Introduction
Usher syndrome (USH), the most common cause of combined deaf-blindness in humans, is a group of autosomal recessive disorders characterized by congenital sensorineural hearing loss (HL), retinitis pigmentosa (RP), and with or without vestibular dysfunction [Nolen et al., 2020]. The prevalence of USH varies in different populations but is estimated to range from 3.5 to 6 per 100,000 people [Yan and Liu, 2010]. Genetically and clinically, USH is a heterogeneous disorder [Yan and Liu, 2010], and according to the OMIM database, up to now, three loci and fifteen genes (fourteen causatives and one modifier) for USH have been reported. Most USH patients, according to the age of onset of the visual and auditory symptoms and the presence/absence of vestibular dysfunction, are categorized into four distinct types [Abad-Morales et al., 2020; Peter et al., 2021], with each type based on the causative gene being divided into several subtypes (Table 1). USH1, the most severe form, is featured by severe to profound congenital HL, childhood RP, and congenital vestibular dysfunction. To date, seven genes and three loci associated with the USH1 type have been reported, including MYO7A, USHIC, CDH23, PCDH15, USHIG, CIB2, ESPN, 21q21, 15q22-q23, and 10p11.21-q21.1, respectively (Table 1) [French et al., 2020]. USH2, the most common form, is characterized by moderate to severe congenital HL, late-onset RP (in the second decade of life), and normal vestibular function, which is the result of damaging variants in the USH2A, ADGRV1, and WHRN genes [Besnard et al., 2014; Toms et al., 2020]. Furthermore, PDZD7 has been suggested as a modifier gene for patients with USH2A, whose damaging variants result in earlier onset and more severe retinal disease [Ebermann et al., 2010]. A variant in CLRN1 or HARS1 genes leads to USH3, which is characterized by progressive HL, variable RP onset age, and varying vestibular abnormalities [Puffenberger et al., 2012; Toms et al., 2020]. USH3 is extremely rare, but it is prevalent in some populations, such as the Finish, Acadian, and Ashkenazi Jewish [Besnard et al., 2014]. The USH4 is a newly introduced type that is caused by a damaging variant in the ARSG gene [Khateb et al., 2018; Peter et al., 2021]. USH4 patients generally showed late-onset HL (∼40) and RP with normal vestibular function [Abad-Morales et al., 2020; Peter et al., 2021]. Some USH genes, including PDZD7, HARS, ABHD12, CIB2, ARSG, and ESPN, due to a few reported affected cases, are called ultra-rare USH genes [Nolen et al., 2020]. In addition to monogenic inheritance, some studies report digenic inheritance in USH, including PCDH15/CDH23 and GPR98/PDZD7 [Zheng et al., 2005; Ebermann et al., 2010]. The classification of USH based on clinical symptoms does not apply to all patients, and there are several reports that patients’ manifestations deviate from the canonical criteria of the USH main types, which is called atypical USH [Aparisi et al., 2014; Igelman et al., 2021]. These deviations may occur in the severity or age of onset of RP, HL, or the presence/absence of vestibular dysfunction. Genetic studies in these patients revealed that they had damaging variants in well-known or ultra-rare USH genes [Nolen et al., 2020].
