Introduction
Acute myocardial infarction (AMI) is a leading cause of mortality worldwide. Percutaneous coronary intervention is used to timely restore coronary blood flow and is the recommended treatment modality for patients with AMI []. While the restoration of blood flow is necessary for inhibiting ischemic cell death, it has been documented that reperfusion activates a range of cellular injuries that are more severe than those caused by ischemia alone []. Injuries due to reperfusion may offset the benefits of coronary blood flow recovery, especially in older patients and in patients with prolonged ischemia []. Mitochondrial dysfunction and the resultant oxidative stress are the key mechanisms for the development of cardiac damage during ischemia and reperfusion []. Studies have established that the mitochondrial quality control system is closely associated with ischemia/reperfusion (I/R) injury []. Effective therapeutic options for I/R injuries have not yet been developed []. This article briefly introduces the molecular mechanisms of the mitochondrial quality control system. In addition, we summarize the possibilities of designing novel pharmacological interventions and new molecular approaches that target the mitochondrial quality control system.
Mitochondrial Quality Control System
Mitochondrial Fusion and Fission
Studies have focused on mitochondrial homeostasis, including the fission/fusion changes that occur in human diseases [], a process known as mitochondrial dynamics []. Mitochondrial dynamics are regulated by a series of dynamin-related large GTPases, including mitofusin 1 (Mfn1), mitofusin 2 (Mfn2), optic atrophy 1 (OPA1), dynamin-related protein 1 (Drp1), and fission protein 1 (Fis1) [, ]. Mfn1 and Mfn2 are anchored to the outer mitochondrial membrane (OMM), with their N-terminal and C-terminal domains protruding into the cytoplasm. The N-terminal has a GTP domain, followed by a hydrophobic heptapeptide repeating domain (HR1) and a transmembrane domain (TM) (TM through OMM). At the C-terminal, there is a second hydrophobic heptapeptide repeat sequence (HR2) [] (Fig. 1a). The C-terminal HR2 induces oligomerization among mitofusins and achieves physiological interactions by forming reverse parallel dimers between their HR2 domains [] (Fig. 2). The GTPase domain mediates mitochondrial fusion that is dependent on the hydrolysis of GTP []. In addition to mitochondrial fusion, Mfn1/2 are involved in various biological processes such as cellular metabolism, apoptosis, proliferation, and mitophagy [].
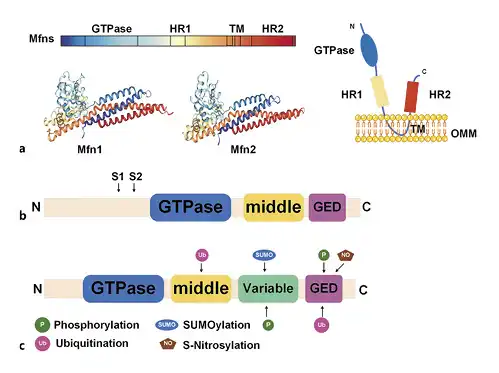
Fig. 1
Schematic illustration of mitochondrial dynamics protein structure. a Structure of mitofusins. b Structure of OPA1. S1 and S2 represent the protease cleavage sites on L-OPA1. c Structure of the dynamin-related protein and post-translational modifications. OPA1, optic atrophy 1; Mfn, mitofusin; OMM, outer mitochondrial membrane; GED, GTPase effector domain.
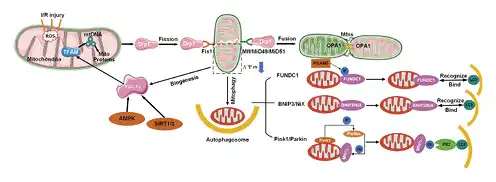
Fig. 2
Schematic illustration of the mitochondrial quality control system, including mitochondrial fission, fusion, mitophagy, and mitochondrial biogenesis. OPA1, optic atrophy 1.
OPA1 is a dynmin-related protein that is located on the inner membrane (IM) that faces the mitochondrial membrane space. This protein consists of a characteristic GTPase intermediate region and GTPase effector domain (Fig. 1b) []. The relative abundance of OPA1 S-form (soluble in the mitochondrial membrane space) and L-form (anchored in the inner mitochondrial membrane) affects subsequent membrane fusion/fission events []. In addition to mitochondrial fusion, OPA1 controls the integrity of the intimal cristae and regulates apoptosis through the regionalization of soluble cytochrome C in the cristae [].
The domain structure of Drp1 is made up of the N-terminal GTPase domain, middle domain, variable domain (also known as B-insert), and the C-terminal GTPase effector domain [] (Fig. 1c). The Drp1 protein is located in the cytoplasm and is recruited by receptors on the mitochondrial outer membrane in response to specific cellular cues []. In mammals, 4 intact outer membrane proteins, Fis1, Mff, MiD51, and MiD49, have been identified as the receptors associated with the recruitment of Drp1 into OMM []. The translocation of Drp1 into OMM is associated with multiple post-translational modification processes such as phosphorylation, ubiquitination, SUMOylation, and S-nitrosylation [] (Fig. 1c). Mitochondrial fission is necessary for mitochondrial biogenesis and for mitophagic quality control (Fig. 2) [].
Mitochondrial Biogenesis
Mitochondrial biogenesis is a process that controls the quality and copy number of mitochondria [, ]. In response to increased energy demands by cells undergoing oxidative stress [], biogenesis is responsible for supplementing new mitochondria and participates in energy balance regulation that is crucial for maintaining normal cellular functions [, ]. This process is regulated by several key transcription factors, of which peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α) is the most important. In addition, mitochondrial biogenesis is induced during the regulation of energy balance by mitochondria. After ischemia/reperfusion injuries, activated AMPK is a key target for initiating myocardial protection [] and further activates PGC-1α through phosphorylation []. It has also been established that sirtuin 1/3 activates PGC-1α through deacetylation by utilizing NAD+ as a co-substrate []. Phosphorylated and deacetylated PGC-1α interacts with the nuclear respiratory factor (NRF1/2) and mitochondrial transcription factor A (TFAM) to activate mitochondrial DNA, protein synthesis, and the production of new mitochondria [] (Fig. 2).
