Introduction
Arteries in the human brain share many features with those of arteries from other organs throughout the body, and recommendations to prevent and treat vascular disorders affecting the brain share features with those used for other organs, such as an emphasis on physical activity, lipid control, and prescription of antithrombotic agents. The brain however is susceptible to several vascular disorders that either are not found in other parts of the body or when found are much less likely to cause clinical syndromes in other organs. This specific vulnerability of the brain may be explained by structural and functional differences between the vessels of the brain and those of vessels in other parts of the body.
Human arteries consist of three layers: intima, media, and adventitia. The intima is the innermost layer and consists of endothelial cells with an underlying thin basement membrane. The intima is separated from the media by the internal elastic lamina (IEL), which is a dense membrane composed of elastic fibers that contributes to the elasticity and integrity of the vessel wall. The media is comprised of smooth muscle cells (SMCs), the predominant matrix-synthesizing cells of the vascular wall, surrounded by elastin fibers and collagen. The media is separated from the adventitia by an external elastic lamina. The adventitia is the outermost layer of the artery and contains fibroblasts, connective tissue, and, in many organ systems, vasa vasorum. Vasa vasorum, meaning “vessels of the vessels,” is a network of small blood vessels that delivers oxygen and nutrition and eliminates wastes from the adventitia and media layers.
The brain’s vascular and neuronal constituents have a uniquely close interaction with one another in the form of the neurovascular unit (NVU). A relatively new concept, the NVU, is believed to be responsible for the maintenance of the blood-brain barrier (BBB) and cerebral homeostasis, as well as regulating cerebral blood flow. The BBB is the term used to describe the highly selective barrier system that separates the brain and cerebrospinal fluid from blood and prevents the nonselective crossing of solutes from the blood into the brain. The NVU is composed of endothelial cells, basement membrane, neurons, microglia, mural cells (comprising vascular SMC and pericytes), and astrocytes []. Endothelial cells line the cerebral vasculature and provide the major anatomical BBB. The tight junction between endothelial cells provides a physical barrier that helps regulate the entry of molecules and ions from blood into the tissue while restricting entry of potentially harmful components from the blood. Astrocytes play a crucial role in the formation and maintenance of the BBB. They are located between neurons and endothelial cells within the NVU, and they extend “end-feet” processes from their cell bodies to almost completely surround the arterioles and capillaries. Pericytes are perivascular cells that play an important role in regulating homeostasis in the BBB. Their unique position surrounding the endothelial cells of the vascular wall allows them to influence vascular diameter and alter local blood flow [].
Vessel wall composition, perivascular cell types, and neurovascular associations vary within the NVU []. Large extracranial and intracranial cerebral arteries have multiple layers of SMCs that encircle the endothelial basement membrane. As the arteries become smaller, the number of layers of SMC decreases. In arterioles, the endothelial and SMC basement membrane join together and are separated from the astrocytic basement membrane by the perivascular space. In smaller arterioles, the perivascular space disappears, and the vascular basement membrane joins the astrocytic basement membrane and the vessel-associated microglia []. In capillaries, SMCs are replaced by pericytes nestled into the endothelial basement membrane. SMCs reappear in the veins and are surrounded by vascular basement membranes and perivascular spaces. Depending on the location and size of the vessel, a wide variety of cells surround the outer vessel wall in the perivascular space. The internal carotid artery is surrounded by fibroblast-like cells embedded into the connective tissue of the adventitia, while arteries entering the subarachnoid space are enveloped by a network of fibrous septations of the arachnoid membrane together with fibroblasts, leptomeningeal cells, mast cells, and meningeal macrophages []. Penetrating arterioles and venules are surrounded by perivascular macrophages and leptomeningeal cells from the pia. Neural elements and vessels are in close contact throughout the neurovascular network, with nerve bundles originating from cranial autonomic ganglia forming a dense plexus around extracranial and intracranial cerebral arteries []. Pial arteriolar branches have a perivascular nerve plexus surrounding the vascular wall which eventually disappears in smaller penetrating arterioles [].
Intracranial arteries display several anatomic and physiological qualities that distinguish them from extracranial arteries, including coronary arteries, as shown in Figure 1. First, the intracranial arterial wall has a thicker IEL, thinner media with lower proportion of elastic fibers and SMC, absent external elastic lamina, the rare presence of vasa vasorum, and less abundant adventitia []. These arterial wall changes occur as the extracranial vertebral and internal carotid arteries enter the skull and become intradural and intracranial []. Additional differences exist even among the large intracranial arteries, with the vertebral and internal carotid arteries having greater wall thickness than the middle cerebral and basilar artery [], perhaps due to their natural extension from the extracranial arteries and serving as transition zones. Further differences exist between the anterior and posterior intracranial arteries, with posterior arteries having a larger IEL proportion, less elastin, and more concentric intimal thickening []. The lower media elastic fiber content in intracranial arteries plays an important role in making these arteries more muscular than the elastic extracranial arteries []. Additionally, the lower proportion of elastic fibers in intracranial arteries may affect the composition of the matrix surrounding the SMCs and their inflammatory and proliferative response.
