Introduction
Fibrosis is the process of excessive deposition of extracellular matrix (ECM) proteins and formation of scar tissue resulting from chronic tissue injury and inflammation. Fibrosis can occur in multiple organs including lung, skin, liver, kidney, and heart and can be triggered by a variety of infectious, toxic, or autoimmune processes [Wynn, 2008]. Accordingly, the clinical implications of fibrosis are of high importance, with some studies attributing up to 45% of deaths in the industrial world to tissue fibrosis [Wynn, 2004]. Understanding the pathogenesis of fibrosis, including shared and unique mediators in the different tissues, is important for developing therapeutic targets and strategies for patients with fibrotic diseases.
Fibrosis shares similar cellular and molecular pathways with wound healing and tissue repair. Wound healing and fibrosis result from a coordinated multicellular process, involving tissue macrophages and fibroblasts as well as tissue specific cellular populations depending on the tissue, such as type II alveolar epithelial cells in the lungs [Wynn, 2011] or hepatic stellate cells (HSCs) in the liver [Friedman, 2000]. During acute endothelial/epithelial damage, the coagulation pathway is initially triggered, followed by other inflammatory mediators to facilitate wound healing and restore homeostasis [Wynn and Ramalingam, 2012]. Release of pro-inflammatory cytokines and growth factors by epithelial cells results in the recruitment of innate immune cells such as macrophages and neutrophils to phagocytose dead tissue and debris. These macrophages also secrete multiple pro-resolutory cytokines and growth factors including transforming growth factor-β (TGF-β) and IL-13 that lead to differentiation of mesenchymal cells such as fibroblasts or liver-specific HSCs into pro-fibrogenic myofibroblasts [Wynn and Ramalingam, 2012; Wilkinson and Hardman, 2020].
TGF-β is a key cytokine that drives the differentiation of local and recruited fibroblasts into activated myofibroblasts. Myofibroblasts are capable of secreting large quantities of ECM components, including, but not limited to, type I collagen and fibronectin [Biernacka et al., 2011]. Myofibroblasts are essential in wound repair as they generate constrictive forces to facilitate wound closure but are later cleared by apoptosis as the ECM is degraded and remodeled during resolution and the restoration of the normal tissue architecture [Hantash et al., 2008]. However, if inflammation or the injury persists, activated myofibroblasts continue to accumulate, resulting in excessive deposition of ECM and disruption of the tissue architecture.
Understanding the cellular and molecular pathways of fibrogenesis is key to identifying novel molecular mediators that might be targeted therapeutically. As discussed in this review, cadherin-11 (CDH11) may be one such mediator. CDH11 expression is increased in the tissue of multiple fibrotic diseases including lung, skin, liver, cardiac, renal, and intestines. Murine models of tissue fibrosis in the lung, skin, liver, heart, and intestines also demonstrate an important mechanistic role for CDH11 in the development of organ fibrosis. More recently, mechanistic insight at the cellular and molecular level is emerging as to how CDH11 may drive fibrosis, which may allow for the development of novel therapeutic approaches. In this review, we will discuss CDH11 and present current evidence on its involvement in fibrosis and proposed mechanisms.
Cadherins
Cadherins are a superfamily of transmembrane cell-surface proteins that mediate calcium-dependent, homophilic, cell-to-cell adhesion. Mammalian cadherins are typically subdivided into 2 classes, type I and type II, and are termed classical cadherins. The presence of a conserved HAV sequence in the EC1 domain differentiates type I from type II cadherins. However, as shown in Figure 1, all classical cadherins contain calcium-binding extracellular cadherin repeats, a single pass transmembrane domain, and a highly conserved cytoplasmic tail that links the cadherin to the actin cytoskeleton through β-catenin and α-catenin [Goodwin and Yap, 2004].
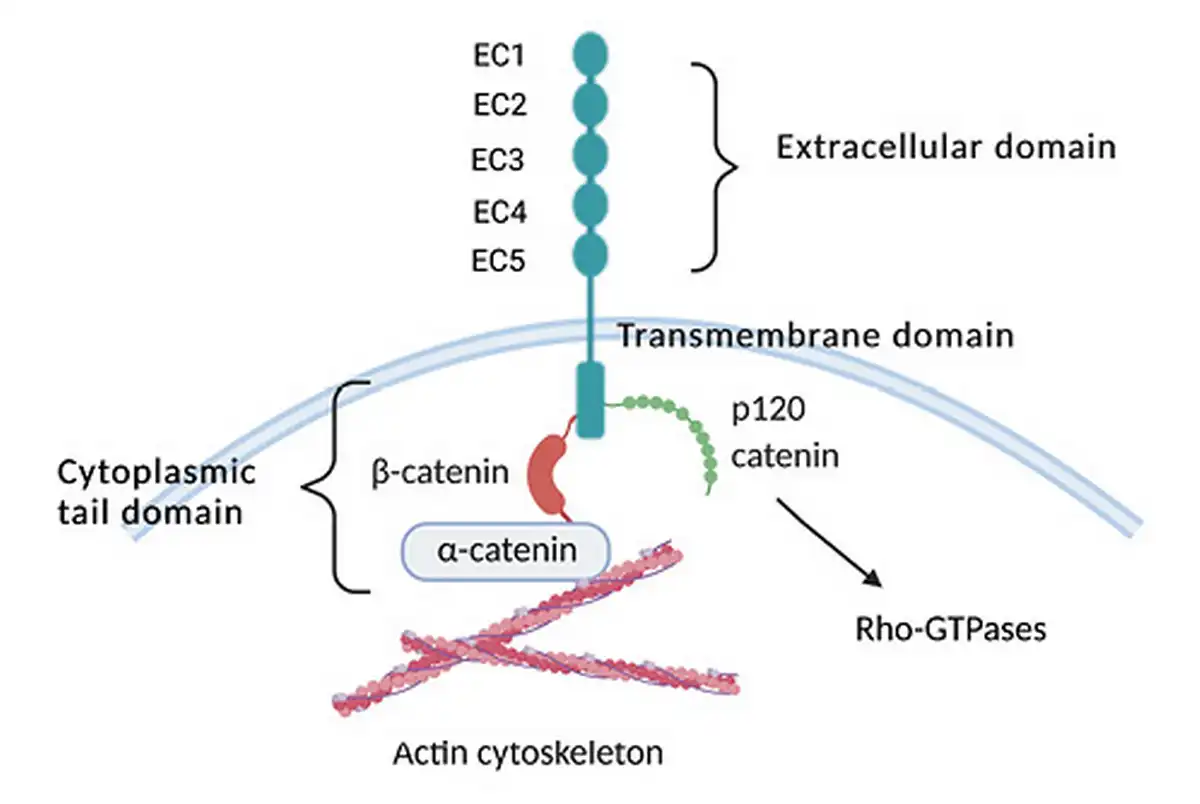
Fig. 1
Schematic diagram of cadherin-11 structure. Cadherins contain 5 calcium-binding extra cellular (EC) repeats, a transmembrane domain, and cytoplasmic tail. Classical cadherins interact with catenins, namely p120 catenin, α-catenin, and β-catenin that regulate the small Rho-GTPases and the actin cytoskeleton.
During embryogenesis and tissue morphogenesis, cadherins are critical regulators of cellular differentiation, adhesion, segregation, and migration. Postnatally, cadherins continue to function in the maintenance of tissue architecture. Many of these functions are mediated through the hοmophilic adhesive properties of cadherins via the interactions with catenins and the cytoskeleton. Cadherins also regulate the Rho-family GTPases through p120 catenin [Cavallaro and Dejana, 2011]. But cadherins likely function in part through adhesion independent pathways, particularly through the WNT, PI-3 kinase/Akt, and FGF pathways [Goodwin and Yap, 2004].