Mitochondrial Autophagy
Under extreme stress conditions such as mitochondrial dysfunctions, the activated quality control mechanism is the mitochondrial autophagy. Autophagy is a highly dynamic and multistep process whose activity is determined by an autophagic flux []. The classical autophagic pathway is the PINK1-PRKN-dependent pathway. PINK1 and Parkin, as the quality control mechanisms of OMM, maintain mitochondrial integrity by mediating the ubiquitination and degradation of the resident proteins of the mitochondrial outer membrane []. Mitofusins (Mfn1 and Mfn2) are the targets for Parkin-mediated ubiquitination. Under hypoxia, FUNDC1 acts as a receptor for mitochondrial autophagy []. Phosphoglycerate mutase family member 5 is a mitochondrial protein that dephosphorylates FUNDC1 at Ser13 and activates mitophagy by binding to LC3 on the phagosome []. BNIP3/NIX are BCL2-associated proteins. When there is a stable membrane potential in mitochondria, BNIP3/NIX can activate mitophagy. The role of BNIP3-mediated mitochondrial autophagy in I/R injury is that ROS induces BNIP3-mediated mitophagy by inhibiting mTOR and promotes autophagosome formation (Fig. 2).
Mitochondrial Quality Control System and I/R Injury
Damaged, abnormal, or dysfunctional mitochondria are destroyed by mitochondrial autophagy while, to meet cellular needs, new mitochondria are replenished through mitochondrial biogenesis. Mitochondrial biogenesis requires adaptation to mitochondrial dynamics []. During ischemia, mitochondrial fission increases while fusion decreases. This situation predisposes mitochondria to autophagy []. Ma et al. [] documented that under hypoxic conditions, the expression of Drp1 and Mfn1/2 are both upregulated and participate in mitochondrial damage. Mitochondrial fission is a key regulator of autophagic efficiency as it separates damaged mitochondria from healthy networks in order to load them into autophagosomes []. Pulmonary endothelial cells exposed to hyperoxia exhibited an elevated rate of Drp1 phosphorylation. Subsequently, the upregulated expression of Drp1 led to a severe fragmentation of mitochondria. In addition, mitochondrial autophagy was activated, and the expression of P62 and LC3B was elevated, while the decreased expression of PINK1 was inhibited. PINK1 reduction in hyperoxic PECs increases the possibility of inefficient autophagy []. Complex interactions occur between mitochondrial biogenesis and mitophagy. During I/R stress, mitochondrial biogenesis (mitochondrial renewal) balances mitophagy []. The regulation of mitochondrial quality selectively eliminates individual damaged mitochondria and promotes mitochondrial turnover in the cell and organs. This process relieves pressure of the intracellular environment when a metabolic energy crisis occurs during disease transition [] (Fig. 2).
Summary of Various Therapeutic Options
Targeting Mitochondrial Fusion and Fission Proteins
Drug Therapy: Inhibit Excessive Fission and Promote Proper Fusion
In the past 2 decades, mitochondrial fission inhibitors have been developed []. They include Mdivi-1 [] (representative inhibitors). Mitochondrial fusion promoters such as BGP-15 [] (representative promoters) have also been developed. Their beneficial effects have been tested in cellular models and MIRI animal models [] (Table 1).
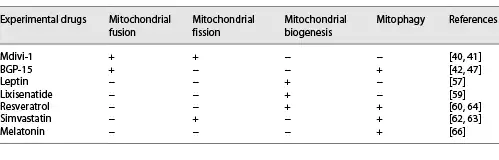
Among mitochondrial fission inhibitors, Mdivi-1 has been extensively studied. It is a Drp1 GTPase inhibitor that selectively inhibits the assembly and GTPase activity of Drp1 in yeast and mammalian cells. These effects inhibit mitochondrial fragmentation []. Manczak et al. [] documented that the expression levels of Drp1 and Fis1 decreased while the expression levels of fusion genes (Mfn1, Mfn2, and OPA1) increased in N2a cells administered with full-length Drp1 + Mdivi-1 and N2a cells administered with Drp1 RNAi + Mdivi-1. The intraperitoneal injection of Drp1 small-molecule inhibitor (Mdivi-1 at 1.2 mg/kg) 15 min before reperfusion significantly inhibits the translocation of Drp1 to the OMM of MIRI in diabetic mice. This reduces mitochondrial fission while mitochondrial dynamics are improved. Immunofluorescence analysis showed that when the mitochondrial network increased [], mitochondrial fusion and intracellular ATP concentration were enhanced []. Mdivi-1 has however not been subjected to clinical trials. In cases where large-animal (pig) AMI models were used, Mdivi-1 administration at the beginning of reperfusion did not preserve the left-ventricular functions or reduce the scope of myocardial infarction. A study done by Ong et al. [] obtained similar results.
Increased mitochondrial fusion events are the main reason for the changes in length []. Activation of mitochondrial fusion provides a new therapeutic strategy for mitochondrial-related diseases such as IR injury []. BGP-15 is a hydroxylamine derivative that has a wide range of cytoprotective effects []. Cell culture experiments have shown that BGP-15 triggers mitochondrial fusion and activates OPA1, Mfn1/2, and Akt (also called protein kinase B) []. In addition, Szabo et al. [] also found that BGP-15 leads to GTP hydrolysis of OPA1, promotes mitochondrial IM fusion, and stabilizes the cristae membrane in the mitochondria [] to protect the mitochondrial membrane system [].
Utilization of Molecular Methods: Double Knockout of Drp1 and OPA1
The immediate consequence of mitochondrial fission and fusion is the change in size []. Mitochondrial fission leads to the production of small mitochondria, while mitochondrial fusion leads to the production of large mitochondria. The balance between fission and fusion maintains mitochondrial sizes in healthy cells []. The relationship between mitochondrial morphology and function is complex. Morphological changes can alter functions depending on the cell type and physiological environment []. The functional plasticity of mitochondria is closely associated to its morphology []. Mitochondrial morphology and integrity are key features that reflect cellular stress, bioenergetics, and survival rate [].
