit fatty acid desaturation in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2019;63: e00136–19. 10.1128/aac.00136-0019
- Riaz M, Mahmood Z, Javed MT, et al Drug resistant strains of Mycobacterium tuberculosis identified through PCR-RFLP from patients of Central Punjab, Pakistan. Int J Immunopathol Pharmacol. 2016;29:443–9. 10.1177/0394632016638100
- Sao Emani C, Reiling N. The efflux pumps Rv1877 and Rv0191 play differential roles in the protection of mycobacterium tuberculosis against chemical stress. Front Microbiol. 2024;15:1359188. 10.3389/fmicb.2024.1359188
- Siddiqi SH, Rüsch-Gerdes S. Foundation for innovative new diagnostics MGIT procedure manual for BACTEC MGIT 960 TB system. Geneva, Switzerland. 2006:41–51.
- Srilohasin P, Prammananan T, Faksri K, et al Genomic evidence supporting the clonal expansion of extensively drug-resistant tuberculosis bacteria belonging to a rare proto-Beijing genotype. Emerg Microbes Infect. 2020;9:2632–41.
- Tilahun M, Wegayehu T, Wondale B, et al Phenotypic and genotypic drug susceptibility patterns of Mycobacterium tuberculosis isolates from pulmonary tuberculosis patients in Central and southern Ethiopia. PLoS One. 2023;18:e0285063. 10.1371/journal.pone.0285063
- Torres Ortiz A, Coronel J, Vidal JR, et al Genomic signatures of pre-resistance in Mycobacterium tuberculosis. Nat Commun. 2021;12:7312. 10.1038/s41467-021-27616-7
- Verboven L, Phelan J, Heupink TH et al. TBProfiler for automated calling of the association with drug resistance of variants in Mycobacterium tuberculosis. PLoS One. 2022;17:e0279644. 10.1371/journal.pone.0279644
- Wan L, Hu P, Zhang L, et al Omics analysis of Mycobacterium tuberculosis isolates uncovers Rv3094c, an ethionamide metabolism-associated gene. Commun Biol. 2023;6:156. 10.1038/s42003-023-04433-w
- Wildner LM, Gould KA, Waddell SJ. Transcriptional profiling mycobacterium tuberculosis from patient sputa. Methods Mol Biol . 2018;1736:117–28. 10.1007/978-1-4939-7638-6_11
- World Health Organization Catalogue of Mutations in Mycobacterium Tuberculosis Complex and Their Association with Drug Resistance, Geneva, Switzerland, World Health Organization, 2021.
- Yang X, Liu D, Liu F, et al HTQC: a fast quality control toolkit for Illumina sequencing data. BMC Bioinf. 2013;14:1–4. 10.1186/1471-2105-14-33
- Yu G. Using ggtree to visualize data on tree-like structures. Curr Protoc Bioinformatics. 2020;69:e96. 10.1002/cpbi.96