Cadherin-11
CDH11, also called OB-Cadherin, is a type II cadherin first discovered in mouse osteoblasts [Okazaki et al., 1994]. Expression was first thought to be restricted to mesenchymal tissue but has been shown in other tissues including the heart, brain, placenta, and lung [Kawaguchi et al., 1999]. At the cellular level, CDH11 expression has been reported on alveolar epithelial cells, macrophages, hepatocytes, HSCs, and keratinocytes [Schneider et al., 2012; Kim et al., 2014; Wu et al., 2014; Lodyga et al., 2019; Pedroza et al., 2019; Schroer et al., 2019].
CDH11 regulates numerous cellular processes including cell-cell adhesion and migration, which are essential for tissue organization during embryogenesis especially in the process of epithelial-mesenchymal transition (EMT) [Goodwin and Yap, 2004; Cavallaro and Dejana, 2011; Yang et al., 2020]. During early development, CDH11 regulates stem cell differentiation and lineage specification including osteogenic and adipogenic lineages [Alimperti and Andreadis, 2015]. By upregulating TGFβ-1 expression, CDH11 also regulates mesenchymal stem cell differentiation into smooth muscle cells [Alimperti et al., 2014]. The role of CDH11 in regulating bone growth and development has been shown postnatally. In support of this, Cdh11 null mice have reduced bone density and mass in the skull and long bones [Kawaguchi et al., 2001; Di Benedetto et al., 2010].
Of significant interest is the emerging evidence for CDH11 in cardiac development. CDH11 expression is restricted to mesenchymal cells of the early embryonic outflow tract and atrio-ventricular cushions during embryogenesis. In the adult heart, CDH11 expression is restricted to cardiac valves but can revert to an embryonic pattern of expression in diseased aortic valves [Zhou et al., 2013]. To that end, deletion of CDH11 disrupts embryonic valve formation, as it is essential for migration and function of mesenchymal cells that form the aortic valves [Bowen et al., 2015].
CDH11 has been shown to play an important role in several pathological processes, in particular in cancer. EMT is a critical process in cancer progression during which carcinoma cells suppress their epithelial features and transition into a mesenchymal phenotype, which includes increased tumor invasiveness and metastatic potential [Thiery, 2002]. Cadherin switch is seen during EMT, with downregulation of epithelial cadherin (E-cadherin, CDH1) and upregulation of CDH11 and neural cadherin (N-cadherin, CDH2) [Christofori and Semb, 1999; Thiery, 2002]. Increased expression of CDH11 and regulation of EMT has been shown in numerous malignancies including invasive breast cancer, metastatic pancreatic cancer, and prostate cancer. The upregulation of CDH11 in metastatic cells in these cancers supports its involvement in tumor cell migration and invasion [Pishvaian et al., 1999; Chu et al., 2008; Huang et al., 2010; Li et al., 2011; Pohlodek et al., 2016; Birtolo et al., 2017]. While CDH11 is linked to increased tumor growth in most cancers, it might also act as a tumor suppressor in other cancers such as retinoblastoma [Marchonget al., 2010].
Finally, CDH11 also has an important role in the development of the joint synovium and can regulate the development of inflammatory arthritis in mouse models of rheumatoid arthritis (RA). Mice deficient of CDH11 have synovial hypoplasia and disrupted architecture. When challenged in the K/BxN serum transfer arthritis model, CDH11 knockout mice have reduced inflammatory arthritis and joint damage [Lee et al., 2007]. Consistent with the potential promotive role of CDH11 in RA, CDH11 mRNA was found in blood from patients with RA, especially those with active disease, compared to healthy controls [Sfikakis et al., 2014]. Furthermore, flow cytometric analyses of circulating cells in RA patients indicated that CDH11 was expressed on both CD45 positive and negative cells.
Cadherin-11 in Tissue Fibrosis
A role for CDH11 in fibrosis has been hypothesized and is supported by multiple publications as will be subsequently discussed. The expression of CDH11 on fibroblast populations and its role in creating a mesenchymal phenotype makes it an intriguing molecule in fibrosis. However, these studies have also highlighted CDH11 expression on multiple cellular populations involved in fibrogenesis, making it a novel therapeutic target for fibrotic diseases.
Pulmonary Fibrosis
Pulmonary fibrosis is the replacement of the normal lung parenchyma with fibrotic tissue. Interstitial lung disease (ILD) is a group of lung diseases that affect the interstitium, leading to the destruction of the normal lung architecture by the deposition of excessive ECM. ILD is a severe clinical manifestation in several autoimmune diseases, including rheumatoid arthritis and systemic sclerosis (SSc). Idiopathic pulmonary fibrosis (IPF) is another form of ILD that affects up to 200,000 Americans [Lee et al., 2014] with a high morbidity and mortality [Olson et al., 2007]. Despite 2 new therapeutics, pirfenidone and nintedanib, IPF still has a poor prognosis [Collard et al., 2012] as these drugs only slow disease progression and are not curative [Gahl et al., 2002; Azuma et al., 2005; Richeldi et al., 2014]. Despite this, progress has been made in understanding some of the pathways involved in disease pathogenesis and highlighting the involvement of CDH11.
The expression of CDH11 is increased in fibrotic sections of lungs of IPF patients compared to unaffected lungs and healthy subjects [Schneider et al., 2012]. CDH11 expression was localized to fibroblasts as expected but was also noted to be expressed by hyperplastic type II alveolar epithelial cells and alveolar macrophages. Interestingly, mice genetically deficient in CDH11 developed less lung fibrosis when challenged with intratracheal bleomycin compared to wildtype mice, consistent with a role of CDH11 in the development of pulmonary fibrosis. These data were confirmed by successful treatment of mice in the bleomycin model using a systemic anti-CDH11 monoclonal antibody, demonstrating its mechanistic role in fibrosis as well as the potential of anti-CDH11 therapy [Schneider et al., 2012].
The mechanisms by which CDH11 regulates the development of lung fibrosis are under active investigation. The expression of CDH11 on fibroblasts, injured epithelial cells, and macrophages in the fibrotic lung suggests that it regulates multiple cellular populations and multiple steps during the development of fibrosis as shown in Figure 2.
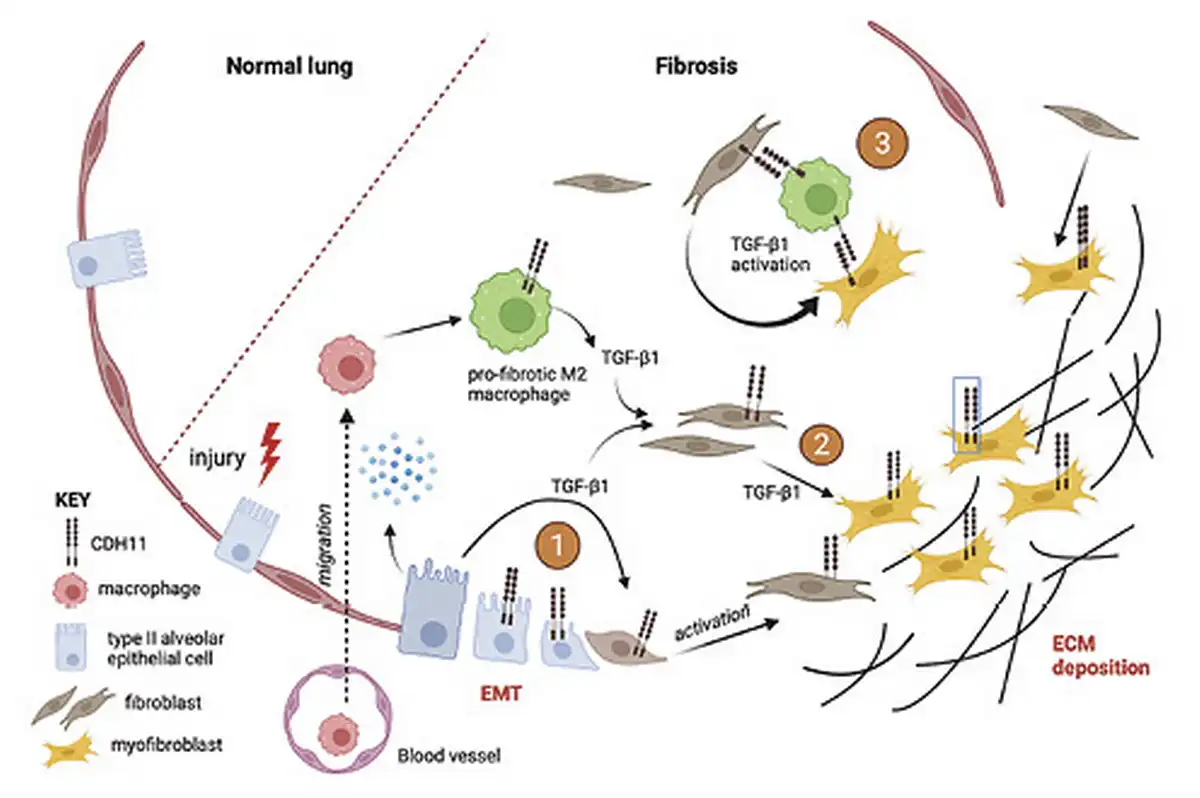
Fig. 2
Mechanism of cadherin-11 in pulmonary fibrosis. CDH11 is expressed on alveolar macrophages, injured type II alveolar epithelial cells, resident fibroblasts, and myofibroblasts. After injury, release of cytokines promotes migration and recruitment of macrophages which are key in secreting TGFβ-1 and other profibrotic cytokines. Three potential mechanisms by which CDH11 contributes to IPF are shown: (1) Regulation of EMT as epithelial cells transition into mesenchymal fibroblasts under the influence of TGFβ-1. (2) Resident fibroblast activation and differentiation into myofibroblasts that secrete ECM. (3) Physical coupling of macrophages and fibroblasts/myofibroblasts which promotes activation of latent TGFβ-1, myofibroblast activation, and creates a pro-fibrotic niche.