To regulate unequal fission and fusion of mitochondria, cells readjust their balance. The mitochondrial size of Drp1KO hepatocytes was shown to be significantly increased due to nonresistant fusion, while the mitochondrial size of Opal KO hepatocytes was shown to be significantly reduced due to nonresistant fission. In the study by Yamada et al. [], mitochondrial stasis was introduced by deleting 2 GTPases associated with motility of fission (Drp1) and fusion (Opa1) in the liver. The size of the mitochondria in Drp1Opa1KO hepatocytes recovered. At the cellular level, mitochondrial stasis reduces the pathological effects caused by a single fission or fusion defect to recover the mitochondrial size. Simultaneous inhibitions of fusion and fission processes reduce the harmful effects associated with unbalanced mitochondrial fusion or fission []. In addition, the significantly low Mfn1 and Mfn2 levels were boosted to their original levels. Meanwhile, Parkin-independent autophagy recovered in hepatocytes, and almost no accumulation of P62, ubiquitin, or LC3 was detected. The balance of mitochondrial size, rather than mitochondrial fission or fusion itself, is the basis for the maintenance of a healthy liver. Yang et al. [] also established that the maintenance of cellular mitochondrial integrity in severe ischemic conditions plays a vital role in anti-apoptosis []. It is important to determine whether the mitochondrial stasis caused by the double knockout of Drp1 and OPA1 in this experiment will show similar outcomes in an I/R injury model.
Drug Therapy: Promote Mitochondrial Biogenesis
To improve bioenergetic parameters and cell quality under pathological conditions, mitochondrial biogenesis is a potential pharmacological target []. Leptin enhances mitochondrial DNA replication by activating the expression of PGC-1α (the main regulator of mitochondrial biogenesis) to increase the efficiency of bioenergy production []. Zhao and Pu [] performed tissue activity testing and found that the inhibition of the activity of the electron transport chain complex reduced the biogenesis of cardiomyocyte mitochondria (Table 1).
Lixisenatide has been approved by the US Food and Drug Administration as a therapeutic choice for type 2 diabetes. Lixisenatide has been shown to significantly increase the expression of PGC-1α and upregulate the expression of the target genes of PGC-1 nuclear respiratory factor-1 and TFAM []. This drug therefore affects mitochondrial biogenesis by stimulating the PGC-1α/NRF-1/TFAM pathway. Resveratrol has cardioprotective properties and has also been found to regulate mitochondrial biogenesis in a SIRT3-dependent manner by activating AMPK-PGC-1α-ERRα signals. This regulation exhibits protective effects against oxidative stress [].
Targeting Intervention of Mitophagy
Drug Therapy: Activate or Inhibit Mitophagy?
Mitophagy is a protective or adaptive mechanism []. There is a basic balance in the control mechanism of mitochondrial autophagy. Determining the threshold of the autophagic regulatory pathway is a valuable target option for the treatment of cardiovascular diseases. Mitochondrial fission is a prerequisite for the occurrence of mitochondrial autophagy [] (Table 1).
Simvastatin-treated HL-1 cardiomyocytes exhibited an activation of their mitochondrial fission and autophagy. This resulted in the translocation of Parkin and p62 to mitochondria. The upregulated Parkin-mediated mitophagy reduced the infarct range of wild-type mice suffering from ischemia-reperfusion injury (IRI). This phenomenon was however not observed in Parkin knockout mice [, ]. In the rat myocardial I/R injury model, a significant mitophagy activity was associated with the significant upregulation of Drp1 and Parkin and the significant downregulation of OPA1 []. Resveratrol, aided by the action of D-Galactose, upregulates the expression of Drp1 and mitochondrial autophagy protein LC3-II in H9c2 cells and increases the mitochondrial translocation of Parkin and PINK1 [].Through the Drp1-Parkin-PINK1 signal, resveratrol regulates mitochondrial fragmentation autophagy to eliminate unhealthy cardiac mitochondria.
Baseline autophagy is an important cell quality-control mechanism []. Moderate autophagy is beneficial to viable cardiomyocytes, while excess autophagy aggravates cardiomyocyte death []. Melatonin reduces the scope of myocardial infarction and protects cardiac functions by restoring peroxisome proliferator-activated receptor γ on platelets and inhibiting FUNDC1-mediated autophagy [] during reperfusion.
Using Molecular Biology Methods: The Introduction of Proteolysis-Targeting Chimeras
Targeted protein degradation (proteolysis-targeting chimeras [PROTAC]) is an emerging area in the field of drug research and development. It is different from the traditional small molecules that function by blocking protein functions (Fig. 3). Sakamoto et al. [] first proposed the concept of PROTAC in 2001. The protein-of-interest (POI) ligand in its structure specifically binds to the corresponding target protein after PROTAC molecule entering cells. The other end of PROTAC can recruit E3 ligase to form a POI-PROTAC-E3 ligase ternary complex, where E3 ubiquitin ligase ubiquitinates POI by ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. After the ternary complex dissociates, the ubiquitin-labeled POI is recognized and degraded by the proteasome to selectively reduce the level of the target protein. In theory, this strategy can be used for any protein (Fig. 3). Recent breakthroughs in PROTAC small molecule research have focused on specific targets. This strategy can target nontargetable targets in the traditional sense and therefore can change the target from “nondrugable” to “medicinal properties.”
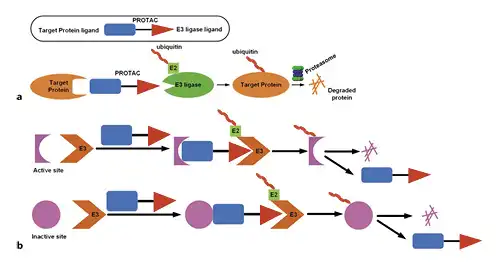
Fig. 3
a Schematic of the PROTAC technology. A small molecule PROTAC simultaneously binds the target protein and an E3-ubiquitin ligase complex. The trimeric complex formation leads to the transfer of ubiquitins to the target protein. Upon dissociation of the complex, the polyubiquitinated target protein is recognized by the proteasome and degraded. b PROTAC molecules can bind to almost any site on the target protein, not limited to the active site. PROTAC, PROteolysis-TArgeting Chimera; OPA1, optic atrophy 1; Mfn, mitofusin.
PROTAC-mediated degradation can be used to regulate the mitochondrial quality control proteins involved in the pathogenesis of I/R injury []. Targeting mitochondrial-associated pathological changes is a promising strategy for resolving I/R injury []. The existing clinical and nonclinical drugs can be used to screen suitable PROTACs, explore the range of existing E3 substrates according to known pathways of mitochondrial autophagy, and design correct clinical programs. For example, based on high-resolution mass spectrometry, thousands of compounds in a pool can be screened and analyzed to improve the screening throughput. When applied to the discovery of ligands, this method can repeatedly identify high-affinity hits from a highly complex compound pool. Utilization of different E3 ligase mutants for multiple rounds of screening can be used to overcome the PROTAC molecular resistance problem associated with E3 ligase mutations. RNAi [] and CRISPR-Cas9 [] as therapeutic tools can enhance the efficacy of traditional drugs. However, RNA-based therapies are limited by RNA instability and immunogenicity, while the efficiency of CRISPR-Cas9 has not been established in clinical practice []. In contrast, the mechanism of action and therapeutic advantages associated with the PROTAC technology means that it has broad application prospects in the field of small molecule drug development.