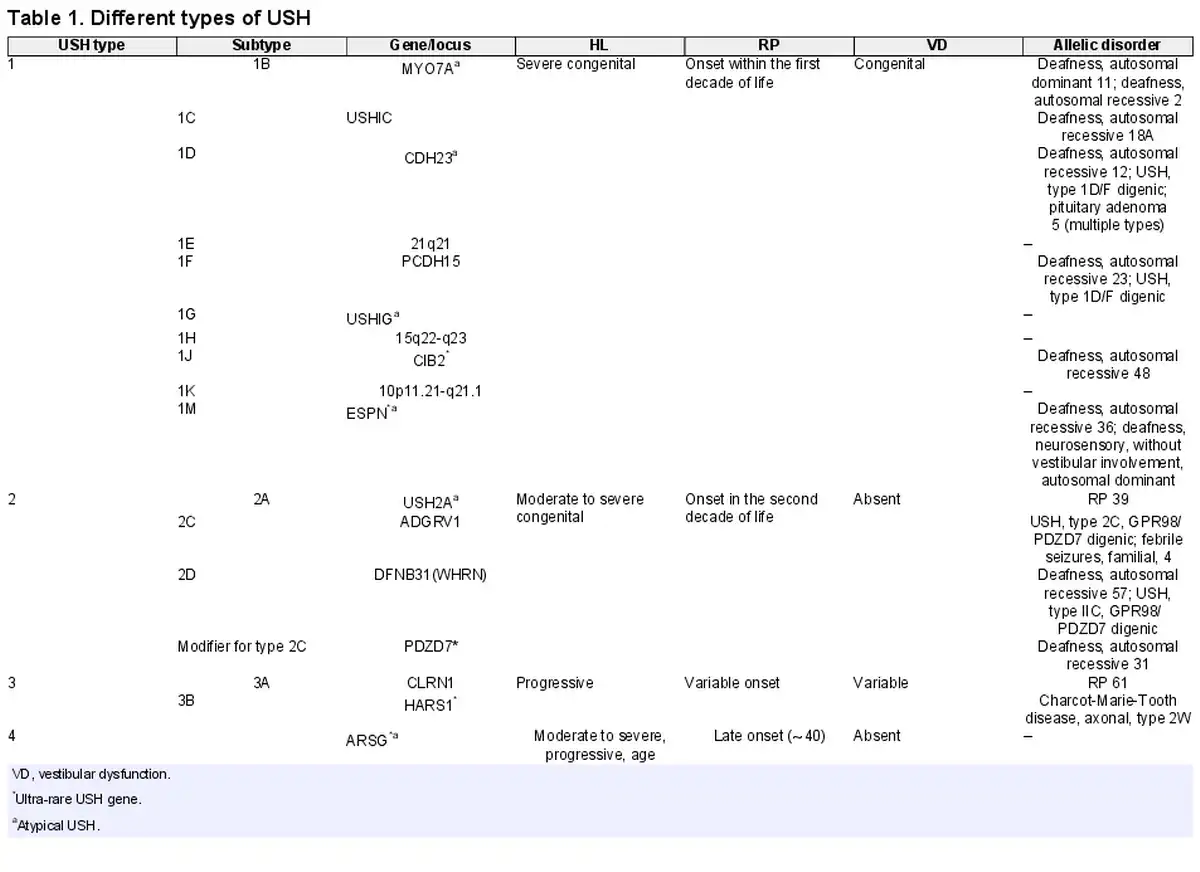
The CDH23 gene is the second most common USH gene, in which few atypical USH patients due to its damaging variants have been reported [Millán et al., 2011; Schultz et al., 2011] (Table 2). CDH23 gene encodes a calcium-dependent cell-cell adhesion glycoprotein with 27 Extracellular (EC) domains, a transmembrane domain, and a cytoplasmic domain [Pan and Zhang, 2012]. CDH23 and other USH proteins interact with each other and form an interaction network, which is called the USH interactome. USH interactome proteins are mainly colonized in stereocilia and the synaptic region of hair cells in the inner ear. The murine model revealed that the USH interactome is crucial for morphogenesis and development of the retina and hair bundle in the inner ear [Yan and Liu, 2010]. In addition to atypical USH, damaging variants in the CDH23 gene cause other allelic disorders, including USH1D and non-syndromic autosomal recessive deafness-12 (DFNB12) [Schultz et al., 2011]. Although it has been suggested that missense and truncating damaging variants in the CDH23 gene result in DFNB12 and USH1D, respectively [Valero et al., 2019], to the best of our knowledge, no genotype-phenotype correlation has yet been established in association with CDH23 damaging variants and atypical USH1D.
In this study, we used whole-exome sequencing (WES) to investigate 2 patients from an Iranian family and identified a novel homozygous nonsense variant (c.6562G>T; p.Glu2188Ter) in the CDH23 gene, which manifested atypical USH1D. Moreover, by collecting all atypical USH1D cases and those that violated the proposed genotype-phenotype correlation for the CDH23 gene allelic disorders, we found that there was no clear-cut genotype-phenotype correlation and nearly all types of genotypes in USH1D, atypical USH1D, and DFNB12 have been reported.
Material and Methods
Subjects
A consanguineous Iranian family with 2 affected patients whose manifesting phenotypes were consistent with USH was referred for genetic counseling. A comprehensive questionnaire was taken regarding family history, pregnancy complications, environmental risks, developmental milestones, and disease progression.
Genomic DNA Preparation
Venous blood samples were collected in ethylenediaminetetraacetic acid-containing tubes from the patients (IV-1 and IV-2), parents (III-1 and III-2), and healthy sibling (IV-3) (shown in Fig. 1). Genomic DNA was extracted following standard procedures (QIAGEN). The concentration and quantity of the extracted DNA were determined by agarose gel electrophoresis and a NanoDrop 2000 Spectrophotometer [NanoDrop 2000, Thermo Scientific, USA].