Activated myofibroblasts are the key cellular source of ECM components during fibrosis and originate from fibroblasts. CDH11 expression increases during fibroblast to myofibroblast activation suggesting that CDH11 may regulate this process [Lodyga et al., 2019]. Additional support of CDH11 regulation of myofibroblast activation has come from studies on FOXF1, an upstream transcriptional regulator of CDH11 expression [Black et al., 2018]. Analysis of publicly available genomic data from human IPF patients reveal downregulated expression of FOXF1 mRNA and decreased FOXF1 protein by immunofluorescence in IPF lung biopsies [Black et al., 2018]. Using transgenic mice with conditional tamoxifen-induced FOXF1 inactivation, depletion of FOXF1 in myofibroblasts promoted CDH11 expression and subsequent myofibroblast activation and collagen secretion after intratracheal bleomycin treatment. Mechanistically, FOXF1 was shown to directly bind CDH2 and CDH11 promoters and up- and downregulate their transcription, respectively. Taken together, these results indicate that FOXF1 is an anti-fibrotic transcription factor and provide supportive evidence for CDH11 in driving pulmonary fibrosis by regulating myofibroblasts.
The regulation of EMT or mesenchymal reprogramming of type II alveolar epithelial cells has been suggested as a possible mechanism through which CDH11 contributes to lung fibrosis. In support of this, CDH11 expression in A549 cells, a type II alveolar epithelial cell line, is increased with TGF-β1 stimulation, a known inducer of EMT [Willis et al., 2005]. Interestingly, silencing of CDH11 expression using siRNA reduced TGF-β1 induced expression of mesenchymal genes and prevented development of the mesenchymal phenotype in A549 cells [Schneider et al., 2012].
Finally, it is likely that CDH11 regulates macrophage behavior to contribute to the development of lung fibrosis. Lodyga et al. [2019] confirmed the expression of CDH11 on macrophages in the lung and demonstrated that CDH11 was enriched at points of contact between macrophages and myofibroblasts in fibrotic lung tissue from mice and human IPF patients. Their work describes a novel mechanism where CDH11 promotes the direct adhesion of macrophages to myofibroblasts to drive activation of latent TGF-β1 and the activation of the myofibroblast, creating a pro-fibrotic niche [Lodyga et al., 2019].
Together, these murine and human studies support an important role for CDH11 in the development of lung fibrosis. The expression of CDH11 on multiple cellular populations, known to be important in the development of lung fibrosis, makes it an intriguing therapeutic target that might be effective in treatment as well as prevention of lung fibrosis in patients with IPF and ILD from autoimmune diseases.
Dermal Fibrosis
Systemic sclerosis (SSc) is a multi-system autoimmune disease affecting connective tissue and the vascular system that is characterized by excessive collagen deposition and fibrosis of the skin and internal organs, including the lung. The pathogenesis of SSc is complex with 3 distinct processes identified: autoimmunity, vasculopathy of small vessels, and excessive ECM deposition in skin, blood vessels, and internal organs [Pattanaik et al., 2015]. Activated fibroblasts, under the influence of TGF-β and other molecular stimuli, differentiate into pro-fibrotic myofibroblasts. Excessive ECM deposition by myofibroblasts causes tissue contraction, which precedes tissue atrophy and organ failure [Abraham and Varga, 2005]. TGF-β and its intracellular signaling pathway plays a crucial role in initiating and sustaining the fibrotic response, and multiple reviews have reported on abnormal TGF-β signaling in SSc [Varga, 2002; Leask and Abraham, 2004; Pannu and Trojanowska, 2004]. Other cytokines, including IL-4, IL-6, and IL-13, are increased in SSc patients and may contribute to the disease process [Hasegawa et al., 1997; Gourh et al., 2009; Nguyen et al., 2020].
Two independent microarrays revealed increased CDH11 mRNA transcripts in the skin of patients with SSc [Whitfield et al., 2003; Gardner et al., 2006]. CDH11 expression has been confirmed in SSc skin biopsies and murine fibrotic skin where it localizes to dermal fibroblasts and macrophages [Wu et al., 2014]. Interestingly, increased CDH11 mRNA transcripts were also observed in the peripheral blood of patients with SSc and correlated with diffuse skin involvement [Christopoulos et al., 2015]. Finally, in the tight skin (Tsk-1) mouse model of hypodermal fibrosis, CDH11 expression was also increased and localized to hypodermal fibroblasts [Pedroza et al., 2017].
Murine studies have extended these findings to support a mechanistic role of CDH11 in fibrosis in the skin. Compared to wildtype mice, mice deficient in CDH11 have decreased fibrosis and expression of profibrotic mediators in the subcutaneous bleomycin-induced dermal fibrosis model [Wu et al., 2014]. These findings were confirmed using anti-CDH11 monoclonal antibodies, which not only demonstrated prevention of the development of dermal fibrosis but also the accelerated resolution of dermal fibrosis in the subcutaneous bleomycin-induced dermal fibrosis model [Wu et al., 2014]. These findings were also confirmed in the Tsk-1 mouse model of fibrosis, where anti-CDH11 monoclonal antibodies reduced hypodermal fibrosis and expression of fibrotic mediators in Tsk-1 mice compared to isotype control [Pedroza et al., 2017]. Together, these studies demonstrate an important role for CDH11 in the development of skin fibrosis, and combined with the human expression data in SSc, support the potential of CDH11 as a therapeutic target in SSc and fibrotic skin diseases.
Liver Fibrosis
Liver fibrosis is a common outcome associated with chronic liver disease and injury. Although potentially reversible if the insulting factor is removed, liver fibrosis has high morbidity and mortality, as no effective treatments to reverse the process in humans have been identified [Unalp-Arida and Ruhl, 2017; Caballeria et al., 2018]. Efforts to understand the pathogenesis of liver fibrosis and identify therapeutic targets to slow its progression have yielded some promising results regarding treatment options, especially in mouse models.
Activation of HSCs towards proliferative, fibrogenic myofibroblasts remains the main theme in liver fibrosis pathogenesis. HSCs are quiescent perivascular mesenchymal cells that normally store retinoids and are activated after liver injury. Once activated, HSCs undergo key phenotypic changes, including proliferation, contractility, and TGF-β mediated fibrogenesis [Friedman, 2000].