Stem-Cell Therapy and Mitochondrial Transplantation
It has always been thought that cardiomyocyte regeneration is impossible. However, studies have begun to use potential progenitor cells for cardiac repair. Clinical trials involving cell therapy in patients with ischemic heart disease has produced beneficial effects that correspond to traditional medicinal therapies []. Newborn mice (<1 week old) can regenerate the apex after excision []. After the first ligation of the left anterior descending (LAD) coronary artery in mice, significant tissue damage could still be observed 4 days after ligation. Newborn mice have complete morphology and functional cardiac regeneration ability after large-scale ischemic cardiac injury [].
Clinical trials involving combined stem-cell therapies such as mesenchymal stem cells and cardiac stem cells are being done [] (Fig. 4a). A meta-analysis of bone-marrow mesenchymal stem-cell therapy from multiple clinical trials concluded that [, ] despite the different treatment and data analysis methods, the ejection fraction of patients with AMI and chronic ischemic heart disease treated with these stem cells exhibited a 2–5% improvement in the first 3 months and had short-term positive effects on cardiac functions []. Human CPCs are an attractive target cell population in regenerative cell therapy []. In vivo studies have shown that human CPCs can regenerate cardiac tissues and improve its functions after myocardial infarction []. Although cell-based therapies are limited by the inability of cells to integrate into the myocardium (implantation rate) and the lack of survival and retention of infused cells (survival rate) [, ], they can be delivered to the infarcted heart by minimally invasive injection of hydrogel [], or they can be seeded into a scaffold and attached for increased cell retention on the heart. Injectable hydrogels and scaffolds provide an environment for cellular attachment, which is necessary for cell survival (Fig. 4a). In order to avoid the Ca2+ ion effect, Abdi et al. [] dissolved hydrogen peroxide in PLGA microspheres and obtained 7 days of oxygen release. From a clinical perspective, safety and effectiveness are still the most important issues. More studies are needed to develop a reliable, long-term, safe, and effective cell-biomaterial delivery system [].
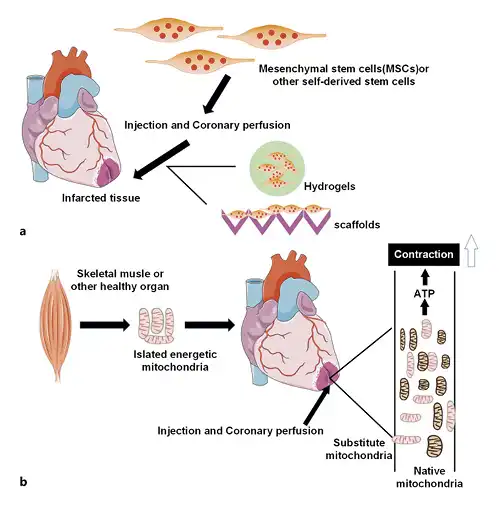
Fig. 4
Stem-cell therapy and mitochondrial transplantation. a Injectable hydrogels and scaffolds can be used as delivery vehicles for cell therapeutics. b Healthy mitochondria isolated from autologous skeletal muscle or other healthy tissues enter the infarcted area by injection or coronary perfusion, which significantly enhances overall and local myocardial function. MSCs, mesenchymal stem cells.
Management for myocardial tissue necrosis and postischemic function by pharmacological interventional treatments and/or exogenous substrates, either in single-use or in combination with multiple techniques, only can bring limited cardioprotection []. Mitochondrial damage and dysfunction events are initiated during ischemia and continue to reperfusion. Mitochondria are the main supplier of energy for heart, and replacement of damaged mitochondria during ischemia and reperfusion is a viable option (Fig. 4b). Mitochondrial transplantation (MT) is now emerging as a direct way targeting mitochondria to achieve effective cardioprotection []. At present, MT has been practiced in models and even in clinical transplantation in patients. During the procedure, the following 3 issues are mostly considered: (1) the source of fresh mitochondria: nonautologous/autologous transplantation; (2) the time for MT: prior to ischemia, before reperfusion, or after reperfusion; (3) the position for MT: direct epicardial MT into the ischemic heart [, ] or MT via coronary arterial vascular perfusion [].There was a study indicating that either autologous or nonautologous MT was effective for IRI []. To improve clinical applicability and reduce immunogenic complications, mitochondria should be isolated from remote nonischemic tissues in the same organism (autologous) for transplantation into the myocardial ischemic area []. In a rabbit model of local myocardial ischemia-reperfusion, autologously derived mitochondria isolated from the pectoral major muscle was immediately transplanted into the myocardium before reperfusion after 29 min of ischemia []. The results demonstrated that management by MT could significantly decrease the area suffering from myocardial infarct and increase cardiomyocyte viability. Besides, the autologous MT tested by multiple laboratory examinations did not induce any autoimmune responses [], and no increase in inflammatory cytokines associated with heart transplantation rejection was observed. Moreover, there showed no remarkable variance regarding the ability in cardioprotection of transplanted mitochondria derived from different tissue types [, ] or subgroups (subsarcolemmal or interfibrillar mitochondria) of the same subject []. Since the porcine heart shares highly similar characteristics in size and anatomy with an adult heart, McCully’s group [, ] conducted multiple experiments to identify their clinical relationship. In the majority of their experiments, Yorkshire pig models of local ischemia-reperfusion were established, and the efficacy of MT via direct myocardial injection versus coronary arterial vascular perfusion was comparatively analyzed [].MT prior to ischemia was given by single administration (MTS) or continuous administration (10 times within 60 min, MTSS), and following 15 min, the LAD artery was managed to be snared to cause temporary heart regional ischemia. As revealed by experimental observations, the MT prior to ischemia was preventively cardioprotective in the presence of local IRI in pigs, notably reduced myocardial infarct-affected area, and brought benefits in the overall as well as local myocardial functions []. MTS and MTSS both manifested good efficacy, while MTS was superior. Additionally, MT also showed delayed efficacy when performed after reperfusion. Following 30 min of ischemia caused by the snare of LAD in female Yorkshire pigs, a heart reperfusion was implemented which lasted 120 min. Autologous mitochondria (1 × 109/5 mL vehicle) were delivered to the left coronary artery as a bolus, followed by a further reperfusion for 120 min. The results displayed that, relative to untreated pigs, pigs of local ischemia-reperfusion undergoing delayed MT after 240 min of management with reperfusion had elevated survival rate of cardiomyocytes and increased postischemic myocardial functions, with statistically significant difference []. This suggested that delayed MT was an effective therapeutic strategy. Besides, varying transplantation approaches for exogenous mitochondria presented a similar cardioprotective effect. At Boston Children’s Hospital, 5 critically ill children on extracorporeal membrane oxygenation support due to myocardial dysfunction caused by ischemia-reperfusion received mitochondrial transplantation. The autologous mitochondria obtained from the rectus abdominis muscle of the patients were injected into 10 different sites with the lowest myocardial dynamics. The myocardial contractile function of all 5 patients was significantly improved; except for 1 patient, the rest were successfully separated from extracorporeal membrane oxygenation support on the second day after mitochondrial transplantation []. The results of this series of experimental studies indicate that mitochondrial transplantation provides a valuable strategy for cardioprotection during ischemic cardiac disease and opens the door for more in-depth understanding of the functions and therapeutic potential of mitochondria [].