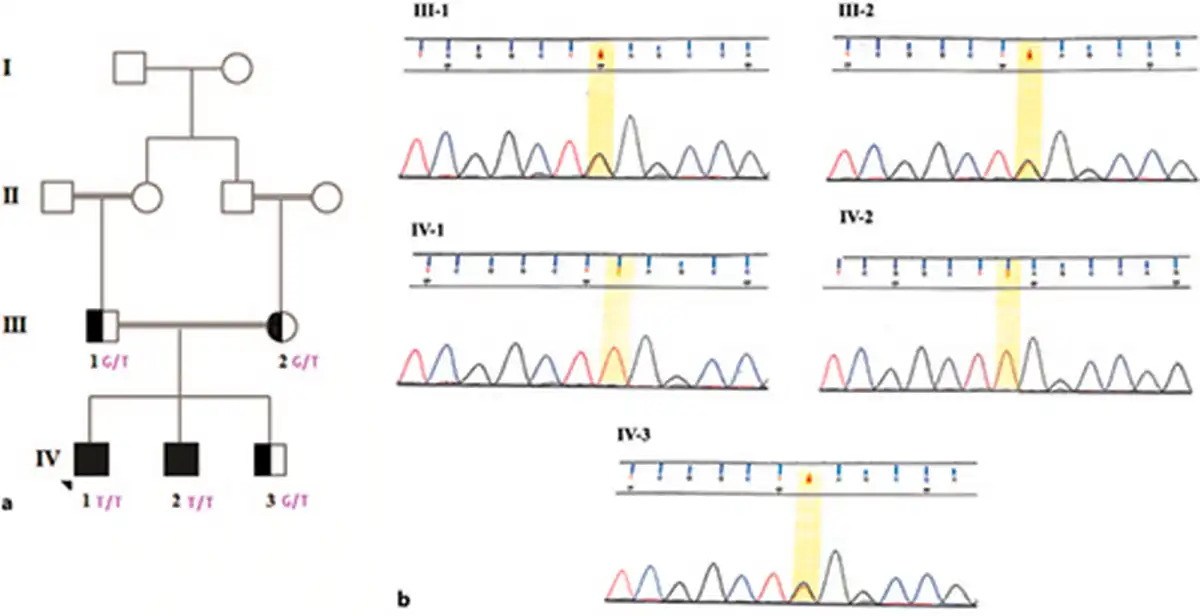
Fig. 1
Pedigree and electropherogram of the family. a Healthy, carrier, and affected individuals are represented as hollow, semi-black, and black circles or squares, respectively. b The father, mother, and healthy siblings were heterozygous for the c.6562G>T variant. The proband and his affected brother were homozygous for the c.6562G>T variant.
WES and in silico Analysis
WES was performed on high quality DNA sample of the IV-1 (shown in Fig. 1), and interpretation of the identified variants was performed according to the ACMG guidelines [Richards et al., 2015]. Exome-enriched genomic libraries was performed using SureSelect Human All Exon V7. Captured DNA fragments were sequenced using Illumina HiSeq 2500 (Illumina, Inc) with coverage of 100 × mean depth. Burrows-Wheeler Aligner software (http://bio-bwa.sourceforge. net/) was used for the alignment of reads with the human reference sequence (hg19 assembly). Variant calling was done by GATK software (https://gatk.broadinstitute.org/), and subsequent annotation was performed using ANNOVAR software. All analytical methods were done in accordance with the previous reports [Khorrami et al., 2021].
Variant Verification and Co-Segregation Analysis
Primer3 (http://primer3.ut.ee) was used for designing specific primers (F: 5′- GTAATGCCACCATCGACAGAG -3′ and R: 5′- GATATTCACAGCAAAGGCGTC - 3′) flanking the candidate variant. The primer pair checked for SNP absence with the BLAT (https://genome.ucsc.edu/cgi-bin/hgBlat). These primers amplify PCR products with 323 bp length, which were visualized using 1% agarose gel. Sanger sequencing was performed by an ABI 3130 sequencer (Applied Biosystems, USA), and the result was compared with the CDH23 gene reference sequence (NM-022124).