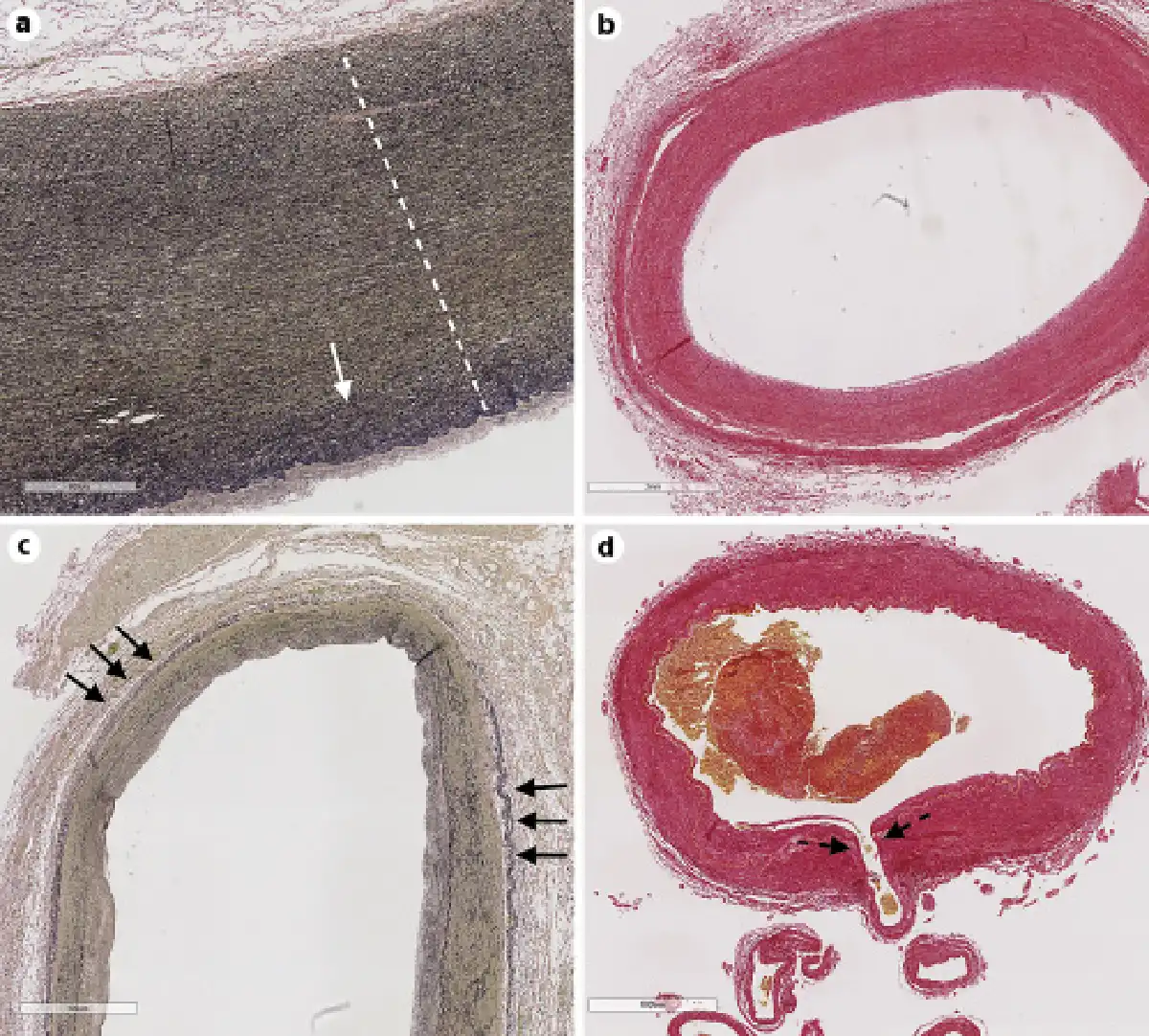
Fig. 1
The aorta (a; stain elastic Van Gieson) is the largest caliber artery in our body and has the thickest arterial wall. The aortic wall is rich in elastin fiber (black threads in the media, delimited by the dotted white line), which tend to increase in density on the luminal side (solid white arrow). The vertebral artery (b) and common carotid artery (c) in their most proximal portion have relatively larger adventitia compared to the aorta and in this case have concentric intima thickening with no apparent intima cholesterol deposition (cholesterol deposits will be white as evidenced by Sirius Red stain in b). These muscular arteries have less elastin compared to the aorta but more elastin than cerebral arteries. In addition, these arteries exhibit external elastic laminae (short black arrows, c). Cerebral arteries, such as the basilar artery (d), often give off penetrating arteries that supply the brainstem and the basal ganglia and that are susceptible disease, causing lacunar infarcts (penetrating artery origin, dotted black arrow).
Second, cerebrospinal fluid surrounds the intracranial arteries and may be relevant to the development of certain intracranial arterial pathologies []. For example, the lack of external elastic lamina, medial elastic fibers, and surrounding connective tissue all play an important role in making cerebral arteries susceptible to saccular aneurysm formation []. Third, compared to the extracranial arteries, there is a relative lack of intracranial artery vasa vasorum. Vasa vasorum are absent in the intracranial arteries of neonates and children and are mostly seen in proximal parts of large intracranial arteries in adults, suggesting they may be an acquired intrusion of extracranial artery vasa vasorum [, ]. The lack of vasa vasorum in more distal intracranial arteries further suggests the outer layers of the arteries may be nurtured through simple diffusion from the cerebrospinal fluid. Fourth, autoregulation, due to arteriolar dilatation and constriction, is a feature of intracranial arteries that allows cerebral blood flow to be maintained at a stable level across a wide systemic blood pressure range. Arteries display varying degrees of autoregulation, with the greatest capacity for autoregulation in cerebral, coronary, and renal arteries. Metabolic, myogenic, and neurogenic mechanisms are all involved in preserving cerebral autoregulation []. Fifth, the BBB is a unique feature of intracranial arteries that controls the content of brain interstitial fluid and only allows certain particles to diffuse from the cerebral vasculature into the brain. Finally, brain vessels display distinctive anatomic features, such as the circle of Willis and the presence of tiny threadlike vessels such as the lenticulostriate arteries that branch off of large parent vessels at right angles. Hypertensive hemorrhage is an example of how systemic pressure can affect penetrating arteries. Chronic hypertension can cause excessive wall tension, medial layer degeneration, and loss of elasticity in the middle and distal perforating arteries (100–600 μm in diameter) []. These changes increase the risk of vessel rupture and subsequent hemorrhage, particularly at bifurcations where the effects of tensile force are strongest [].