Summary and Outlook
Mitochondria adapt to a variety of physiological needs, converge stress responses, and maintain the balance of intracellular environment [, ]. It is a key regulator of cell fate during disease [] and is a potential therapeutic target for I/R injury []. The review here describes the efficacy evaluation of pharmacology-based interventional treatment with the mitochondria quality control system as functional target, which tends to be presented in biomedical models and focuses on studying the effects of drugs on anatomic injury, biochemical alterations, and other pathological changes, with the evaluation methods individualized to different experimental models. In the future, genomic and clinical studies should be considered to facilitate the conventional drugs in new use and speed up the development of new drugs, finally to provide myocardial IRI treatment with more options. Due to the fact that drug treatment remains the mainstay and universal treatment by now, the application of PROTAC must provide a broad application future for the development of small molecular drugs on the basis of a whole new state of pharmacology. The mitochondrial division protein Drp1 and fusion protein OPA1 can induce mitochondrial stagnation when they are both knocked out. Such conception helps realize mitochondrial reconstruction and again, prove the function of the balance between mitochondrial fusion and division in organic injury and repair. Nevertheless, this is limited in wide application for a lack of clinical applicability. MT is a new way of clinical applicability emerging in recent decades that is promising and expected to be an alternative of conventional treatment method. By extracting mitochondria from their own healthy areas and delivering them to the myocardial ischemic injury area, mitochondrial transplantation solves the problem of finding a donor for transplantation in the past. The existing animal and clinical experiments have shown good therapeutic effects, making mitochondrial transplantation full of hope for the future treatment of myocardial ischemia and reperfusion.
Acknowledgments
Those who contributed to the work and meet the authorship criteria are listed as authors of the article.
Conflict of Interest Statement
The authors have no conflicts of interest to declare in relation to this article.
Funding Sources
This work was supported by the National Natural Science Foundation of China (No. 81973753 to W. Jian and No. 81774207 to W. Jian) and the Natural Science Foundation of Hunan Province (No. 2018JJ2291 to W. Jian).
Author Contributions
All the authors contributed to this article. Manli Zhou and Yunfeng Yu conceptualized and designed the study and drafted the initial manuscript. Weixiong Jian reviewed and revised the manuscript. Manli Zhou, Xiaoxin Luo, Jianzhang Wang, and Xiaodong Lan collected references and data and carried out the initial analyses. Weixiong Jian, Yunfeng Yu, Pei Liu, and Yu Feng co-ordinated and supervised data collection and critically reviewed the manuscript for important intellectual content. All the authors approved the final version submitted for publication.
References
- 1. Arslan F, Smeets MB, O’Neill LA, Keogh B, McGuirk P, Timmers L, et al. Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody. Circulation. 2010;121(1):80–90. http://dx.doi.org/10.1161/CIRCULATIONAHA.109.880187.
- 2. Shin B, Cowan DB, Emani SM, Del Nido PJ, McCully JD. Mitochondrial transplantation in myocardial ischemia and reperfusion injury. Adv Exp Med Biol. 2017;982:595–619. http://dx.doi.org/10.1007/978-3-319-55330-6_31.
- 3. Betgem RP, de Waard GA, Nijveldt R, Beek AM, Escaned J, van Royen N. Intramyocardial haemorrhage after acute myocardial infarction. Nat Rev Cardiol. 2015;12(3):156–67. http://dx.doi.org/10.1038/nrcardio.2014.188.
- 4. Lesnefsky EJ, Chen Q, Tandler B, Hoppel CL. Mitochondrial dysfunction and myocardial ischemia-reperfusion: implications for novel therapies. Annu Rev Pharmacol Toxicol. 2017;57:535–65.
- 5. Yang M, Linn BS, Zhang Y, Ren J. Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochim Biophys Acta Mol Basis Dis. 2019;1865(9):2293–302. http://dx.doi.org/10.1016/j.bbadis.2019.05.007.
- 6. Deng Y, Yang M, Xu F, Zhang Q, Zhao Q, Yu H, et al. Combined salvianolic acid B and ginsenoside Rg1 exerts cardioprotection against ischemia/reperfusion injury in rats. PLoS One. 2015;10(8):e0135435. http://dx.doi.org/10.1371/journal.pone.0135435.
- 7. Park KS, Wiederkehr A, Wollheim CB. Defective mitochondrial function and motility due to mitofusin 1 overexpression in insulin secreting cells. Korean J Physiol Pharmacol. 2012;16(1):71–7. http://dx.doi.org/10.4196/kjpp.2012.16.1.71.
- 8. Meyer JN, Leuthner TC, Luz AL. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology. 2017;391:42–53. http://dx.doi.org/10.1016/j.tox.2017.07.019.
- 9. Sas K, Szabó E, Vécsei L. Mitochondria, oxidative stress and the kynurenine system, with a focus on ageing and neuroprotection. Molecules. 2018;23(1):191. http://dx.doi.org/10.3390/molecules23010191.
- 10. Belenguer P, Pellegrini L. The dynamin GTPase OPA1: more than mitochondria?Biochim Biophys Acta. 2013;1833(1):176–83.
