1. Introduction
The two central components of the circulatory system are the cardiovascular, comprised of the heart, blood vessels, and blood; and the lymphovascular, comprised of vessels, lymph nodes, and lymph. The majority of interstitial fluid, consisting of the filtered blood plasma from between cells, enters the initial capillaries of the lymphovascular system where it becomes lymph. Whilst the cardiovascular system is a closed network of blood vessels and capillaries, the lymphatic system is an open system, which by a series of blind-ended capillaries, vessels, trunks, and ducts returns the drained fluid back into the bloodstream at the subclavian vein.
Diseases of the cardiovascular system are the leading cause of death globally. Of these, congenital heart disease (CHD) is the most common birth defect that arises due to abnormalities in heart and great vessel development early during embryonic life. Disorders of lymphatic vessels, such as lymphoedema, lymphangitis, lymphangiectasia, and lymphatic malformations, are less common than cardiovascular diseases but can have severe effects on well-being and life expectancy. Lymphoedemas can be primary, caused by genetic factors, or secondary, as the result of accident or other disease.
Vascular endothelial growth factor receptors (VEGFRs) are essential in orchestrating the development and lifelong maintenance of both cardiovascular and lymphovascular systems. Their aberrant expression or dysfunction is associated with a range of human diseases (Table 1). VEGFR signalling is highly complex; the reader is referred to excellent recent reviews dealing in particular with VEGFR1 and VEGFR2 signalling. In this review, we will focus on the established and emerging roles of VEGFR3, encoded by the FLT4 gene in humans and the Vegfr3 gene in the mouse, in both lymphatic and cardiac congenital disorders. By exploring the cell biology, animal models, and human conditions associated with Vegfr3/FLT4, we aim to highlight the diverse developmental and physiological roles played by the receptor.

2. Vascular endothelial growth factor receptors and their ligands
2.1. Signalling via VEGFR1 and VEGFR2
Since the discovery of VEGF over 30 years ago by Leung et al. the variety of roles VEGFs and VEGFRs play in development and maintenance of the vasculature and their function in health and disease have been extensively studied. We commence with a necessarily brief overview of the broader signalling pathway, directing the reader to appropriate authoritative articles, to provide a degree of context for our more in-depth consideration of VEGFR3/FLT4. The family consists of five peptide ligands (VEGFA-D and placental growth factor) and three receptors (VEGFR1-3), which can act together or antagonistically during development and throughout life in the circulatory system but also in other tissues (Figure 1)., VEGFRs all homodimerize; in addition, VEGFR2 can heterodimerize with either VEGFR1 or VEGFR3, resulting in receptor activation. VEGFRs can also interact with distinct coreceptors; and different VEGF cleavage products and isoforms add further complexity to the regulation of VEGFR signalling.,
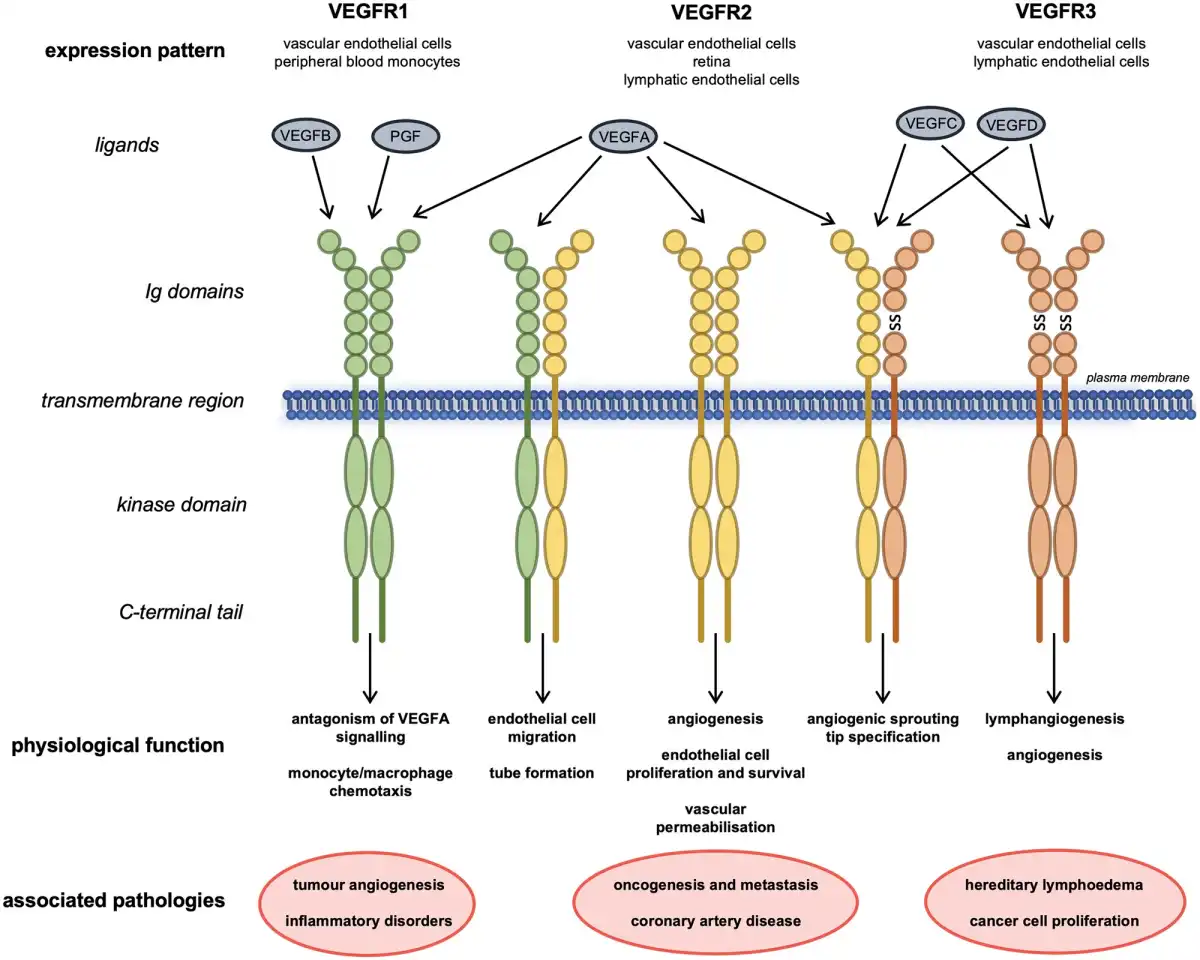
Figure 1
Structures, interactions, physiological roles, and associated diseases of VEGFR family members. Schematic showing the interactions of the three VEGFRs with each other and their ligands at the plasma membrane. Their expression pattern, physiological roles and pathologies associated with their levels or dysfunction are also shown. Ig, immunoglobulin-like domain; SS, disulphide bond.