Although vascular disease is a systemic issue, it is unclear why certain diseases almost exclusively affect the brain. We aim to address how cerebrovascular anatomy and physiology may make the brain and its vessels more susceptible to vascular pathologies that are not seen, or rarely seen, in other organs. We focus on five diseases and syndromes that more commonly manifest in the intracranial vasculature. For each, we highlight the specific characteristics of the intracranial arteries that make them susceptible to these diseases, while noting areas of uncertainty requiring further research.
Posterior Reversible Encephalopathy Syndrome
Posterior Reversible Encephalopathy Syndrome (PRES) is a clinico-radiographic syndrome defined by reversible vasogenic edema and a constellation of neurological symptoms, including cortical visual loss, headache, and seizures []. While traditionally associated with acutely elevated blood pressure, PRES can result from a number of medical conditions including eclampsia, cytotoxic or immunosuppressive medication use, renal failure, and autoimmune conditions []. PRES does not seem to have a correlate in other organs.
PRES affects the intracranial arteries and is probably due to a failure of cerebral autoregulation and endothelial dysfunction [, ]. Rapid and severe hypertension can cause the systemic blood pressure to exceed the blood pressure range at which cerebral autoregulation functions to maintain stable cerebral blood flow. This causes the arterioles to continue to dilate with excessive cerebral blood flow, leading to cerebral hyperperfusion, breakdown of the BBB, and subsequent vasogenic edema. Endothelial dysfunction is also thought to play a significant role in development of PRES from autoimmune conditions and cytotoxic medication use. Endothelial dysfunction from cytotoxic or immunogenic causes may ultimately lead to BBB dysfunction, impaired autoregulation, and vasogenic edema []. The presence of endothelial dysfunction with or without systemic hypertension and blood pressure fluctuation is currently the main hypothesized mechanism of PRES development.
Bilateral cortical blindness was long considered a first symptom of acute renal failure, awareness of which led to the description of syndromes of biparietal and bioccipital dysfunction now subsumed under PRES. Modern imaging has now shown broader involvement which can include frontal and temporal lobes []. The preferential involvement of the posterior brain regions may be related to the relatively lower level of sympathetic innervation in the posterior circulation compared to the anterior circulation []. Such differences might not explain the biparietal prevalence, presumably from involvement of distal middle cerebral artery branches. Sympathetic innervation is part of the neurogenic mechanism of cerebral autoregulation and may protect the autoregulatory system from rapid and severe increases in systemic blood pressure [, ].
Reversible Cerebral Vasoconstriction Syndrome
Reversible Cerebral Vasoconstriction Syndrome (RCVS) is a reversible multifocal narrowing of intracranial arteries that can cause stroke (hemorrhagic or ischemic), seizures, and intracerebral edema []. RCVS, a frequently underdiagnosed disease, may occur spontaneously or with pregnancy, medication (specifically vasoactive drugs), and illicit drug use [].
Primary angiitis of the central nervous system (PACNS) and secondary central nervous system vasculitis are included in the differential diagnosis of RCVS. A detailed clinical history and review of the imaging findings can help distinguish between RCVS and PACNS. While both diseases cause vessel irregularities, RCVS usually causes a smooth, tapering arterial constriction and dilatation affecting the proximal arteries, whereas PACNS causes an irregular eccentric notched appearance with distal cutoffs and neovascularization []. Additionally, neuroimaging in PACNS is more likely to reveal deep or brainstem infarcts. Recurrent thunderclap headache and single thunderclap headache combined with either normal neuroimaging, border zone infarcts, or vasogenic edema has been reported to have a 100% positive predictive value for diagnosing RCVS [].
RCVS does not typically cause inflammation, vasculitis, or micro-thrombosis in the vessel walls, although prolonged vasoconstriction may rarely cause a secondary vessel inflammation [].
The pathophysiology of RCVS is unclear, but there are several proposed mechanisms. The main hypothesis supports the role of transient cerebral autoregulatory disruption and sympathetic overactivity, leading to impaired arterial tone []. The sympathetic nervous system innervates the proximal portions of the large intracerebral arteries and can cause constriction in these vessels. A strong sympathetic response to endogenous or exogenous factors, or an abnormal antidromic discharge of sympathetic afferents, can cause vasoconstriction and disruption of vascular tone []. Endothelial dysfunction has also been implicated in the disease process and is supported by an association between RCVS and a polymorphism in a brain-derived neurotrophic factor gene Val66Met involved in sympathetic overactivity and endothelial dysfunction []. Additionally, endothelial progenitor cells involved in repairing and maintaining endothelium are reduced in RCVS, particularly in more severe cases of vasoconstriction []. RCVS is frequently associated with PRES, perhaps due to the role of endothelial dysfunction in the pathogenesis of both disorders. Independent of the mechanism, the resulting vasoconstriction can lead to ischemic stroke through distal hypoperfusion, particularly in watershed territories.
Although RCVS was initially considered restricted to intracranial arteries, in some patients, there may be extracranial involvement, as well. RCVS has been linked to cervical artery dissection [], although it is unclear whether these two pathologies occur simultaneously or if one leads to another. Case series and case reports have also illustrated cardiac and renal abnormalities in RCVS patients [, ]. The well-developed autoregulatory mechanisms in these three organs may contribute to their common susceptibility to the effects of vasoconstriction. Further investigations are needed to better illustrate the pathophysiology of this frequently underdiagnosed condition.
Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is a small-vessel arteriopathy causing migraines, lacunar infarcts, and vascular cognitive impairment []. CADASIL is a known cause of stroke in the young and accounts for 11% of lacunar stroke and leukoariosis in patients less than 50 years of age [].