- 11. Rojo M, Legros F, Chateau D, Lombès A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci. 2002;115(Pt 8):1663–74. http://dx.doi.org/10.1242/jcs.115.8.1663.
- 12. Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305(5685):858–62. http://dx.doi.org/10.1126/science.1099793.
- 13. Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24(5):659–67.
- 14. El-Hattab AW, Suleiman J, Almannai M, Scaglia F. Mitochondrial dynamics: biological roles, molecular machinery, and related diseases. Mol Genet Metab. 2018;125(4):315–21.
- 15. Quintana-Cabrera R, Mehrotra A, Rigoni G, Soriano ME. Who and how in the regulation of mitochondrial cristae shape and function. Biochem Biophys Res Commun. 2018;500(1):94–101. http://dx.doi.org/10.1016/j.bbrc.2017.04.088.
- 16. Son JM, Sarsour EH, Kakkerla Balaraju A, Fussell J, Kalen AL, Wagner BA, et al. Mitofusin 1 and optic atrophy 1 shift metabolism to mitochondrial respiration during aging. Aging Cell. 2017;16(5):1136–45. http://dx.doi.org/10.1111/acel.12649.
- 17. Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126(1):177–89. http://dx.doi.org/10.1016/j.cell.2006.06.025.
- 18. Jhun BS, O-Uchi J, Adaniya SM, Cypress MW, Yoon Y. Adrenergic regulation of Drp1-driven mitochondrial fission in cardiac physio-pathology. Antioxidants. 2018;7(12):195. http://dx.doi.org/10.3390/antiox7120195.
- 19. Bui HT, Shaw JM. Dynamin assembly strategies and adaptor proteins in mitochondrial fission. Curr Biol. 2013;23(19):R891–9. http://dx.doi.org/10.1016/j.cub.2013.08.040.
- 20. Ma J, Zhai Y, Chen M, Zhang K, Chen Q, Pang X, et al. New interfaces on MiD51 for Drp1 recruitment and regulation. PLoS One. 2019;14(1):e0211459. http://dx.doi.org/10.1371/journal.pone.0211459.
- 21. Hu Q, Zhang H, Gutiérrez Cortés N, Wu D, Wang P, Zhang J, et al. Increased Drp1 acetylation by lipid overload induces cardiomyocyte death and heart dysfunction. Circ Res. 2020;126(4):456–70. http://dx.doi.org/10.1161/CIRCRESAHA.119.315252.
- 22. Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337(6098):1062–5. http://dx.doi.org/10.1126/science.1219855.
- 23. Sanchis-Gomar F, Garcia-Gimenez JL, Gomez-Cabrera MC, Pallardo FV. Mitochondrial biogenesis in health and disease. Molecular and therapeutic approaches. Curr Pharm Des. 2014;20(35):5619–33.
- 24. Valero T. Mitochondrial biogenesis: pharmacological approaches. Curr Pharm Des. 2014;20(35):5507–9.
- 25. Lahera V, de Las Heras N, López-Farré A, Manucha W, Ferder L. Role of mitochondrial dysfunction in hypertension and obesity. Curr Hypertens Rep. 2017;19(2):11. http://dx.doi.org/10.1007/s11906-017-0710-9.
- 26. Ren J, Pulakat L, Whaley-Connell A, Sowers JR. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J Mol Med. 2010;88(10):993–1001. http://dx.doi.org/10.1007/s00109-010-0663-9.
- 27. Jiang S, Li T, Ji T, Yi W, Yang Z, Wang S, et al. AMPK: potential therapeutic target for ischemic stroke. Theranostics. 2018;8(16):4535–51.
- 28. Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104(29):12017–22.
- 29. Gurd BJ. Deacetylation of PGC-1alpha by SIRT1: importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl Physiol Nutr Metab. 2011;36(5):589–97.
- 30. Suliman HB, Piantadosi CA. Mitochondrial quality control as a therapeutic target. Pharmacol Rev. 2016;68(1):20–48. http://dx.doi.org/10.1124/pr.115.011502.
- 31. Hao M, Zhu S, Hu L, Zhu H, Wu X, Li Q. Myocardial ischemic postconditioning promotes autophagy against ischemia reperfusion injury via the activation of the nNOS/AMPK/mTOR pathway. Int J Mol Sci. 2017;18(3):614. http://dx.doi.org/10.3390/ijms18030614.
- 32. Chen Y, Dorn GW 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340(6131):471–5. http://dx.doi.org/10.1126/science.1231031.
- 33. Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014;54(3):362–77. http://dx.doi.org/10.1016/j.molcel.2014.02.034.
- 34. Reshma PL, Sainu NS, Mathew AK, Raghu KG. Mitochondrial dysfunction in H9c2 cells during ischemia and amelioration with Tribulus terrestris L. Life Sci. 2016;152:220–30. http://dx.doi.org/10.1016/j.lfs.2016.03.055.
- 35. Ma C, Zhang C, Ma M, Zhang L, Zhang L, Zhang F, et al. MiR-125a regulates mitochondrial homeostasis through targeting mitofusin 1 to control hypoxic pulmonary vascular remodeling. J Mol Med. 2017;95(9):977–93. http://dx.doi.org/10.1007/s00109-017-1541-5.
- 36. Ni HM, Williams JA, Ding WX. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015;4:6–13. http://dx.doi.org/10.1016/j.redox.2014.11.006.
- 37. Ma C, Beyer AM, Durand M, Clough AV, Zhu D, Norwood Toro L, et al. Hyperoxia causes mitochondrial fragmentation in pulmonary endothelial cells by increasing expression of pro-fission proteins. Arterioscler Thromb Vasc Biol. 2018;38(3):622–35. http://dx.doi.org/10.1161/ATVBAHA.117.310605.
- 38. Andres AM, Tucker KC, Thomas A, Taylor DJ, Sengstock D, Jahania SM, et al. Mitophagy and mitochondrial biogenesis in atrial tissue of patients undergoing heart surgery with cardiopulmonary bypass. JCI Insight. 2017;2(4):e89303. http://dx.doi.org/10.1172/jci.insight.89303.
- 39. Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. 2015;350(6265):aad2459. http://dx.doi.org/10.1126/science.aad2459.
- 40. Manczak M, Kandimalla R, Yin X, Reddy PH. Mitochondrial division inhibitor 1 reduces dynamin-related protein 1 and mitochondrial fission activity. Hum Mol Genet. 2019;28(2):177–99. http://dx.doi.org/10.1093/hmg/ddy335.