Results
Clinical Presentation
Parents stated that both patients had not responded to sounds since birth. Audiometry showed the proband and his affected brother both had severe HL at the age of 4 and 3 years old, respectively. Currently, the proband and his affected brother have 28 and 27 years old, respectively, and their audiometry showed that they both had severe sensorineural hearing loss (SNHL). Videonystagmography (VNG) test results indicated normal balance systems in both cases. Furthermore, the proband, at the age of 27 years, and his affected brother, at the age of 26 years, both showed night blindness (nyctalopia), and fundus photography revealed that both patients had mild RP (shown in Fig. 2).
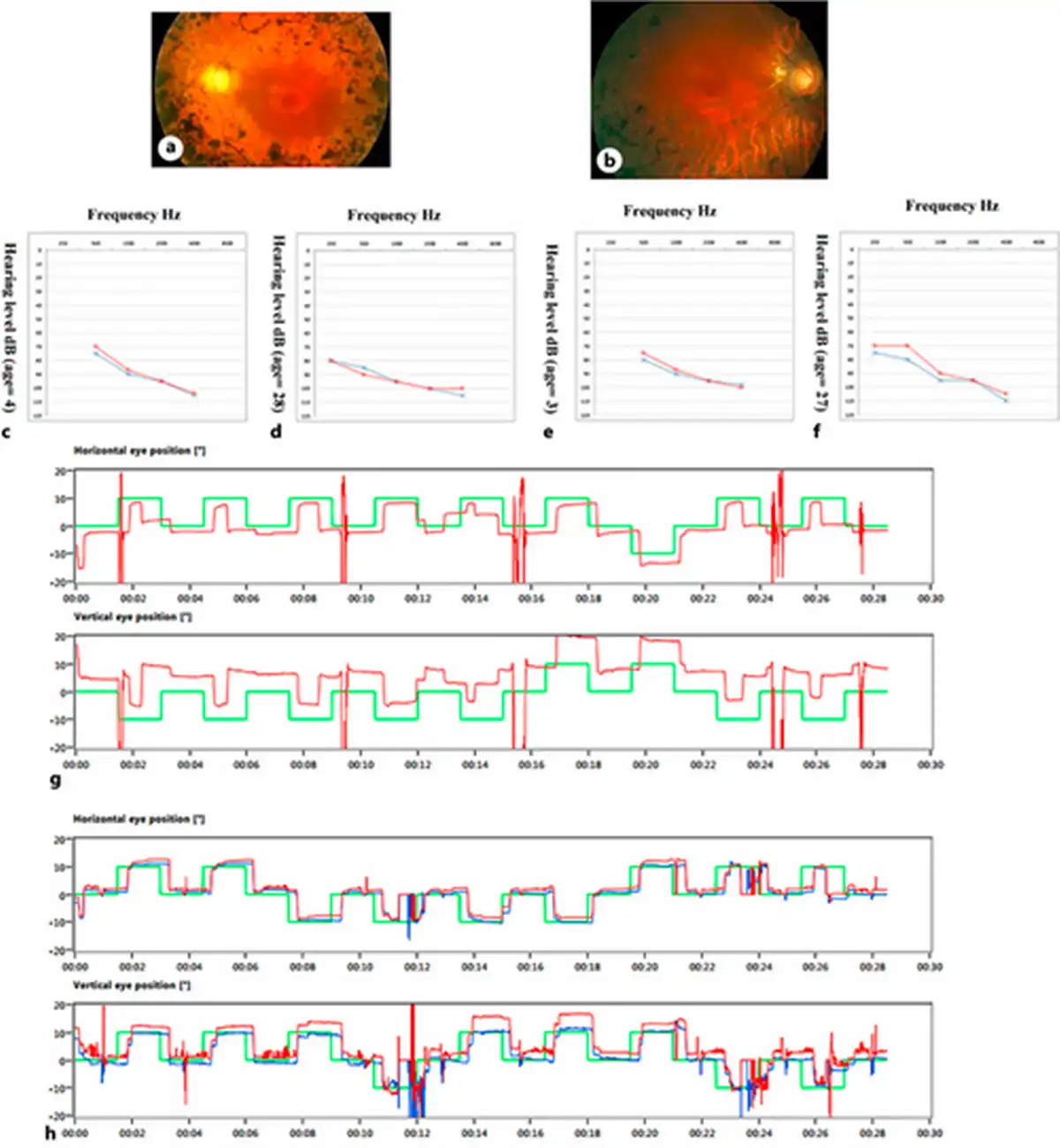
Fig. 2
Audiometry, VNG, and fundus photography of the patients. Fundus photography of the proband (a) and his affected brother (b) showed the RP diagnostic triad, including disc pallor, bone spicule view, and arterial attenuation. c, d Audiograms of the proband at 4 and 28 years which show the patients had severe SNHL. e, f Audiograms of the other affected brother, at ages 3 and 27 years, which show the patient had severe SNHL. g, h Proband’s and the affected brother’s VNG test results, which showed that their balance systems were normal.