CADASIL is due to inherited and sporadic pathogenic mutations in the NOTCH3 gene on chromosome 19p13.2-p13.1. The NOTCH genes code for receptors involved in cell fate decisions during embryonic development. NOTCH3 is a large transmembrane receptor with extracellular and intracellular domains. Notch3 is the protein product of the NOTCH3 gene and a member of a family of four Notch receptors in mammals []. Over 200 different pathogenic variants in NOTCH3 have been reported in patients with CADASIL and include missense variants, splice site variants, and in-frame deletions []. All reported pathogenic variants affect highly conserved cysteine residues within epidermal growth factor-like repeats in the extracellular domain of the Notch3 transmembrane receptor []. The protein product Notch3 is mostly expressed in capillary pericytes and vascular SMCs of small arteries, where it is involved in SMC differentiation and functional development []. NOTCH3 is a large transmembrane receptor with extracellular and intracellular domains. A mutation in the gene leads to accumulation of the NOTCH3 extracellular domain around the vascular SMCs and pericytes of small arteries and capillaries [, ]. The changes in the vascular SMCs cause an arteriopathy in the small penetrating cerebral and leptomeningeal arteries, including many in the centrum semiovale not typically affected by hypertension. These affected arteries display prominent vascular SMC morphological changes and nonamyloid granular osmiophilic material within the media, causing thickening of the arterial wall and resulting in intraluminal stenosis [-]. Arterial thickening mostly occurs at an arteriole wall internal diameter of 20–30 µm and an external diameter of 100–130 µm []. Thrombus can form in these stenotic lumens and cause distal infarcts. Likewise, NOTCH3 accumulation in pericytes causes BBB dysfunction through reductions in pericyte number, detachment of astrocytic end-feet from cerebral microvessels, and loss of endothelial junction proteins []. Ultimately, these significant changes in the NVU compromise the integrity of the BBB and allow the leakage of proteins into the parenchyma, leading to neuronal damage [].
CADASIL is a systemic arteriopathy with involvement of arteries in the spleen, liver, heart, kidneys, muscle, aorta, and skin [, ]. Yet, for unclear reasons, the disease is primarily symptomatic in the brain and rarely affects function of other organs. This may be due to the brain’s higher blood flow demand in addition to the relative thickening and stenosis of the intracranial arteries. Further research on the varying phenotypic expression of CADASIL is warranted.
Moyamoya Disease
Moyamoya disease (MMD) is a rare cerebrovascular condition characterized by progressive bilateral stenosis of the proximal intracranial arteries with resultant compensation through a network of collaterals. The pathogenesis of MMD is unclear but may be related in at least some cases to the recently identified RNF213 susceptibility gene or elevated growth factors involved in angiogenesis, intimal thickening, and elastin synthesis []. Importantly, idiopathic MMD is distinct from moyamoya syndrome. Patients with moyamoya vasculopathy with an associated medical condition (intracranial atherosclerosis, sickle cell disease, neurofibromatosis type 1, cranial radiation therapy) are categorized as moyamoya syndrome, while patients with no known associated conditions and a possible genetic susceptibility are categorized as MMD [].
Conventional digital substraction angiography is the gold standard used to diagnose MMD. Disease severity is classified into six progressive stages using the Suzuki angiographic grading system, with grade I and II referring to narrowing of ICA apex and initiation of moyamoya collaterals, grade IV including the development of external carotid artery collaterals, and grade VI including total occlusion of ICA and disappearance of moyamoya-associated collaterals [].
The pathological changes of MMD affect both the proximal intracranial arteries (distal internal carotid artery and proximal middle, anterior, and posterior cerebral arteries) and the small compensating collateral “moyamoya” vessels []. MMD primarily affects the intima, causing multilayered fibrocellular thickening with newly formed elastic lamina between the layers []. Additional pathological changes include smooth muscle hyperplasia, attenuation of the media, irregular IEL, and luminal thrombosis [, ]. These changes cause progressive stenosis of the large intracranial arteries and can lead to IS. The irregular IEL and thinning media may also cause aneurysm formation and subsequent rupture.
To prevent reduced cerebral blood flow and hemodynamic insufficiency from the intracranial stenosis, collateralization forms via perforators, extracranial-intracranial pathways, and leptomeningeal and transdural anastomoses, as shown in Figure 2 []. The most commonly seen collaterals are new and preexisting perforating arteries, also called “moyamoya” vessels. These perforating arteries can become thin-walled and dilated as a response to hemodynamic stress and more readily form microaneurysms with the potential to rupture []. Other vessels can develop intimal thickening with IEL duplication, causing vessel stenosis with collapse of the lumen and subsequent thrombosis [, ].