- 41. Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14(2):193–204. http://dx.doi.org/10.1016/j.devcel.2007.11.019.
- 42. Szabo A, Sumegi K, Fekete K, Hocsak E, Debreceni B, Setalo G, et al. Activation of mitochondrial fusion provides a new treatment for mitochondria-related diseases. Biochem Pharmacol. 2018;150:86–96. http://dx.doi.org/10.1016/j.bcp.2018.01.038.
- 43. Oliver D, Reddy PH. Dynamics of dynamin-related protein 1 in Alzheimer’s disease and other neurodegenerative diseases. Cells. 2019;8(9):961. http://dx.doi.org/10.3390/cells8090961.
- 44. Valenti D, Rossi L, Marzulli D, Bellomo F, De Rasmo D, Signorile A, et al. Inhibition of Drp1-mediated mitochondrial fission improves mitochondrial dynamics and bioenergetics stimulating neurogenesis in hippocampal progenitor cells from a Down syndrome mouse model. Biochim Biophys Acta Mol Basis Dis. 2017;1863(12):3117–27. http://dx.doi.org/10.1016/j.bbadis.2017.09.014.
- 45. Ong SB, Kwek XY, Katwadi K, Hernandez-Resendiz S, Crespo-Avilan GE, Ismail NI, et al. Targeting mitochondrial fission using Mdivi-1 in a clinically relevant large animal model of acute myocardial infarction: a pilot study. Int J Mol Sci. 2019;20(16):3972.
- 46. Martinez J, Tarallo D, Martinez-Palma L, Victoria S, Bresque M, Rodríguez-Bottero S, et al. Mitofusins modulate the increase in mitochondrial length, bioenergetics and secretory phenotype in therapy-induced senescent melanoma cells. Biochem J. 2019;476(17):2463–86.
- 47. Sapra G, Tham YK, Cemerlang N, Matsumoto A, Kiriazis H, Bernardo BC, et al. The small-molecule BGP-15 protects against heart failure and atrial fibrillation in mice. Nat Commun. 2014;5:5705. http://dx.doi.org/10.1038/ncomms6705.
- 48. Nagy G, Szarka A, Lotz G, Dóczi J, Wunderlich L, Kiss A, et al. BGP-15 inhibits caspase-independent programmed cell death in acetaminophen-induced liver injury. Toxicol Appl Pharmacol. 2010;243(1):96–103. http://dx.doi.org/10.1016/j.taap.2009.11.017.
- 49. Pernas L, Scorrano L. Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu Rev Physiol. 2016;78:505–31.
- 50. Yamada T, Murata D, Adachi Y, Itoh K, Kameoka S, Igarashi A, et al. Mitochondrial stasis reveals p62-mediated ubiquitination in parkin-independent mitophagy and mitigates nonalcoholic fatty liver disease. Cell Metab. 2018;28(4):588–604.e5. http://dx.doi.org/10.1016/j.cmet.2018.06.014.
- 51. Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15(7):397–408. http://dx.doi.org/10.1038/nrc3960.
- 52. Tilokani L, Nagashima S, Paupe V, Prudent J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. 2018;62(3):341–60.
- 53. Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148(6):1145–59.
- 54. Romanello V, Scalabrin M, Albiero M, Blaauw B, Scorrano L, Sandri M. Inhibition of the fission machinery mitigates OPA1 impairment in adult skeletal muscles. Cells. 2019;8(6):597. http://dx.doi.org/10.3390/cells8060597.
- 55. Yang F, Wu R, Jiang Z, Chen J, Nan J, Su S, et al. Leptin increases mitochondrial OPA1 via GSK3-mediated OMA1 ubiquitination to enhance therapeutic effects of mesenchymal stem cell transplantation. Cell Death Dis. 2018;9(5):556. http://dx.doi.org/10.1038/s41419-018-0579-9.
- 56. Das S, Mitrovsky G, Vasanthi HR, Das DK. Antiaging properties of a grape-derived antioxidant are regulated by mitochondrial balance of fusion and fission leading to mitophagy triggered by a signaling network of Sirt1-Sirt3-Foxo3-PINK1-PARKIN. Oxid Med Cell Longev. 2014;2014:345105. http://dx.doi.org/10.1155/2014/345105.
- 57. Blanquer-Rossello MM, Santandreu FM, Oliver J, Roca P, Valle A. Leptin modulates mitochondrial function, dynamics and biogenesis in MCF-7 cells. J Cell Biochem. 2015;116(9):2039–48.
- 58. Zhao Z, Pu Y. Lixisenatide enhances mitochondrial biogenesis and function through regulating the CREB/PGC-1α pathway. Biochem Biophys Res Commun. 2019;508(4):1120–5. http://dx.doi.org/10.1016/j.bbrc.2018.11.135.
- 59. Villena JA. New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015;282(4):647–72.
- 60. de Oliveira MR, Nabavi SF, Manayi A, Daglia M, Hajheydari Z, Nabavi SM. Resveratrol and the mitochondria: from triggering the intrinsic apoptotic pathway to inducing mitochondrial biogenesis, a mechanistic view. Biochim Biophys Acta. 2016;1860(4):727–45.
- 61. Jimenez RE, Kubli DA, Gustafsson AB. Autophagy and mitophagy in the myocardium: therapeutic potential and concerns. Br J Pharmacol. 2014;171(8):1907–16.
- 62. Andres AM, Hernandez G, Lee P, Huang C, Ratliff EP, Sin J, et al. Mitophagy is required for acute cardioprotection by simvastatin. Antioxid Redox Signal. 2014;21(14):1960–73. http://dx.doi.org/10.1089/ars.2013.5416.
- 63. Yu P, Zhang J, Yu S, Luo Z, Hua F, Yuan L, et al. Protective effect of sevoflurane postconditioning against cardiac ischemia/reperfusion injury via ameliorating mitochondrial impairment, oxidative stress and rescuing autophagic clearance. PLoS One. 2015;10(8):e0134666. http://dx.doi.org/10.1371/journal.pone.0134666.
- 64. Ren X, Chen L, Xie J, Zhang Z, Dong G, Liang J, et al. Resveratrol ameliorates mitochondrial elongation via Drp1/Parkin/PINK1 signaling in senescent-like cardiomyocytes. Oxid Med Cell Longev. 2017;2017:4175353. http://dx.doi.org/10.1155/2017/4175353.