WES Detected a Novel Nonsense Variant (c.6562G>T; p.Glu2188Ter)
Analysis of the proband’s (IV-1) annotated VCF file revealed 70,145 variants, of which 1,865 occurred in coding regions with MAF ≤0.01. The mean depth of coverage was 100X for greater than 93.3% of the variants. Subsequent data filtering determined a nonsense variant c.6562G>T (p.Glu2188Ter) in the CDH23 gene, which results in a truncated protein (PVS1). The identified variant using Sanger sequencing was co-segregated with the disease (PP1). The variant was not reported in any population databases like ExAC, gnomAD and was not submitted in the ClinVar (PM2). Also, this variant was neither present in 50 healthy, ethnically matched controls nor in the Iranom project, in which 800 healthy individuals from eight major ethnic groups in Iran went through WES. No other pathogenic or likely pathogenic variant matching the clinical manifestation of the studied patient was found. The Combined Annotation Dependent Depletion score (CADD) of this variant was 46. Therefore, the detected variant was evaluated as pathogen according to ACMG guidelines.
Discussion
Atypical USH is a poorly defined clinical type in which patients’ symptoms do not meet the canonical criteria for main types of USH. In atypical USH, patients generally show a discrepancy in severity or age of onset of RP, HL, or the presence/absence of vestibular dysfunction [Nolen et al., 2020]. In this paper, we studied an Iranian family with two affected siblings who manifested congenital bilateral HL, late-onset nyctalopia and RP (in their late twenties), and normal vestibular function, indicating that their clinical symptoms are consistent with USH2. Using WES, we found a novel homozygous nonsense damaging variant (c.6562G>T; p.Glu2188Ter) in the CDH23 gene, which was confirmed by Sanger sequencing and co-segregated with the disease. Despite having the damaging variants in the CDH23 gene, our patient’s clinical symptoms deviated from typical manifestation of the USH1D; therefore, both patients are classified as having atypical USH1D. Patients with a damaging variant in the CDH23 gene may show significant variability in their phenotypes. In other words, damaging variants in the CDH23 gene cause different allelic disorders, including USH1D, atypical USH1D, and DFNB12 [Astuto et al., 2002]. Furthermore, there is a report of pituitary adenoma due to a missense damaging variant in the CDH23 gene [Zhang et al., 2017]. Since no genotype-phenotype correlation has yet been reported for atypical USH1D, we collected all the reported atypical USH1D cases in order to find a definite genotype-phenotype correlation. In previous reported atypical USH cases harboring damaging variants in the CDH23 gene, clinical deviations such as mild/late-onset RP, progressive HL, and normal vestibular function have been described (Table 2). In the analysis of damaging variants revealed in atypical USH1D, different types of genotypes, including missense/missense, nonsense/missense, missense/duplication, frameshift/frameshift, and splice-site/splice-site, have been reported. Also, these damaging variants do not cluster in a specific domain and distribute from the N-terminal to the C-terminal of the CDH23 gene (Table 2). Therefore, we found no correlation between the severity or clinical spectrum of the symptoms and the type and location of the damaging variants. Although no genotype-phenotype correlation for atypical USH1D has yet been reported, several papers have proposed a genotype-phenotype correlation for DFNB12 and USH1D, which are other allelic disorders for the CDH23 gene, i.e., a truncating damaging variant causes USH1D and a missense damaging variant results in DFNB12 [Fuster-García et al., 2021]. By reviewing the literature, we found many cases that refute this correlation (Table 2). As can be deduced from Table 2, there are many reports of homozygous or compound heterozygous missense damaging variants in patients with USH1D manifestation. Some of these missense damaging variants are located in the first or last nucleotide of exons, which may be involved in splicing (indicated by the asterisk in Table 2). Exon trapping experiments established that some of these damaging variants impaired splicing (indicated by a in Table 2), while others did not affect the splicing pattern of the CDH23 gene transcripts. Moreover, other types of genotypes such as missense/splice-site, missense/frameshift, and missense/nonsense in USH1D patients have been reported. On the other hand, there are several DFNB12 patients with genotypes such as frameshift/frameshift, homozygous in-frame deletion, missense/nonsense, and missense/frameshift (Table 2). Our investigations also show that some CDH23 gene damaging variants are involved in a wide range of phenotypes. For example, the c.5237G>A (p.R1746Q) damaging variants in the compound heterozygous state with the c.3016G>A (p.E1006K) missense damaging variants result in USH1D [Schultz et al., 2011]. It can also cause atypical USH1D in the homozygous state or in the compound heterozygous state with the c.7466G>A (p.R2489H) missense damaging variants [Bolz et al., 2001; Astuto et al., 2002; Zhao et al., 2015]. Also, the homozygous c.6050-9G>A splice site damaging variants occur in many patients, resulting in USH, but there is also a report that they cause a non-syndromic HL phenotype [Besnard et al., 2014; Valero et al., 2019]. By and large, there is no clear-cut genotype-phenotype correlation, and we suppose that there are unknown modifier genes or epigenetic factors that alter the phenotypic consequences of damaging variants and cause different clinical manifestations. We recommend that clinicians and medical geneticists consider this issue and inform families about the possibility of RP occurrence in patients with CDH23 gene missense damaging variants.