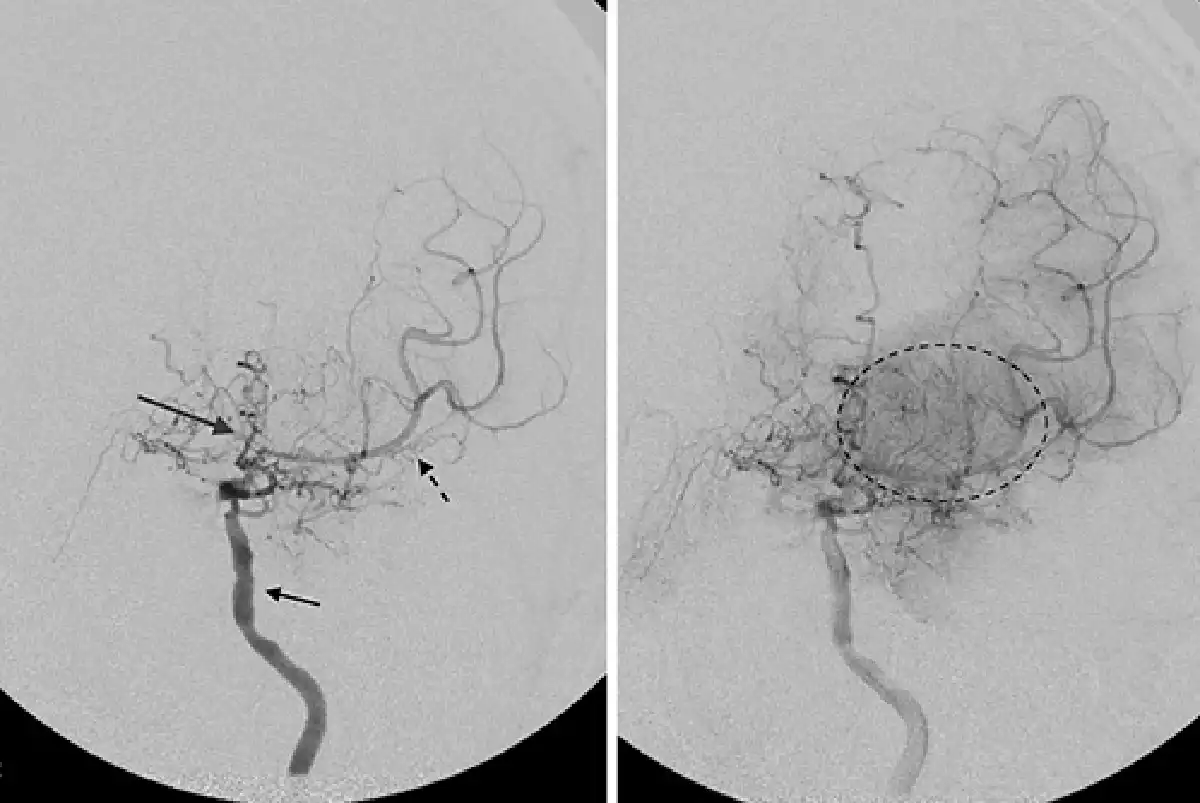
Fig. 2
Angiogram of the left internal carotid artery in a patient with chronic MMD. There is multifocal irregularity and stenosis of the left internal carotid artery proximal to the origin of the ophthalmic artery (short black arrow). There is severe stenosis of the proximal left middle cerebral artery (black dotted arrow) and anterior cerebral artery (long gray arrow). Abnormal network of collateral blood vessels, the “moyamoya vessels,” are seen in the basal ganglia (dotted black circle).
Although much less common, MMD may affect systemic arteries [-]. Most notably, 1 in 12 cases develop renal artery pathology, including proximal renal artery stenosis, leading to renovascular hypertension [-]. Similar to intracranial arteries, the involved extracranial vessels develop intimal fibrous thickening and collaterals with thrombi or occlusive lesions in them []. These changes suggest that MMD may be a more systemic process than previously appreciated, although the extent of extracranial involvement and the preference for intracranial vessels need to be further investigated.
Varicella Zoster Virus Vasculopathy
Varicella zoster virus (VZV) vasculopathy is a predominantly intracerebral artery infection from productive varicella or zoster in both immunocompetent and immunocompromised individuals []. Although seen in all age-groups, VZV is a cause of ischemic stroke in the young and may account for 31% of all childhood strokes and 44% of transient cerebral arteriopathies of childhood [, ]. Other clinical manifestations of VZV vasculopathy include both ischemic and hemorrhagic stroke, subarachnoid hemorrhage secondary to development and rupture of cerebral aneurysms, arterial dissections, dolichoectasia, spinal cord infarct, and sinus venous thrombosis [].
VZV is a DNA virus that belongs to the family of human herpesviruses. The virus is seen worldwide and is considered highly infectious. While most older adults were infected with the virus during childhood, the advent of a live virus vaccine has resulted in the majority of young adults and children having been vaccinated against the disease []. Primary VZV infection occurs via aerosols from skin vesicles of infected individuals and results in varicella. Varicella typically manifests with a fever and a vesicular, pruritic, widely disseminated rash that primarily involves the face and trunk. Once the primary infection resolves, VZV becomes latent in the dorsal root and autonomic ganglia along the neuraxis. This noninfectious phase of VZV is very common, with more than 95% of the general population harboring the latent form of the disease []. VZV reactivation may occur later in life (manifested as zoster or shingles), at which point, the virus may cause an exanthem of vesicular lesions within a dermatomal distribution. Occasionally, it can spread from the dorsal root ganglia to adjacent blood vessels and cause VZV vasculopathy [].
VZV vasculopathy is typically multifocal and can involve both small and large arteries []. Large-vessel disease results from a spread of either trigeminal- or cervical-distribution VZV within weeks to months after the reactivation of the virus []. Reactivated cranial VZV in immunocompetent patients typically involves the ophthalmic division of the trigeminal nerve []. Branches of the ophthalmic division of the trigeminal nerve provide afferent fibers to the ipsilateral intracranial portions of the internal carotid artery, the proximal anterior and middle cerebral arteries, and the dura [, ]. Similarly, the upper cervical dorsal root ganglia are thought to innervate the vertebrobasilar arteries []. These connections provide a pathway for the transaxonal spread of the reactivated virus from the ophthalmic division of the trigeminal nerve to the large anterior intracranial vessels and from the cervical dorsal root ganglia to the large posterior intracranial vessels. This transaxonal spread goes from the outermost adventitia toward the intima, ultimately causing intimal thickening, disruption of the IEL, and the presence of inflammatory cells in the adventitia and intima []. VZV can also cause a small-vessel vasculopathy, leading to a combination of necrosis and demyelination []. The small-vessel disease is characterized by multifocal small lesions in the deep or gray-white junction with involvement of white matter more than gray matter and intranuclear Cowdry A inclusions in more significant demyelination []. The presence of both large- and small-vessel arteriopathy should alert the clinician to the possibility of VZV vasculopathy as rapid diagnosis can lead to effective treatment and improvement [].