VEGFRs are receptor tyrosine kinases that associate as homo- or heterodimers at the plasma membrane. They have an extracellular region comprising of seven immunoglobulin-like domains where their respective ligands bind, in addition to a transmembrane region and an intracellular kinase domain. Ligand binding to VEGFRs promotes dimerization and auto- or transphosphorylation of tyrosine residues in their intracellular regions. The phosphotyrosines then act as docking sites for cytoplasmic signalling molecules, which, depending on biological context, activate distinct downstream signalling cascades that allow them to mediate their physiological functions., Evidence also suggests that c-SRC-mediated VEGFR2 activation can occur in the absence of ligand, inducing ligand-independent phosphorylation and downstream signalling of VEGFR2.
Homodimeric VEGFR2 is a key regulator of angiogenesis; endothelial cell proliferation and survival; and vascular permeability in response to the canonical VEGF ligand VEGFA. VEGFR1 has a key function antagonizing VEGFR2 signalling mediated by a greater binding affinity for VEGFA that allows for sequestration of the ligand. VEGFR1 demonstrates immunomodulatory functions, driving monocyte, and macrophage chemotaxis. VEGFR1/VEGFR2 heterodimers orchestrate endothelial cell migration and tube formation. VEGFR2 also cooperates with VEGFR3 during cell tip specification in angiogenic sprouting. VEGFA can also interact directly with the PDGF (platelet-derived growth factor) signalling pathway during mesenchymal cell proliferation and migration.,
Following ligand binding, VEGFRs are rapidly internalized into endocytotic vesicles as a means to control their activity. Vesicular VEGFRs can still actively signal, be recycled back to the plasma membrane, targeted for lysosomal degradation or returned to the Golgi maturation pathway. The internalization process is highly important for the control of VEGF/VEGFR signalling, for example, internalization of VEGFR2 is required for ERK (extracellular signal-regulated kinase) and AKT activation. Post-translational modification, cleavage or degradation have also been described as regulatory mechanisms for controlling signalling through VEGFRs.
Due to their ability to control and stimulate growth of new vasculature, the VEGFR1 and VEGFR2 signalling pathways participate in a wide variety of physiological and pathological processes that lie beyond the scope of this focused review. For excellent reviews providing greater detail regarding the biology of the VEGF ligand family, their dysfunction in disease, and their therapeutic potential see Karaman et al., Park et al., and Uccelli et al. The role of VEGFRs in tumour angiogenesis,,,, Alzheimer’s disease, and vascular dysfunction,,,, has also been reviewed.
2.2. Signalling via VEGFR3 homodimers
VEGFR3 is a key regulator of lymphatic system development and establishment. Unlike the other VEGFRs, VEGFR3 is proteolytically cleaved within its fifth extracellular immunoglobulin-like domain and the two resulting peptides are then disulphide bonded as part of its maturation in the extracellular matrix (Figure 1). The mechanism and function of this cleavage step have not been extensively studied but is thought to be important for ligand binding and stability of the receptor when at the cell membrane.
Structural examination of VEGFR3 ligand binding propensity identified the first three immunoglobulin-like domains as the direct interaction site for VEGFC; however, the majority of the extracellular region is required for proper ligand-induced receptor activation and subsequent downstream signalling (Figure 2A)., In humans, at least three isoforms of VEGFR3 are expressed that function differently and have distinct physiological roles. The full-length isoform is well-characterized and discussed hereafter; a second isoform is shorter from the C-terminus by sixty-five amino acids including tyrosine residues whose phosphorylation can function to activate signalling downstream of the receptor. A third VEGFR3 isoform lacks a much larger C-terminal coding region including the transmembrane domain and is a secreted soluble protein that acts to sequester VEGFC in the retina.