Analysis of murine and human fibrotic liver revealed expression of CDH11 on multiple cell lineages. Using the carbon tetrachloride (CCl4)-induced liver fibrosis model, Pedroza et al. [2019] showed increased CDH11 expression in fibrotic livers of CCl4-treated wildtype mice localized to hepatocytes, HSCs, and macrophages. To demonstrate the involvement of CDH11 in liver fibrosis, Cdh11−/− mice challenged with CCl4 had markedly decreased liver fibrosis and expression of profibrotic mediators compared to wildtype mice [Pedroza et al., 2019]. In vitro cell culture studies further confirmed the expression of CDH11 in human HSCs and murine hepatocyte cell lines, suggesting that CDH11 may regulate liver fibrosis through modulation of HSC behavior and hepatocyte injury [Pedroza et al., 2019].
The importance of CDH11 in liver fibrosis was also demonstrated in a study by Ruan et al. [2019]. Analysis of gene expression datasets revealed increased CDH11 in both plasma and liver samples from patients with liver fibrosis. Upregulation of CDH11 was also seen in fibrotic livers from CCl4-treated mice. In vitro studies elucidated that CDH11 promotes HSC activation and proliferation, providing further insight into the contribution of CDH11 in liver fibrosis. Finally, inhibition of CDH11 expression using adeno-associated virus (AAV) vectors targeting CDH11 resulted in attenuated fibrosis in mice after CCl4 treatment [Ruan et al., 2019]. These studies demonstrate an important role for CDH11 in the development of liver fibrosis and suggest the regulation of the HSC as one potential mechanism by which CDH11 contributes to liver fibrosis as shown in Figure 3.
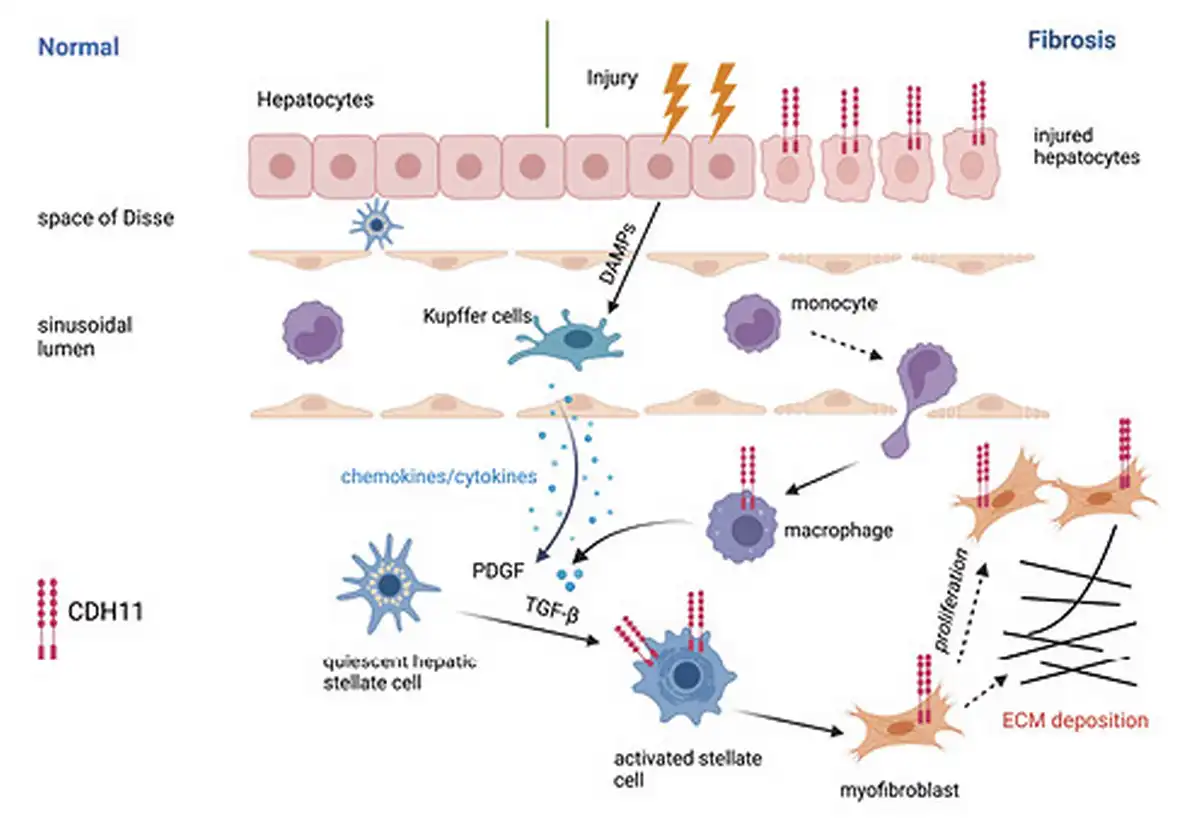
Fig. 3
Cadherin-11 regulation of HSC activation in liver fibrosis. During liver injury, release of damage associated molecular patterns (DAMPs) promotes activation of resident macrophages (Kupffer cells). Release of cytokines and chemokines drives recruitment of inflammatory monocytes that differentiate into macrophages. Macrophage-derived growth factors, including TGF-β and platelet-derived growth factor (PDGF), stimulate activation of HSCs into pro-fibrotic myofibroblasts. In human and murine fibrotic liver, CDH11 expression is shown on injured hepatocytes, HSCs, myofibroblasts and macrophages. In HSCs, TGF-β promotes CDH11 expression, which regulates the activation of HSCs and differentiation into myofibroblasts that secrete ECM.
Cardiac Fibrosis
Cardiac disease is a leading cause of mortality with nearly 800,000 annual deaths in the USA attributed to cardiovascular disease. Excessive deposition of ECM and remodeling of the cardiac basement membrane by cardiac fibroblasts (CFs) is the key mechanism in cardiac fibrosis. The process of EMT is considered one of the main sources of CFs in the neonatal and adult heart, with these fibroblasts forming the cardiac valves [Potts and Runyan, 1989; Visconti et al., 2006]. The origins of CFs have been comprehensively detailed in other reviews [Bellini and Mattoli, 2007; Krenning et al., 2010].
In response to injury, CFs transform into myofibroblasts which are normally absent in the normal heart. Myofibroblasts are necessary for secretion of ECM proteins and scar tissue formation during the wound healing process, especially since the heart has limited regenerative capacity after injury [Senyo et al., 2013]. However, excessive secretion of ECM proteins and subsequent cardiac tissue remodeling leads to compromised cardiac function, which underlies the pathology in cardiac fibrosis. At the molecular level, TGF-β and the WNT/β-catenin signaling pathways, among others, play a major role in cardiac fibrogenesis [Abraityte et al., 2017; Blyszczuk et al., 2017; Dzialo et al., 2019; Yousefi et al., 2020].
In the human heart, CDH11 expression is observed in fibroblasts [Hutcheson et al., 2013], myofibroblasts [Hinz et al., 2004], and vascular smooth muscle cells [Monahan et al., 2007]. Murine studies confirm the expression of CDH11 by CFs, epithelial, and mesenchymal cells. Interestingly, CDH11 expression has also been shown in myeloid cells including neutrophils and macrophages during acute cardiac injury [Schroer et al., 2019].
The potential mechanisms by which CDH11 drives cardiac pathology are highlighted by multiple studies. Overexpression of CDH11 in adult mice leads to calcific lesion formation on aortic valves with resultant valvular and ventricular degeneration [Sung et al., 2016]. CDH11 overexpression also upregulates GTP-RhoA and Sox9 to mediate migration, stress fiber formation, and compaction in vitro. In porcine aortic valve interstitial cells (AVICs), TGF-β-induced CDH11 expression regulates intercellular tension necessary for calcific nodule formation. Knockdown of CDH11 and α-SMA inhibits formation of nodules, signifying the importance of both proteins in calcific nodule formation [Hutcheson et al., 2013]. In murine AVICs, CDH11 promotes contractility by upregulating IL-6 secretion, thus potentiating the myofibroblast phenotype [Bowler et al., 2018].