In summary, we used WES and Sanger sequencing to investigate two siblings manifesting clinical symptoms consistent with USH2 and identified a novel nonsense damaging variant (c.6562G>T; p.Glu2188Ter) in the CDH23 gene. Since the patient’s symptoms deviated from those of USH1D, they were categorized as atypical USH1D. By reviewing the literature, we endeavored to find a genotype-phenotype correlation between the type or position of the CDH23 gene damaging variants and atypical USH1D, but we figured out there is no correlation. Furthermore, we demonstrate that different genotypes may occur in other allelic disorders of the CDH23 gene, and there is no definite genotype-phenotype correlation.
Acknowledgments
Hereby, we would like to express our special thanks to our patients and their family for participating in this study.
Statement of Ethics
The study was performed according to the Declaration of Helsinki and with the approval of the Institutional Review Board of Isfahan University of Medical Sciences (IR.MUI.MED.REC.1398.186). Also, written informed consent was obtained from all participants. Indeed, for children under the age of 18 years, written informed consent was obtained from the participants’ legal guardian.
Conflict of Interest Statement
The authors report no conflict of interest.
Funding Sources
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author Contributions
Erfan Khorram, Mehdi Khorrami, and Omid Irvani performed in silico analysis, investigation, visualization, and writing the draft. Sara Jahanian examined the patients and provided their clinical information. Mohammad Hossein Nilforoush conducted patients’ audiological evaluation. Masoomeh Amini, Seyyed Reza Mousavi, and Mahsa Ehsanifard contributed to the isolation of genomic DNA and Sanger sequencing. Majid Kheirollahi supervised the study. All authors have read, edited, and approved the final version of the manuscript.
Data Availability Statement
All data generated or analyzed during this study are included in this article. Further inquiries can be directed to the corresponding author.
References
- 1. Abad-Morales V, Navarro R, Burés-Jelstrup A, Pomares E Identification of a novel homozygous ARSG mutation as the second cause of Usher syndrome type 4 Am J Ophthalmol Case Rep 2020 Sep 19 100736
- 2. Aparisi MJ, Aller E, Fuster-García C, García-García G, Rodrigo R, Vázquez-Manrique RP, et al Targeted next generation sequencing for molecular diagnosis of Usher syndrome Orphanet J Rare Dis 2014 Nov 18 9 1 168
- 3. Astuto LM, Bork JM, Weston MD, Askew JW, Fields RR, Orten DJ, et al CDH23 mutation and phenotype heterogeneity: a profile of 107 diverse families with Usher syndrome and nonsyndromic deafness Am J Hum Genet 2002 Aug 71 2 262–75
- 4. Bademci G, Foster J 2nd, Mahdieh N, Bonyadi M, Duman D, Cengiz FB, et al Comprehensive analysis via exome sequencing uncovers genetic etiology in autosomal recessive nonsyndromic deafness in a large multiethnic cohort Genet Med 2016 Apr 18 4 364–71
- 5. Becirovic E, Ebermann I, Nagy D, Zrenner E, Seeliger MW, Bolz HJ Usher syndrome type 1 due to missense mutations on both CDH23 alleles: investigation of mRNA splicing Hum Mutat 2008 Mar 29 3 452
- 6. Besnard T, García-García G, Baux D, Vaché C, Faugère V, Larrieu L, et al Experience of targeted Usher exome sequencing as a clinical test Mol Genet Genomic Med 2014 Jan 2 1 30–43
- 7. Bolz H, von Brederlow B, Ramírez A, Bryda EC, Kutsche K, Nothwang HG, et al Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D Nat Genet 2001 Jan 27 1 108–12