VZV’s predilection for intracranial and spinal arteries is most likely a result of local infection of small vessels near the dorsal root ganglia. However, VZV can also rarely cause an extracranial, systemic vasculopathy by affecting the central retinal [], radial [], femoral, and iliac arteries []. This raises the possibility that VZV may be implicated rarely in systemic vasculopathy.
Conclusion
Although significant advances have been made in the treatment and management of intracranial arteriopathies, many issues and questions related to pathophysiology remain unanswered. Specifically, it is unclear why many of these disease states preferentially affect the intracranial over the extracranial arteries. We suspect that much of this specificity is related to the different anatomy and composition of the intracranial arterial wall, lack of defined vasa vasorum, and the significant role of autoregulation in the cerebral vessels. By highlighting areas of uncertainty, we hope to help further the path for more clinical research in cerebral arteriopathies.
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
Funding Sources
Dr. Jose Gutierrez is supported by NIH Grant No. R01AG057709.
Author Contributions
Dr. Mitchell S.V. Elkind and Dr. Setareh Salehi Omran participated in research conception and design. Dr. Setareh Salehi Omran, Dr. Jose Gutierrez, Dr. Jay P. Mohr, and Dr. Mitchell S.V. Elkind contributed to interpretation of data for the work. Dr. Setareh Salehi Omran drafted the article. Dr. Jose Gutierrez, Dr. Mitchell S.V. Elkind, and Dr. Jay P. Mohr revised the article critically. Dr. Setareh Salehi Omran, Dr. Jose Gutierrez, Dr. Jay P. Mohr, and Dr. Mitchell S.V. Elkind contributed to final approval. All the authors agree to be accountable for all aspects of the work.
References
- 1. Schaeffer S, Iadecola C. Revisiting the neurovascular unit. Nat Neurosci. 2021;24(9):1198–209. http://dx.doi.org/10.1038/s41593-021-00904-7.
- 2. Bell AH, Miller SL, Castillo-Melendez M, Malhotra A. The neurovascular unit: effects of brain insults during the perinatal period. Front Neurosci. 2020;13:1452. http://dx.doi.org/10.3389/fnins.2019.01452.
- 3. Roth W, Morgello S, Goldman J, Mohr JP, Elkind MS, Marshall RS, et al. Histopathological differences between the anterior and posterior brain arteries as a function of aging. Stroke. 2017;48(3):638–44.
- 4. Ratinov G. Extradural intracranial portion of carotid artery; a clinicopathologic study. Arch Neurol. 1964;10:66–73. http://dx.doi.org/10.1001/archneur.1964.00460130070010.
- 5. Gutierrez J, Rosoklija G, Murray J, Chon C, Elkind MS, Goldman J, et al. A quantitative perspective to the study of brain arterial remodeling of donors with and without HIV in the Brain Arterial Remodeling Study (BARS). Front Physiol. 2014;5:56.
- 6. Ritz K, Denswil NP, Stam OC, van Lieshout JJ, Daemen MJ. Cause and mechanisms of intracranial atherosclerosis. Circulation. 2014;130(16):1407–14. http://dx.doi.org/10.1161/CIRCULATIONAHA.114.011147.
- 7. Etminan N, Rinkel GJ. Unruptured intracranial aneurysms: development, rupture and preventive management. Nat Rev Neurol. 2016;12(12):699–713. http://dx.doi.org/10.1038/nrneurol.2016.150.
- 8. Humphrey JD, Taylor CA. Intracranial and abdominal aortic aneurysms: similarities, differences, and need for a new class of computational models. Annu Rev Biomed Eng. 2008;10:221–46. http://dx.doi.org/10.1146/annurev.bioeng.10.061807.160439.
- 9. Aydin F. Do human intracranial arteries lack vasa vasorum? A comparative immunohistochemical study of intracranial and systemic arteries. Acta Neuropathol. 1998;96(1):22–8. http://dx.doi.org/10.1007/s004010050856.
- 10. Hamner JW, Tan CO. Relative contributions of sympathetic, cholinergic, and myogenic mechanisms to cerebral autoregulation. Stroke. 2014;45(6):1771–7. http://dx.doi.org/10.1161/STROKEAHA.114.005293.
- 11. Takebayashi S, Kaneko M. Electron microscopic studies of ruptured arteries in hypertensive intracerebral hemorrhage. Stroke. 1983;14(1):28–36. http://dx.doi.org/10.1161/01.str.14.1.28.
- 12. Feske SK. Posterior reversible encephalopathy syndrome: a review. Semin Neurol. 2011;31(2):202–15. http://dx.doi.org/10.1055/s-0031-1277990.
- 13. Fugate JE, Rabinstein AA. Posterior reversible encephalopathy syndrome: clinical and radiological manifestations, pathophysiology, and outstanding questions. Lancet Neurol. 2015;14(9):914–25. http://dx.doi.org/10.1016/S1474-4422(15)00111-8.
- 14. Fugate JE, Claassen DO, Cloft HJ, Kallmes DF, Kozak OS, Rabinstein AA. Posterior reversible encephalopathy syndrome: associated clinical and radiologic findings. Mayo Clin Proc. 2010;85(5):427–32. http://dx.doi.org/10.4065/mcp.2009.0590.
- 15. Edvinsson L, Owman C, Siesjö B. Physiological role of cerebrovascular sympathetic nerves in the autoregulation of cerebral blood flow. Brain Res. 1976;117(3):519–23. http://dx.doi.org/10.1016/0006-8993(76)90760-5.
- 16. ter Laan M, van Dijk JM, Elting JW, Staal MJ, Absalom AR. Sympathetic regulation of cerebral blood flow in humans: a review. Br J Anaesth. 2013;111(3):361–7. http://dx.doi.org/10.1093/bja/aet122.