Studies in other cardiac disease models present a multi-cellular role of CDH11. CDH11 has been shown to regulate adipose tissue formation [Chang et al., 2017], and obesity is a known risk factor for atrial fibrillation (AF). Work by Fang et al. [2021] revealed increased CDH11 expression in AF patients with obesity and in high-fat diet mice. CDH11 was shown to regulate atrial fibroblast activation and cytokine-mediated ECM secretion. Furthermore, CDH11 deficiency in vivo protected against high fat diet-induced atrial disease and is seen as a potential therapeutic target in AF. A recent study supports the involvement of CDH11 in atrial fibrosis associated with AF. Increased CDH11 expression was found in fibrotic atrial tissue in patients with AF. Mice deficient in CDH11 had reduced atrial fibrosis compared to wildtype after angiotensin II (Ang-II) infusion in the Ang-II induced atrial fibrosis model. RNA-sequencing (RNA-seq) analysis and validation by qRT-PCR revealed decreased expression of pro-fibrotic genes in atrial fibroblasts isolated from CDH11-deficient mice after Ang-II treatment compared to wildtype. Mechanistically, CDH11 deletion disrupted Ang-II mediated atrial fibroblast activation, proliferation, and migration. These results suggest that CDH11 may contribute to atrial fibrosis by regulation atrial fibroblasts [Cao et al., 2021].
Finally, during acute injury like myocardial infarction (MI), CDH11 deficiency through genetic deletion and chemical inhibition protected against MI-induced inflammation and fibrotic remodeling and improved clinical disease in mice [Schroer et al., 2019]. Taken together, these studies provide compelling evidence for CDH11 in cardiac fibrotic disease and strengthen its potential as a common therapeutic target in heart failure resulting from multiple etiologies and involving multiple pathways.
Kidney Fibrosis
Chronic kidney disease (CKD) is a major public health concern and affects upto 15% of the US adult population according to the CDC. Kidney fibrosis is a common pathological finding in CKD and nearly all chronic nephropathies. Fibrosis has long been suggested as a useful tool in assessing renal function as it correlates well with renal function [Schainuck et al., 1970]. Although fibrosis can provide useful insights into renal function, it has also been shown to be limited in prognosing end stage renal disease in a subset of patients [Menn-Josephy et al., 2016]. Kidney fibrosis, like in other tissues, is characterized by myofibroblast proliferation and secretion of excessive ECM under conditions of chronic inflammation.
Assessment and diagnosis of renal fibrosis in CKD is a challenge as renal biopsy is the current gold standard. However, the invasive nature and potential complication of renal biopsy makes repeat biopsies for disease follow-up impractical [Jiang et al., 2019]. Additionally, commonly used histological assessment techniques such as Mason’s trichome are insensitive in mild fibrosis. Imaging techniques such as magnetic resonance imaging and ultrasound elastography show promise as noninvasive tools for assessing renal fibrosis but their limited specificity and practicality makes them less ideal for clinical use [Jiang et al., 2019]. Traditional markers of CKD such as serum creatinine and urine protein are also not ideal as they are not direct measures of fibrosis [Jiang et al., 2019].
Given the difficulty of obtaining human tissue samples, few studies have comprehensively defined the molecular and cellular factors that drive renal fibrosis in human patients. The use of animal models overcomes this challenge, and potential targets for reversing renal fibrosis in vivo have been identified [Boffa et al., 2001; Adamczak et al., 2003; El Chaar et al., 2007; Wang et al., 2010]. However, more work still needs to be done to translate these studies to human patients.
Few studies have highlighted CDH11 in renal fibrosis. Using the folic acid-induced fibrosis mouse model and RNA-seq analysis, Craciun et al. [2016] identified a panel of genes upregulated in renal fibrosis. Further analysis in a cohort of kidney fibrosis patients and in vitro molecular assays confirmed increased protein expression and highlighted CDH11 among the most significantly upregulated. Treatment with angiotensin converting enzyme (ACE) inhibitors has been shown to potentially regress renal fibrosis [Adamczak et al., 2003]. To that end, ACE antagonism resulted in improved clinical disease in vivo with subsequent decrease in CDH11 expression in the study by Craciun et al. [2016].
More recently, CDH11 has been identified as a promising non-invasive biomarker for renal fibrosis [Schmidt et al., 2021]. Analysis of murine fibrosis single cell RNA-seq and human CKD single nucleus RNA-seq datasets revealed increased expression of CDH11 in fibroblast populations. Relative to healthy human kidney, CDH11 was strongly expressed in CKD kidney. Clinical analysis of urine and plasma from CKD patients in 2 large cohorts also revealed increased protein expression of CDH11 that strongly was associated with severity of interstitial fibrosis and progression to end stage kidney disease [Schmidt et al., 2021]. Further studies of CDH11 in renal fibrosis are needed to determine if CDH11 is indeed a useful biomarker for fibrosis in patients with CKD and if CDH11 inhibition is a potential therapeutic strategy in kidney fibrosis.
Intestinal Fibrosis
Intestinal fibrosis is a common complication of inflammatory bowel disease (IBD). The involvement of CDH11 in intestinal fibrosis has been highlighted in patients with IBD and in murine colitis models. Genome wide microarrays revealed upregulation of CDH11 in patients with IBD [Costello et al., 2005; Van der Goten et al., 2014]. Increased levels of CDH11 have been confirmed in colonic biopsies at the mRNA and protein level in patients with ulcerative colitis and Crohn’s disease (CD) [Franze et al., 2020]. Interestingly, the levels of CDH11 were highest in patients with fibrostricturing CD, and immunohistological studies confirmed that the expression of CDH11 in IBD colonic biopsies was mostly localized to intestinal fibroblasts. Murine studies using the 2,4,6-trinitrobenzene sulfonic acid-driven colitis model demonstrated that CDH11 deficient mice developed less intestinal fibrosis with comparable levels of inflammation compared to wildtype mice [Franze et al., 2020]. Lastly, inhibition of CDH11 expression in intestinal fibroblasts resulted in decreased type 1 and 3 collagen production, TGF-β signaling, and activation of RhoA/ROCK pathways. Together, these studies implicate CDH11 in the development of intestinal fibrosis and emphasize its importance in regulating fibrosis in multiple tissues.
Conclusion
Fibrosis is an important feature of many diseases and results in high morbidity and mortality. Significant progress has been made in identifying molecular and cellular factors that contribute to pathogenesis of tissue fibrosis. Animal models and analysis of human patient samples have been key in identifying CDH11 as a common mediator of fibrosis in different tissues. The challenge remains in translating the plethora of in vitro and in vivo data that support CDH11 inhibition as an effective treatment to human research. The multi-cellular expression of CDH11 in mesenchymal and myeloid populations and the regulation of multiple pathways present a unique challenge in designing a novel therapeutic. Crucial to future studies would be to determine how CDH11 blockade interplays with ongoing inflammation.
Conflict of Interest Statement
Sandeep K. Agarwal has a patent related to targeting cadherin-11 in non-dermal fibrosis. No royalty payments have been received in more than 5 years.
Funding Source
The authors did not receive any funding support for this manuscript.
Author Contributions
Thandiwe Chavula, Sarah To, and Sandeep K. Agarwal: concept and design of manuscript content, drafting the article, critical revision of the article.
Thandiwe Chavula: original design of the figures. All figures were created with BioRender.com.
All authors were involved in the final approval of the version to be published.
References
- 1. Abraham DJ, Varga J. Scleroderma: from cell and molecular mechanisms to disease models. Trends Immunol. 2005;26(11):587–95. http://dx.doi.org/10.1016/j.it.2005.09.004.
- 2. Abraityte A, Vinge LE, Askevold ET, Lekva T, Michelsen AE, Ranheim T, et al. Wnt5a is elevated in heart failure and affects cardiac fibroblast function. J Mol Med (Berl). 2017 Jul;95(7):767–77. http://dx.doi.org/10.1007/s00109-017-1529-1.