- 8. Bonnet C, Grati M, Marlin S, Levilliers J, Hardelin JP, Parodi M, et al Complete exon sequencing of all known Usher syndrome genes greatly improves molecular diagnosis Orphanet J Rare Dis 2011 May 11 6 21
- 9. Bravo-Gil N, Méndez-Vidal C, Romero-Pérez L, González-del Pozo M, Rodríguez-de la Rúa E, Dopazo J, et al Improving the management of inherited retinal dystrophies by targeted sequencing of a population-specific gene panel Sci Rep 2016 Apr 1 6 23910
- 10. Dad S, Rendtorff ND, Tranebjærg L, Grønskov K, Karstensen HG, Brox V, et al Usher syndrome in Denmark: mutation spectrum and some clinical observations Mol Genet Genomic Med 2016 Sep 4 5 527–39
- 11. Ebermann I, Phillips JB, Liebau MC, Koenekoop RK, Schermer B, Lopez I, et al PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome J Clin Invest 2010 Jun 120 6 1812–23
- 12. French LS, Mellough CB, Chen FK, Carvalho LS A review of gene, drug and cell-based therapies for usher syndrome Front Cel Neurosci 2020 14 183
- 13. Fuster-García C, García-Bohórquez B, Rodríguez-Muñoz A, Aller E, Jaijo T, Millán JM, et al Usher syndrome: genetics of a human ciliopathy Int J Mol Sci 2021 Jun 23 22 13 6723
- 14. Ganapathy A, Pandey N, Srisailapathy CRS, Jalvi R, Malhotra V, Venkatappa M, et al Non-syndromic hearing impairment in India: high allelic heterogeneity among mutations in TMPRSS3, TMC1, USHIC, CDH23 and TMIE PloS One 2014 9 1 e84773
- 15. Igelman AD, Ku C, da Palma MM, Georgiou M, Schiff ER, Lam BL, et al Expanding the clinical phenotype in patients with disease causing variants associated with atypical Usher syndrome Ophthalmic Genet 2021 Dec 42 6 664–73
- 16. Khateb S, Kowalewski B, Bedoni N, Damme M, Pollack N, Saada A, et al A homozygous founder missense variant in arylsulfatase G abolishes its enzymatic activity causing atypical Usher syndrome in humans Genet Med 2018 Sep 20 9 1004–12
- 17. Khorrami M, Khorram E, Yaghini O, Rezaei M, Hejazifar A, Iravani O, et al Identification of a missense variant in the EIF2B3 gene causing vanishing white matter disease with antenatal-onset but mild symptoms and long-term survival J Mol Neurosci 2021 Nov 71 11 2405–14
- 18. Kim SY, Kim AR, Kim NKD, Kim MY, Jeon EH, Kim BJ, et al Strong founder effect of p.P240L in CDH23 in Koreans and its significant contribution to severe-to-profound nonsyndromic hearing loss in a Korean pediatric population J Transl Med 2015 Aug 13 13 1 263
- 19. Le Quesne Stabej P, Saihan Z, Rangesh N, Steele-Stallard HB, Ambrose J, Coffey A, et al Comprehensive sequence analysis of nine usher syndrome genes in the UK national collaborative usher study J Med Genet 2012 Jan 49 1 27–36
- 20. Lenarduzzi S, Vozzi D, Morgan A, Rubinato E, D’Eustacchio A, Osland TM, et al Usher syndrome: an effective sequencing approach to establish a genetic and clinical diagnosis Hear Res 2015 Feb 320 18–23
- 21. Millán JM, Aller E, Jaijo T, Blanco-Kelly F, Gimenez-Pardo A, Ayuso C An update on the genetics of usher syndrome J Ophthalmol 2011 2011 417217
- 22. Mizutari K, Mutai H, Namba K, Miyanaga Y, Nakano A, Arimoto Y, et al High prevalence of CDH23 mutations in patients with congenital high-frequency sporadic or recessively inherited hearing loss Orphanet J Rare Dis 2015 May 13 10 60
- 23. Nolen RM, Hufnagel RB, Friedman TB, Turriff AE, Brewer CC, Zalewski CK, et al Atypical and ultra-rare Usher syndrome: a review Ophthalmic Genet 2020 Oct 41 5 401–12
- 24. Oshima A, Jaijo T, Aller E, Millan JM, Carney C, Usami S, et al Mutation profile of the CDH23 gene in 56 probands with Usher syndrome type I Hum Mutat 2008 Jun 29 6 E37–46