- 17. Beausang-Linder M, Bill A. Cerebral circulation in acute arterial hypertension: protective effects of sympathetic nervous activity. Acta physiol Scand. 1981;111(2):193–9. http://dx.doi.org/10.1111/j.1748-1716.1981.tb06724.x.
- 18. Singhal AB, Topcuoglu MA, Fok JW, Kursun O, Nogueira RG, Frosch MP, et al. Reversible cerebral vasoconstriction syndromes and primary angiitis of the central nervous system: clinical, imaging, and angiographic comparison. Ann Neurol. 2016;79(6):882–94.
- 19. Ducros A. Reversible cerebral vasoconstriction syndrome. Lancet Neurol. 2012;11(10):906–17. http://dx.doi.org/10.1016/S1474-4422(12)70135-7.
- 20. Calado S, Vale-Santos J, Lima C, Viana-Baptista M. Postpartum cerebral angiopathy: vasospasm, vasculitis or both?Cerebrovasc Dis. 2004;18(4):340–1. http://dx.doi.org/10.1159/000080976.
- 21. Chen SP, Yang AC, Fuh JL, Wang SJ. Autonomic dysfunction in reversible cerebral vasoconstriction syndromes. J Headache Pain. 2013;14:94. http://dx.doi.org/10.1186/1129-2377-14-94.
- 22. Schwedt TJ, Matharu MS, Dodick DW. Thunderclap headache. Lancet Neurol. 2006;5(7):621–31. http://dx.doi.org/10.1016/S1474-4422(06)70497-5.
- 23. Chen SP, Fuh JL, Wang SJ, Tsai SJ, Hong CJ, Yang AC. Brain-derived neurotrophic factor gene Val66Met polymorphism modulates reversible cerebral vasoconstriction syndromes. PLoS One. 2011;6(3):e18024. http://dx.doi.org/10.1371/journal.pone.0018024.
- 24. Chen SP, Wang YF, Huang PH, Chi CW, Fuh JL, Wang SJ. Reduced circulating endothelial progenitor cells in reversible cerebral vasoconstriction syndrome. J Headache Pain. 2014;15:82. http://dx.doi.org/10.1186/1129-2377-15-82.
- 25. Mawet J, Boukobza M, Franc J, Sarov M, Arnold M, Bousser MG, et al. Reversible cerebral vasoconstriction syndrome and cervical artery dissection in 20 patients. Neurology. 2013;81(9):821–4.
- 26. John S, Hajj-Ali RA, Min D, Calabrese LH, Cerejo R, Uchino K. Reversible cerebral vasoconstriction syndrome: is it more than just cerebral vasoconstriction?Cephalalgia. 2015;35(7):631–4. http://dx.doi.org/10.1177/0333102414547139.
- 27. Mukerji SS, Buchbinder BR, Singhal AB. Reversible cerebral vasoconstriction syndrome with reversible renal artery stenosis. Neurology. 2015;85(2):201–2. http://dx.doi.org/10.1212/WNL.0000000000001740.
- 28. Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG. Cadasil. Lancet Neurol. 2009;8(7):643–53. http://dx.doi.org/10.1016/s1474-4422(09)70127-9.
- 29. Dong Y, Hassan A, Zhang Z, Huber D, Dalageorgou C, Markus HS. Yield of screening for CADASIL mutations in lacunar stroke and leukoaraiosis. Stroke. 2003;34(1):203–5. http://dx.doi.org/10.1161/01.str.0000048162.16852.88.
- 30. Coupland K, Lendahl U, Karlström H. Role of NOTCH3 mutations in the cerebral small vessel disease cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke. 2018;49(11):2793–800. http://dx.doi.org/10.1161/STROKEAHA.118.021560.
- 31. Peters N, Opherk C, Bergmann T, Castro M, Herzog J, Dichgans M. Spectrum of mutations in biopsy-proven CADASIL: implications for diagnostic strategies. Arch Neurol. 2005;62(7):1091–4. http://dx.doi.org/10.1001/archneur.62.7.1091.
- 32. Joutel A, Andreux F, Gaulis S, Domenga V, Cecillon M, Battail N, et al. The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J Clin Invest. 2000;105(5):597–605.
- 33. Joutel A, Favrole P, Labauge P, Chabriat H, Lescoat C, Andreux F, et al. Skin biopsy immunostaining with a Notch3 monoclonal antibody for CADASIL diagnosis. Lancet. 2001;358(9298):2049–51.
- 34. Kalimo H, Ruchoux MM, Viitanen M, Kalaria RN. CADASIL: a common form of hereditary arteriopathy causing brain infarcts and dementia. Brain Pathol. 2002;12(3):371–84. http://dx.doi.org/10.1111/j.1750-3639.2002.tb00451.x.
- 35. Ruchoux MM, Maurage CA. CADASIL: cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. J Neuropathol Exp Neurol. 1997;56(9):947–64. http://dx.doi.org/10.1097/00005072-199709000-00001.
- 36. Miao Q, Paloneva T, Tuominen S, Poyhonen M, Tuisku S, Viitanen M, et al. Fibrosis and stenosis of the long penetrating cerebral arteries: the cause of the white matter pathology in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Brain Pathol. 2004;14(4):358–64.
- 37. Ghosh M, Balbi M, Hellal F, Dichgans M, Lindauer U, Plesnila N. Pericytes are involved in the pathogenesis of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Ann Neurol. 2015;78(6):887–900. http://dx.doi.org/10.1002/ana.24512.
- 38. Ruchoux MM, Guerouaou D, Vandenhaute B, Pruvo JP, Vermersch P, Leys D. Systemic vascular smooth muscle cell impairment in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Acta Neuropathol. 1995;89(6):500–12. http://dx.doi.org/10.1007/BF00571504.