- 3. Adamczak M, Gross ML, Krtil J, Koch A, Tyralla K, Amann K, et al. Reversal of glomerulosclerosis after high-dose enalapril treatment in subtotally nephrectomized rats. J Am Soc Nephrol. 2003 Nov;14(11):2833–42. http://dx.doi.org/10.1097/01.asn.0000095248.91994.d3.
- 4. Alimperti S, Andreadis ST. CDH2 and CDH11 act as regulators of stem cell fate decisions. Stem Cell Res. 2015 May;14(3):270–82. http://dx.doi.org/10.1016/j.scr.2015.02.002.
- 5. Alimperti S, You H, George T, Agarwal SK, Andreadis ST. Cadherin-11 regulates both mesenchymal stem cell differentiation into smooth muscle cells and the development of contractile function in vivo. J Cell Sci. 2014 Jun;127(Pt 12):2627–38. http://dx.doi.org/10.1242/jcs.134833.
- 6. Azuma A, Nukiwa T, Tsuboi E, Suga M, Abe S, Nakata K, et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005 May;171(9):1040–7. http://dx.doi.org/10.1164/rccm.200404-571OC.
- 7. Bellini A, Mattoli S. The role of the fibrocyte, a bone marrow-derived mesenchymal progenitor, in reactive and reparative fibroses. Lab Invest. 2007 Sep;87(9):858–70. http://dx.doi.org/10.1038/labinvest.3700654.
- 8. Biernacka A, Dobaczewski M, Frangogiannis NG. TGF-β signaling in fibrosis. Growth Factors. 2011 Oct;29(5):196–202. http://dx.doi.org/10.3109/08977194.2011.595714.
- 9. Birtolo C, Pham H, Morvaridi S, Chheda C, Go VL, Ptasznik A, et al. Cadherin-11 Is a Cell Surface Marker Up-Regulated in Activated Pancreatic Stellate Cells and Is Involved in Pancreatic Cancer Cell Migration. Am J Pathol. 2017 Jan;187(1):146–55. http://dx.doi.org/10.1016/j.ajpath.2016.09.012.
- 10. Black M, Milewski D, Le T, Ren X, Xu Y, Kalinichenko VV, et al. FOXF1 Inhibits Pulmonary Fibrosis by Preventing CDH2-CDH11 Cadherin Switch in Myofibroblasts. Cell Rep. 2018 Apr;23(2):442–58. http://dx.doi.org/10.1016/j.celrep.2018.03.067.
- 11. Blyszczuk P, Müller-Edenborn B, Valenta T, Osto E, Stellato M, Behnke S, et al. Transforming growth factor-β-dependent Wnt secretion controls myofibroblast formation and myocardial fibrosis progression in experimental autoimmune myocarditis. Eur Heart J. 2017 May;38(18):1413–25. http://dx.doi.org/10.1093/eurheartj/ehw116.
- 12. Boffa JJ, Tharaux PL, Dussaule JC, Chatziantoniou C. Regression of renal vascular fibrosis by endothelin receptor antagonism. Hypertension. 2001 Feb;37(2 Pt 2):490–6. http://dx.doi.org/10.1161/01.hyp.37.2.490.
- 13. Bowen CJ, Zhou J, Sung DC, Butcher JT. Cadherin-11 coordinates cellular migration and extracellular matrix remodeling during aortic valve maturation. Dev Biol. 2015 Nov;407(1):145–57. http://dx.doi.org/10.1016/j.ydbio.2015.07.012.
- 14. Bowler MA, Bersi MR, Ryzhova LM, Jerrell RJ, Parekh A, Merryman WD. Cadherin-11 as a regulator of valve myofibroblast mechanobiology. Am J Physiol Heart Circ Physiol. 2018 Dec;315(6):H1614–H26. http://dx.doi.org/10.1152/ajpheart.00277.2018.
- 15. Caballería L, Pera G, Arteaga I, Rodríguez L, Alumà A, Morillas RM, et al. High Prevalence of Liver Fibrosis Among European Adults With Unknown Liver Disease: A Population-Based Study. Clin Gastroenterol Hepatol. 2018 Jul;16(7):1138–e5. http://dx.doi.org/10.1016/j.cgh.2017.12.048.
- 16. Cao W, Song S, Fang G, Li Y, Wang Y, Wang QS. Cadherin-11 Deficiency Attenuates Ang-II-Induced Atrial Fibrosis and Susceptibility to Atrial Fibrillation. J Inflamm Res. 2021;14:2897–911. http://dx.doi.org/10.2147/JIR.S306073.
- 17. Cavallaro U, Dejana E. Adhesion molecule signalling: not always a sticky business. Nat Rev Mol Cell Biol. 2011 Mar;12(3):189–97. http://dx.doi.org/10.1038/nrm3068.
- 18. Chang SK, Kohlgruber AC, Mizoguchi F, Michelet X, Wolf BJ, Wei K, et al. Stromal cell cadherin-11 regulates adipose tissue inflammation and diabetes. J Clin Invest. 2017 Sep;127(9):3300–12. http://dx.doi.org/10.1172/JCI86881.
- 19. Christofori G, Semb H. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends Biochem Sci. 1999 Feb;24(2):73–6. http://dx.doi.org/10.1016/s0968-0004(98)01343-7.
- 20. Christopoulos PF, Bournia VK, Panopoulos S, Vaiopoulos A, Koutsilieris M, Sfikakis PP. Increased messenger RNA levels of the mesenchymal cadherin-11 in the peripheral blood of systemic sclerosis patients correlate with diffuse skin involvement. Clin Exp Rheumatol. 2015 Jul-Aug;33(4 Suppl 91):S36–9.
- 21. Chu K, Cheng CJ, Ye X, Lee YC, Zurita AJ, Chen DT, et al. Cadherin-11 promotes the metastasis of prostate cancer cells to bone. Mol Cancer Res. 2008 Aug;6(8):1259–67. http://dx.doi.org/10.1158/1541-7786.MCR-08-0077.
- 22. Collard HR, Ward AJ, Lanes S, Cortney Hayflinger D, Rosenberg DM, Hunsche E. Burden of illness in idiopathic pulmonary fibrosis. J Med Econ. 2012;15(5):829–35. http://dx.doi.org/10.3111/13696998.2012.680553.
- 23. Costello CM, Mah N, Häsler R, Rosenstiel P, Waetzig GH, Hahn A, et al. Dissection of the inflammatory bowel disease transcriptome using genome-wide cDNA microarrays. PLoS Med. 2005 Aug;2(8):e199. http://dx.doi.org/10.1371/journal.pmed.0020199.
- 24. Craciun FL, Bijol V, Ajay AK, Rao P, Kumar RK, Hutchinson J, et al. RNA Sequencing Identifies Novel Translational Biomarkers of Kidney Fibrosis. J Am Soc Nephrol. 2016 Jun;27(6):1702–13. http://dx.doi.org/10.1681/ASN.2015020225.
- 25. Di Benedetto A, Watkins M, Grimston S, Salazar V, Donsante C, Mbalaviele G, et al. N-cadherin and cadherin 11 modulate postnatal bone growth and osteoblast differentiation by distinct mechanisms. J Cell Sci. 2010 Aug;123(Pt 15):2640–8. http://dx.doi.org/10.1242/jcs.067777.
- 26. Działo E, Rudnik M, Koning RI, Czepiel M, Tkacz K, Baj-Krzyworzeka M, et al. WNT3a and WNT5a Transported by Exosomes Activate WNT Signaling Pathways in Human Cardiac Fibroblasts. Int J Mol Sci. 2019 Mar;20(6):1436. http://dx.doi.org/10.3390/ijms20061436.
- 27. El Chaar M, Chen J, Seshan SV, Jha S, Richardson I, Ledbetter SR, et al. Effect of combination therapy with enalapril and the TGF-beta antagonist 1D11 in unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2007 Apr;292(4):F1291–301. http://dx.doi.org/10.1152/ajprenal.00327.2005.
- 28. Fang G, Cao W, Chen L, Song S, Li Y, Yuan J, et al. Cadherin-11 deficiency mitigates high-fat diet-induced inflammatory atrial remodeling and vulnerability to atrial fibrillation. J Cell Physiol. 2021 Aug;236(8):5725–41. http://dx.doi.org/10.1002/jcp.30257.