- 65. Broughton KM, Sussman MA. Empowering adult stem cells for myocardial regeneration V2.0: success in small steps. Circ Res. 2016;118(5):867–80.
- 66.
- 67. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98(15):8554–9.
- 68. Li MX, Yang Y, Zhao Q, Wu Y, Song L, Yang H, et al. Degradation versus inhibition: development of proteolysis-targeting chimeras for overcoming statin-induced compensatory upregulation of 3-hydroxy-3-methylglutaryl coenzyme a reductase. J Med Chem. 2020;63(9):4908–28.
- 69. Yan W, Zhang H, Liu P, Wang H, Liu J, Gao C, et al. Impaired mitochondrial biogenesis due to dysfunctional adiponectin-AMPK-PGC-1α signaling contributing to increased vulnerability in diabetic heart. Basic Res Cardiol. 2013;108(3):329. http://dx.doi.org/10.1007/s00395-013-0329-1.
- 70. Kubowicz P, Żelaszczyk D, Pękala E. RNAi in clinical studies. Curr Med Chem. 2013;20(14):1801–16. http://dx.doi.org/10.2174/09298673113209990118.
- 71. Fellmann C, Gowen BG, Lin PC, Doudna JA, Corn JE. Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat Rev Drug Discov. 2017;16(2):89–100. http://dx.doi.org/10.1038/nrd.2016.238.
- 72. Fisher SA, Doree C, Mathur A, Martin-Rendon E. Meta-analysis of cell therapy trials for patients with heart failure. Circ Res. 2015;116(8):1361–77. http://dx.doi.org/10.1161/CIRCRESAHA.116.304386.
- 73. Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331(6020):1078–80. http://dx.doi.org/10.1126/science.1200708.
- 74. Haubner BJ, Adamowicz-Brice M, Khadayate S, Tiefenthaler V, Metzler B, Aitman T, et al. Complete cardiac regeneration in a mouse model of myocardial infarction. Aging. 2012;4(12):966–77. http://dx.doi.org/10.18632/aging.100526.
- 75. Zhang S-N, Sun AJ, Ge J-B, Yao K, Huang Z-Y, Wang K-Q, et al. Intracoronary autologous bone marrow stem cells transfer for patients with acute myocardial infarction: a meta-analysis of randomised controlled trials. Int J Cardiol. 2009;136(2):178–85.
- 76. Abdel-Latif A, Bolli R, Tleyjeh IM, Montori VM, Perin EC, Hornung CA, et al. Adult bone marrow-derived cells for cardiac repair: a systematic review and meta-analysis. Arch Intern Med. 2007;167(10):989–97.
- 77. Chavakis E, Koyanagi M, Dimmeler S. Enhancing the outcome of cell therapy for cardiac repair: progress from bench to bedside and back. Circulation. 2010;121(2):325–35.
- 78. Hosoda T, D’Amario D, Cabral-Da-Silva MC, Zheng H, Padin-Iruegas ME, Ogorek B, et al. Clonality of mouse and human cardiomyogenesis in vivo. Proc Natl Acad Sci U S A. 2009;106(40):17169–74. http://dx.doi.org/10.1073/pnas.0903089106.
- 79. Leor J, Aboulafia-Etzion S, Dar A, Shapiro L, Barbash IM, Battler A, et al. Bioengineered cardiac grafts: a new approach to repair the infarcted myocardium?Circulation. 2000;102(19 Suppl 3):III56–61.
- 80. Christman KL, Lee RJ. Biomaterials for the treatment of myocardial infarction. J Am Coll Cardiol. 2006;48(5):907–13. http://dx.doi.org/10.1016/j.jacc.2006.06.005.
- 81. Abdi SI, Ng SM, Lim JO. An enzyme-modulated oxygen-producing micro-system for regenerative therapeutics. Int J Pharm. 2011;409(1–2):203–5. http://dx.doi.org/10.1016/j.ijpharm.2011.02.041.
- 82. Li X, Tamama K, Xie X, Guan J. Improving cell engraftment in cardiac stem cell therapy. Stem Cells Int. 2016;2016:7168797. http://dx.doi.org/10.1155/2016/7168797.
- 83. Masuzawa A, Black KM, Pacak CA, Ericsson M, Barnett RJ, Drumm C, et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2013;304(7):H966–82. http://dx.doi.org/10.1152/ajpheart.00883.2012.
- 84. Blitzer D, Guariento A, Doulamis IP, Shin B, Moskowitzova K, Barbieri GR, et al. Delayed transplantation of autologous mitochondria for cardioprotection in a porcine model. Ann Thorac Surg. 2020;109(3):711–9. http://dx.doi.org/10.1016/j.athoracsur.2019.06.075.
- 85. McCully JD, Cowan DB, Pacak CA, Toumpoulis IK, Dayalan H, Levitsky S. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am J Physiol Heart Circ Physiol. 2009;296(1):H94–H105. http://dx.doi.org/10.1152/ajpheart.00567.2008.
- 86. Cowan DB, Yao R, Akurathi V, Snay ER, Thedsanamoorthy JK, Zurakowski D, et al. Intracoronary delivery of mitochondria to the ischemic heart for cardioprotection. PLoS One. 2016;11(8):e0160889. http://dx.doi.org/10.1371/journal.pone.0160889.
- 87. Lightowlers RN, Chrzanowska-Lightowlers ZM, Russell OM. Mitochondrial transplantation-a possible therapeutic for mitochondrial dysfunction?: mitochondrial transfer is a potential cure for many diseases but proof of efficacy and safety is still lacking. EMBO Rep. 2020;21(9):e50964.
- 88. Guariento A, Blitzer D, Doulamis I, Shin B, Moskowitzova K, Orfany A, et al. Preischemic autologous mitochondrial transplantation by intracoronary injection for myocardial protection. J Thorac Cardiovasc Surg. 2020;160(2):e15–29. http://dx.doi.org/10.1016/j.jtcvs.2019.06.111.
- 89. Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–87.
- 90. Shutt TE, McBride HM. Staying cool in difficult times: mitochondrial dynamics, quality control and the stress response. Biochim Biophys Acta. 2013;1833(2):417–24.
- 91. Anzell AR, Maizy R, Przyklenk K, Sanderson TH. Mitochondrial quality control and disease: insights into ischemia-reperfusion injury. Mol Neurobiol. 2018;55(3):2547–64.
Manli Zhou and Yunfeng Yu contributed equally to this work.