- 39. Kamada F, Aoki Y, Narisawa A, Abe Y, Komatsuzaki S, Kikuchi A, et al. A genome-wide association study identifies RNF213 as the first moyamoya disease gene. J Hum Genet. 2011;56(1):34–40.
- 40. Scott RM, Smith ER. Moyamoya disease and moyamoya syndrome. N Engl J Med. 2009;360(12):1226–37. http://dx.doi.org/10.1056/NEJMra0804622.
- 41. Hosoda Y, Ikeda E, Hirose S. Histopathological studies on spontaneous occlusion of the circle of Willis (cerebrovascular moyamoya disease). Clin Neurol Neurosurg. 1997;99(Suppl 2):S203–8. http://dx.doi.org/10.1016/s0303-8467(97)00044-9.
- 42. Yamashita M, Oka K, Tanaka K. Histopathology of the brain vascular network in moyamoya disease. Stroke. 1983;14(1):50–8. http://dx.doi.org/10.1161/01.str.14.1.50.
- 43. Takagi Y, Kikuta K, Nozaki K, Hashimoto N. Histological features of middle cerebral arteries from patients treated for moyamoya disease. Neurol Med Chir. 2007;47(1):1–4. http://dx.doi.org/10.2176/nmc.47.1.
- 44. Kono S, Oka K, Sueishi K. Histopathologic and morphometric studies of leptomeningeal vessels in moyamoya disease. Stroke. 1990;21(7):1044–50. http://dx.doi.org/10.1161/01.str.21.7.1044.
- 45. Ikeda E. Systemic vascular changes in spontaneous occlusion of the circle of Willis. Stroke. 1991;22(11):1358–62. http://dx.doi.org/10.1161/01.str.22.11.1358.
- 46. Togao O, Mihara F, Yoshiura T, Tanaka A, Kuwabara Y, Morioka T, et al. Prevalence of stenoocclusive lesions in the renal and abdominal arteries in moyamoya disease. AJR Am J Roentgenol. 2004;183(1):119–22.
- 47. Yamada I, Himeno Y, Matsushima Y, Shibuya H. Renal artery lesions in patients with moyamoya disease: angiographic findings. Stroke. 2000;31(3):733–7. http://dx.doi.org/10.1161/01.str.31.3.733.
- 48. Nagel MA, Gilden D. Developments in varicella zoster virus vasculopathy. Curr Neurol Neurosci Rep. 2016;16(2):12. http://dx.doi.org/10.1007/s11910-015-0614-5.
- 49. Askalan R, Laughlin S, Mayank S, Chan A, MacGregor D, Andrew M, et al. Chickenpox and stroke in childhood: a study of frequency and causation. Stroke. 2001;32(6):1257–62.
- 50. Braun KP, Bulder MM, Chabrier S, Kirkham FJ, Uiterwaal CS, Tardieu M, et al. The course and outcome of unilateral intracranial arteriopathy in 79 children with ischaemic stroke. Brain. 2009;132(Pt 2):544–57.
- 51. Heininger U, Seward JF. Varicella. Lancet. 2006;368(9544):1365–76. http://dx.doi.org/10.1016/S0140-6736(06)69561-5.
- 52. Nagel MA, Gilden D. Update on varicella zoster virus vasculopathy. Curr Infect Dis Rep. 2014;16(6):407. http://dx.doi.org/10.1007/s11908-014-0407-z.
- 53. Nagel MA, Cohrs RJ, Mahalingam R, Wellish MC, Forghani B, Schiller A, et al. The varicella zoster virus vasculopathies: clinical, CSF, imaging, and virologic features. Neurology. 2008;70(11):853–60.
- 54. Reshef E, Greenberg SB, Jankovic J. Herpes zoster ophthalmicus followed by contralateral hemiparesis: report of two cases and review of literature. J Neurol Neurosurg Psychiatry. 1985;48(2):122–7. http://dx.doi.org/10.1136/jnnp.48.2.122.
- 55. Mayberg M, Langer RS, Zervas NT, Moskowitz MA. Perivascular meningeal projections from cat trigeminal ganglia: possible pathway for vascular headaches in man. Science. 1981;213(4504):228–30. http://dx.doi.org/10.1126/science.6166046.
- 56. Saito K, Moskowitz MA. Contributions from the upper cervical dorsal roots and trigeminal ganglia to the feline circle of Willis. Stroke. 1989;20(4):524–6. http://dx.doi.org/10.1161/01.str.20.4.524.
- 57. Nagel MA, Traktinskiy I, Azarkh Y, Kleinschmidt-DeMasters B, Hedley-Whyte T, Russman A, et al. Varicella zoster virus vasculopathy: analysis of virus-infected arteries. Neurology. 2011;77(4):364–70.
- 58. Amlie-Lefond C, Kleinschmidt-DeMasters BK, Mahalingam R, Davis LE, Gilden DH. The vasculopathy of varicella-zoster virus encephalitis. Ann Neurol. 1995;37(6):784–90. http://dx.doi.org/10.1002/ana.410370612.
- 59. Hall S, Carlin L, Roach ES, McLean WT Jr.. Herpes zoster and central retinal artery occlusion. Ann Neurol. 1983;13(2):217–8. http://dx.doi.org/10.1002/ana.410130226.
- 60. Tezcan ME, Teksut TK, Onal AB, Oztürk MA. Reactivated varicella zoster virus may cause peripheral arterial thrombosis. J Rheumatol. 2010;37(8):1785–6. http://dx.doi.org/10.3899/jrheum.100232.
- 61. Massano J, Ferreira D, Toledo T, Mansilha A, Azevedo E, Carvalho M. Stroke and multiple peripheral thrombotic events in an adult with varicella. Eur J Neurol. 2008;15(10):e90–1. http://dx.doi.org/10.1111/j.1468-1331.2008.02267.x.