- 25. Ouyang XM, Yan D, Du LL, Hejtmancik JF, Jacobson SG, Nance WE, et al Characterization of Usher syndrome type I gene mutations in an Usher syndrome patient population Hum Genet 2005 Mar 116 4 292–9
- 26. Pan L, Zhang M Structures of usher syndrome 1 proteins and their complexes Physiology 2012 Feb 27 1 25–42
- 27. Pennings RJE, Topsakal V, Astuto L, de Brouwer APM, Wagenaar M, Huygen PLM, et al Variable clinical features in patients with CDH23 mutations (USH1D-DFNB12) Otol Neurotol 2004 Sep 25 5 699–706
- 28. Peter VG, Quinodoz M, Sadio S, Held S, Rodrigues M, Soares M, et al New clinical and molecular evidence linking mutations in ARSG to Usher syndrome type IV Hum Mutat 2021 Mar 42 3 261–71
- 29. Puffenberger EG, Jinks RN, Sougnez C, Cibulskis K, Willert RA, Achilly NP, et al Genetic mapping and exome sequencing identify variants associated with five novel diseases PloS One 2012 7 1 e28936
- 30. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology Genet Med 2015 May 17 5 405–24
- 31. Roux AF, Faugère V, Le Guédard S, Pallares-Ruiz N, Vielle A, Chambert S, et al Survey of the frequency of USH1 gene mutations in a cohort of Usher patients shows the importance of cadherin 23 and protocadherin 15 genes and establishes a detection rate of above 90% J Med Genet 2006 Sep 43 9 763–8
- 32. Roux AF, Faugère V, Vaché C, Baux D, Besnard T, Léonard S, et al Four-year follow-up of diagnostic service in USH1 patients Invest Ophthalmol Vis Sci 2011 Jun 8 52 7 4063–71
- 33. Schultz JM, Bhatti R, Madeo AC, Turriff A, Muskett JA, Zalewski CK, et al Allelic hierarchy of CDH23 mutations causing non-syndromic deafness DFNB12 or Usher syndrome USH1D in compound heterozygotes J Med Genet 2011 Nov 48 11 767–75
- 34. Toms M, Pagarkar W, Moosajee M Usher syndrome: clinical features, molecular genetics and advancing therapeutics Ther Adv Ophthalmol 2020 Jan-Dec 12 2515841420952194
- 35. Valero R, de Castro-Miró M, Jiménez-Ochoa S, Rodríguez-Ezcurra JJ, Marfany G, Gonzàlez-Duarte R Aberrant splicing events associated to CDH23 noncanonical splice site mutations in a proband with atypical usher syndrome 1 Genes 2019 Sep 21 10 10 732
- 36. Wafa TT, Faridi R, King KA, Zalewski C, Yousaf R, Schultz JM, et al Vestibular phenotype-genotype correlation in a cohort of 90 patients with Usher syndrome Clin Genet 2021 Feb 99 2 226–35
- 37. Yan D, Liu XZ Genetics and pathological mechanisms of Usher syndrome J Hum Genet 2010 Jun 55 6 327–35
- 38. Zein WM, Falsini B, Tsilou ET, Turriff AE, Schultz JM, Friedman TB, et al Cone responses in Usher syndrome types 1 and 2 by microvolt electroretinography Invest Ophthalmol Vis Sci 2014 Nov 25 56 1 107–14
- 39. Zhang L, Cheng J, Zhou Q, Khan MA, Fu J, Duan C, et al Targeted next-generation sequencing identified novel compound heterozygous variants in the CDH23 gene causing usher syndrome type ID in a Chinese patient Front Genet 2020 11 422
- 40. Zhang Q, Peng C, Song J, Zhang Y, Chen J, Song Z, et al Germline mutations in CDH23, encoding cadherin-related 23, are associated with both familial and sporadic pituitary adenomas Am J Hum Genet 2017 May 4 100 5 817–23
- 41. Zhao L, Wang F, Wang H, Li Y, Alexander S, Wang K, et al Next-generation sequencing-based molecular diagnosis of 82 retinitis pigmentosa probands from Northern Ireland Hum Genet 2015 Feb 134 2 217–30
- 42. Zheng QY, Yan D, Ouyang XM, Du LL, Yu H, Chang B, et al Digenic inheritance of deafness caused by mutations in genes encoding cadherin 23 and protocadherin 15 in mice and humans Hum Mol Genet 2005 Jan 1 14 1 103–11