- 29. Franzè E, Monteleone I, Laudisi F, Rizzo A, Dinallo V, Di Fusco D, et al. Cadherin-11 Is a Regulator of Intestinal Fibrosis. J Crohns Colitis. 2020 Mar;14(3):406–17. http://dx.doi.org/10.1093/ecco-jcc/jjz147.
- 30. Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000 Jan;275(4):2247–50. http://dx.doi.org/10.1074/jbc.275.4.2247.
- 31. Gahl WA, Brantly M, Troendle J, Avila NA, Padua A, Montalvo C, et al. Effect of pirfenidone on the pulmonary fibrosis of Hermansky-Pudlak syndrome. Mol Genet Metab. 2002 Jul;76(3):234–42. http://dx.doi.org/10.1016/s1096-7192(02)00044-6.
- 32. Gardner H, Shearstone JR, Bandaru R, Crowell T, Lynes M, Trojanowska M, et al. Gene profiling of scleroderma skin reveals robust signatures of disease that are imperfectly reflected in the transcript profiles of explanted fibroblasts. Arthritis Rheum. 2006;54(6):1961–73. http://dx.doi.org/10.1002/art.21894.
- 33. Goodwin M, Yap AS. Classical cadherin adhesion molecules: coordinating cell adhesion, signaling and the cytoskeleton. J Mol Histol. 2004;35(8‐9):839–44. http://dx.doi.org/10.1007/s10735-004-1833-2.
- 34. Gourh P, Arnett FC, Assassi S, Tan FK, Huang M, Diekman L, et al. Plasma cytokine profiles in systemic sclerosis: associations with autoantibody subsets and clinical manifestations. Arthritis Res Ther. 2009;11(5):R147. http://dx.doi.org/10.1186/ar2821.
- 35. Hantash BM, Zhao L, Knowles JA, Lorenz HP. Adult and fetal wound healing. Front Biosci. 2008 Jan;13:51–61. http://dx.doi.org/10.2741/2559.
- 36. Hasegawa M, Fujimoto M, Kikuchi K, Takehara K. Elevated serum levels of interleukin 4 (IL-4), IL-10, and IL-13 in patients with systemic sclerosis. J Rheumatol. 1997;24(2):328–32.
- 37. Hinz B, Pittet P, Smith-Clerc J, Chaponnier C, Meister JJ. Myofibroblast development is characterized by specific cell-cell adherens junctions. Mol Biol Cell. 2004;15(9):4310–20. http://dx.doi.org/10.1091/mbc.e04-05-0386.
- 38. Huang CF, Lira C, Chu K, Bilen MA, Lee YC, Ye X, et al. Cadherin-11 increases migration and invasion of prostate cancer cells and enhances their interaction with osteoblasts. Cancer Res. 2010 Jun;70(11):4580–9. http://dx.doi.org/10.1158/0008-5472.CAN-09-3016.
- 39. Hutcheson JD, Chen J, Sewell-Loftin MK, Ryzhova LM, Fisher CI, Su YR, et al. Cadherin-11 regulates cell-cell tension necessary for calcific nodule formation by valvular myofibroblasts. Arterioscler Thromb Vasc Biol. 2013 Jan;33(1):114–20. http://dx.doi.org/10.1161/ATVBAHA.112.300278.
- 40. Jiang K, Ferguson CM, Lerman LO. Noninvasive assessment of renal fibrosis by magnetic resonance imaging and ultrasound techniques. Transl Res. 2019 Jul;209:105–20. http://dx.doi.org/10.1016/j.trsl.2019.02.009.
- 41. Kawaguchi J, Takeshita S, Kashima T, Imai T, Machinami R, Kudo A. Expression and function of the splice variant of the human cadherin-11 gene in subordination to intact cadherin-11. J Bone Miner Res. 1999;14(5):764–75. http://dx.doi.org/10.1359/jbmr.1999.14.5.764.
- 42. Kawaguchi J, Azuma Y, Hoshi K, Kii I, Takeshita S, Ohta T, et al. Targeted disruption of cadherin-11 leads to a reduction in bone density in calvaria and long bone metaphyses. J Bone Miner Res. 2001;16(7):1265–71. http://dx.doi.org/10.1359/jbmr.2001.16.7.1265.
- 43. Kim NH, Choi SH, Lee TR, Lee CH, Lee AY. Cadherin 11, a miR-675 target, induces N-cadherin expression and epithelial-mesenchymal transition in melasma. J Invest Dermatol. 2014 Dec;134(12):2967–76. http://dx.doi.org/10.1038/jid.2014.257.
- 44. Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. 2010 Nov;225(3):631–7. http://dx.doi.org/10.1002/jcp.22322.
- 45. Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004 May;18(7):816–27. http://dx.doi.org/10.1096/fj.03-1273rev.
- 46. Lee DM, Kiener HP, Agarwal SK, Noss EH, Watts GF, Chisaka O, et al. Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007;315(5814):1006–10. http://dx.doi.org/10.1126/science.1137306.
- 47. Lee AS, Mira-Avendano I, Ryu JH, Daniels CE. The burden of idiopathic pulmonary fibrosis: an unmet public health need. Respir Med. 2014 Jul;108(7):955–67. http://dx.doi.org/10.1016/j.rmed.2014.03.015.
- 48. Li Y, Guo Z, Chen H, Dong Z, Pan ZK, Ding H, et al. HOXC8-Dependent Cadherin 11 Expression Facilitates Breast Cancer Cell Migration through Trio and Rac. Genes Cancer. 2011 Sep;2(9):880–8. http://dx.doi.org/10.1177/1947601911433129.
- 49. Lodyga M, Cambridge E, Karvonen HM, Pakshir P, Wu B, Boo S, et al. Cadherin-11-mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-β. Sci Signal. 2019 Jan;12(564):eaao3469. http://dx.doi.org/10.1126/scisignal.aao3469.
- 50. Marchong MN, Yurkowski C, Ma C, Spencer C, Pajovic S, Gallie BL. Cdh11 acts as a tumor suppressor in a murine retinoblastoma model by facilitating tumor cell death. PLoS Genet. 2010 Apr;6(4):e1000923. http://dx.doi.org/10.1371/journal.pgen.1000923.
- 51. Menn-Josephy H, Lee CS, Nolin A, Christov M, Rybin DV, Weinberg JM, et al. Renal Interstitial Fibrosis: An Imperfect Predictor of Kidney Disease Progression in Some Patient Cohorts. Am J Nephrol. 2016;44(4):289–99. http://dx.doi.org/10.1159/000449511.
- 52. Monahan TS, Andersen ND, Panossian H, Kalish JA, Daniel S, Shrikhande GV, et al. A novel function for cadherin 11/osteoblast-cadherin in vascular smooth muscle cells: modulation of cell migration and proliferation. J Vasc Surg. 2007 Mar;45(3):581–9. http://dx.doi.org/10.1016/j.jvs.2006.12.016.
- 53. Nguyen JK, Austin E, Huang A, Mamalis A, Jagdeo J. The IL-4/IL-13 axis in skin fibrosis and scarring: mechanistic concepts and therapeutic targets. Arch Dermatol Res. 2020 Mar;312(2):81–92. http://dx.doi.org/10.1007/s00403-019-01972-3.
- 54. Okazaki M, Takeshita S, Kawai S, Kikuno R, Tsujimura A, Kudo A, et al. Molecular cloning and characterization of OB-cadherin, a new member of cadherin family expressed in osteoblasts. J Biol Chem. 1994;269(16):12092–8. http://dx.doi.org/10.1016/s0021-9258(17)32685-6.
- 55. Olson AL, Swigris JJ, Lezotte DC, Norris JM, Wilson CG, Brown KK. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. 2007;176(3):277–84. http://dx.doi.org/10.1164/rccm.200701-044OC.
- 56. Pannu J, Trojanowska M. Recent advances in fibroblast signaling and biology in scleroderma. Curr Opin Rheumatol. 2004;16(6):739–45. http://dx.doi.org/10.1097/01.bor.0000137894.63091.1a.
- 57. Pattanaik D, Brown M, Postlethwaite BC, Postlethwaite AE. Pathogenesis of Systemic Sclerosis. Front Immunol. 2015;6:272. http://dx.doi.org/10.3389/fimmu.2015.00272.
- 58. Pedroza M, Welschhans RL, Agarwal SK. Targeting of cadherin-11 decreases skin fibrosis in the tight skin-1 mouse model. PLoS One. 2017;12(11):e0187109. http://dx.doi.org/10.1371/journal.pone.0187109.
- 59. Pedroza M, To S, Smith J, Agarwal SK. Cadherin-11 contributes to liver fibrosis induced by carbon tetrachloride. PLoS One. 2019;14(7):e0218971. http://dx.doi.org/10.1371/journal.pone.0218971.
- 60. Pishvaian MJ, Feltes CM, Thompson P, Bussemakers MJ, Schalken JA, Byers SW. Cadherin-11 is expressed in invasive breast cancer cell lines. Cancer Res. 1999;59(4):947–52.
- 61. Pohlodek K, Tan YY, Singer CF, Gschwantler-Kaulich D. Cadherin-11 expression is upregulated in invasive human breast cancer. Oncol Lett. 2016 Dec;12(6):4393–8. http://dx.doi.org/10.3892/ol.2016.5236.
- 62. Potts JD, Runyan RB. Epithelial-mesenchymal cell transformation in the embryonic heart can be mediated, in part, by transforming growth factor beta. Dev Biol. 1989 Aug;134(2):392–401. http://dx.doi.org/10.1016/0012-1606(89)90111-5.
- 63. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014 May;370(22):2071–82. http://dx.doi.org/10.1056/NEJMoa1402584.
- 64. Ruan W, Pan R, Shen X, Nie Y, Wu Y. CDH11 promotes liver fibrosis via activation of hepatic stellate cells. Biochem Biophys Res Commun. 2019 Jan;508(2):543–9. http://dx.doi.org/10.1016/j.bbrc.2018.11.153.
- 65. Schainuck LI, Striker GE, Cutler RE, Benditt EP. Structural-functional correlations in renal disease. II. The correlations. Hum Pathol. 1970 Dec;1(4):631–41. http://dx.doi.org/10.1016/s0046-8177(70)80061-2.
- 66. Schmidt IM, Colona MR, Kestenbaum BR, Alexopoulos LG, Palsson R, Srivastava A, et al. Cadherin-11, Sparc-related modular calcium binding protein-2, and Pigment epithelium-derived factor are promising non-invasive biomarkers of kidney fibrosis. Kidney Int. 2021 Sep;100(3):672–83. http://dx.doi.org/10.1016/j.kint.2021.04.037.
- 67. Schneider DJ, Wu M, Le TT, Cho SH, Brenner MB, Blackburn MR, et al. Cadherin-11 contributes to pulmonary fibrosis: potential role in TGF-β production and epithelial to mesenchymal transition. FASEB J. 2012 Feb;26(2):503–12. http://dx.doi.org/10.1096/fj.11-186098.
- 68. Schroer AK, Bersi MR, Clark CR, Zhang Q, Sanders LH, Hatzopoulos AK, et al. Cadherin-11 blockade reduces inflammation-driven fibrotic remodeling and improves outcomes after myocardial infarction. JCI Insight. 2019 Sep;4(18):e131545. http://dx.doi.org/10.1172/jci.insight.131545.
- 69. Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013 Jan;493(7432):433–6. http://dx.doi.org/10.1038/nature11682.
- 70. Sfikakis PP, Christopoulos PF, Vaiopoulos AG, Fragiadaki K, Katsiari C, Kapsimali V, et al. Cadherin-11 mRNA transcripts are frequently found in rheumatoid arthritis peripheral blood and correlate with established polyarthritis. Clin Immunol. 2014 Nov;155(1):33–41. http://dx.doi.org/10.1016/j.clim.2014.08.008.
- 71. Sung DC, Bowen CJ, Vaidya KA, Zhou J, Chapurin N, Recknagel A, et al. Cadherin-11 Overexpression Induces Extracellular Matrix Remodeling and Calcification in Mature Aortic Valves. Arterioscler Thromb Vasc Biol. 2016 Aug;36(8):1627–37. http://dx.doi.org/10.1161/ATVBAHA.116.307812.
- 72. Thiery J. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54. http://dx.doi.org/10.1038/nrc822.
- 73. Unalp-Arida A, Ruhl CE. Liver fibrosis scores predict liver disease mortality in the United States population. Hepatology. 2017 Jul;66(1):84–95. http://dx.doi.org/10.1002/hep.29113.
- 74. Van der Goten J, Vanhove W, Lemaire K, Van Lommel L, Machiels K, Wollants WJ, et al. Integrated miRNA and mRNA expression profiling in inflamed colon of patients with ulcerative colitis. PLoS One. 2014;9(12):e116117. http://dx.doi.org/10.1371/journal.pone.0116117.
- 75. Varga J. Scleroderma and Smads: dysfunctional Smad family dynamics culminating in fibrosis. Arthritis Rheum. 2002;46(7):1703–13. http://dx.doi.org/10.1002/art.10413.
- 76. Visconti RP, Ebihara Y, LaRue AC, Fleming PA, McQuinn TC, Masuya M, et al. An in vivo analysis of hematopoietic stem cell potential: hematopoietic origin of cardiac valve interstitial cells. Circ Res. 2006 Mar;98(5):690–6. http://dx.doi.org/10.1161/01.RES.0000207384.81818.d4.
- 77. Wang S, Wilkes MC, Leof EB, Hirschberg R. Noncanonical TGF-beta pathways, mTORC1 and Abl, in renal interstitial fibrogenesis. Am J Physiol Renal Physiol. 2010 Jan;298(1):F142–9. http://dx.doi.org/10.1152/ajprenal.00320.2009.
- 78. Whitfield ML, Finlay DR, Murray JI, Troyanskaya OG, Chi JT, Pergamenschikov A, et al. Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc Natl Acad Sci U S A. 2003;100(21):12319–24. http://dx.doi.org/10.1073/pnas.1635114100.
- 79. Wilkinson HN, Hardman MJ. Wound healing: cellular mechanisms and pathological outcomes. Open Biol. 2020 Sep;10(9):200223. http://dx.doi.org/10.1098/rsob.200223.
- 80. Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, et al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166(5):1321–32. http://dx.doi.org/10.1016/s0002-9440(10)62351-6.
- 81. Wu M, Pedroza M, Lafyatis R, George AT, Mayes MD, Assassi S, et al. Identification of cadherin 11 as a mediator of dermal fibrosis and possible role in systemic sclerosis. Arthritis Rheumatol. 2014 Apr;66(4):1010–21. http://dx.doi.org/10.1002/art.38275.
- 82. Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. 2004 Aug;4(8):583–94. http://dx.doi.org/10.1038/nri1412.
- 83. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214(2):199–210. http://dx.doi.org/10.1002/path.2277.
- 84. Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011 Jul;208(7):1339–50.
- 85. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012 Jul;18(7):1028–40. http://dx.doi.org/10.1038/nm.2807.
- 86. Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2020;21:341–52. http://dx.doi.org/10.1038/s41580-020-0237-9.
- 87. Yousefi F, Shabaninejad Z, Vakili S, Derakhshan M, Movahedpour A, Dabiri H, et al. TGF-β and WNT signaling pathways in cardiac fibrosis: non-coding RNAs come into focus. Cell Commun Signal. 2020 Jun;18(1):87. http://dx.doi.org/10.1186/s12964-020-00555-4.
- 88. Zhou J, Bowen C, Lu G, Knapp Iii C Iii, Recknagel A, Norris RA, et al. Cadherin-11 expression patterns in heart valves associate with key functions during embryonic cushion formation, valve maturation and calcification. Cells Tissues Organs. 2013;198(4):300–10. http://dx.doi.org/10.1159/000356762.