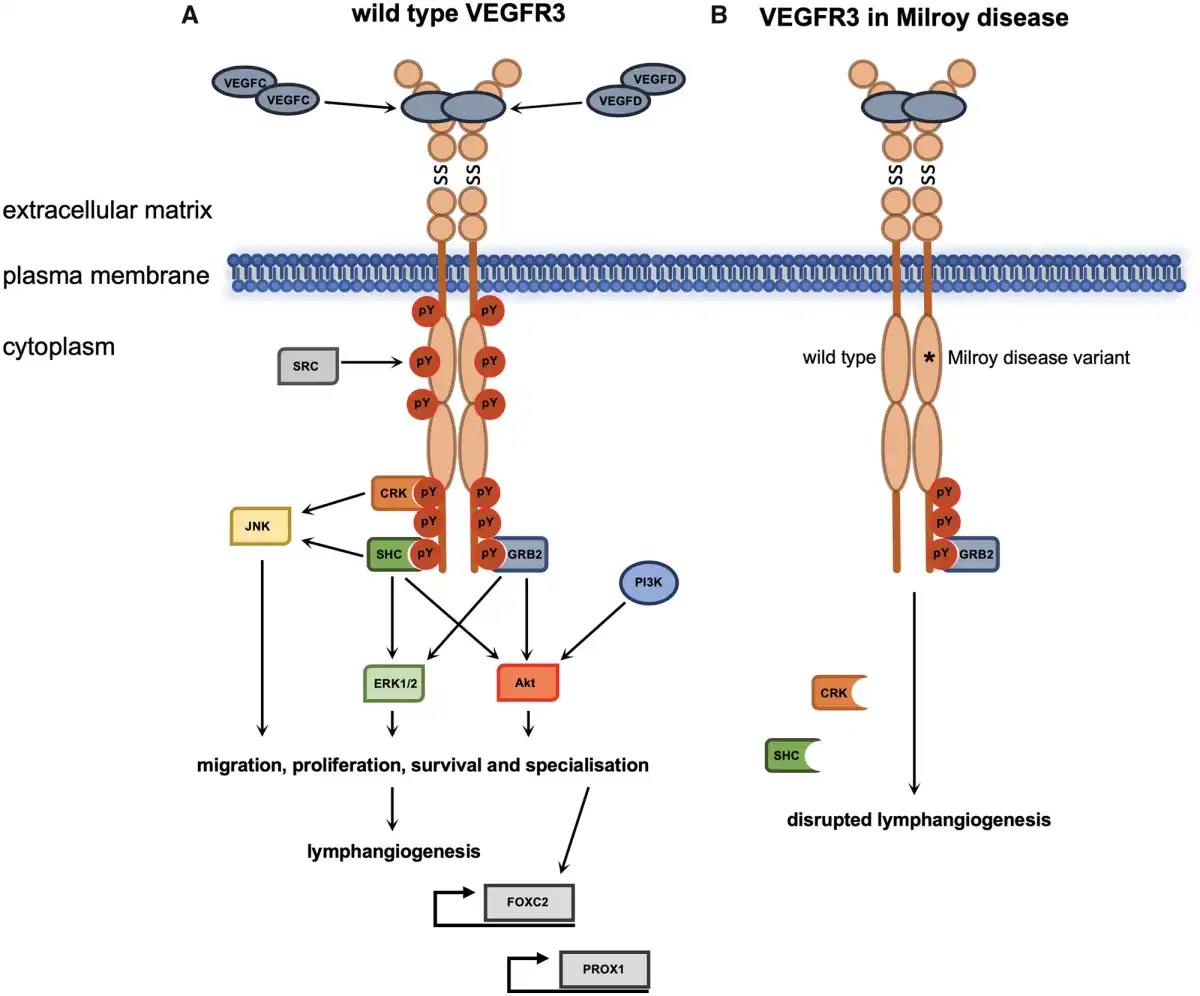
Figure 2
VEGFR3 signalling during lymphangiogenesis and Milroy disease. Ligand binding and VEGFR3 homodimerization during lymphangiogenesis results in activation of downstream signalling pathways in LECs or precursor endothelial cells (A). The dominant negative mutations of the kinase domain of VEGFR3 in Milroy disease result in reduction of downstream signalling following ligand binding due to loss of autophosphorylation and thus consequent disruption of lymphangiogenesis (B). *Mutations in the kinase domain of VEGFR3 that inactivate the receptor’s catalytic activity; pY, phosphotyrosine; SS, disulphide bond; description of protein abbreviations in main text.
The receptor is expressed in lymphatic endothelial cells (LECs) where it acts as a homodimer that responds to the extracellular ligands VEGFC and VEGFD., The binding of the ligands activates the intracellular kinase domains which then trans-autophosphorylate each other. The specific phosphotyrosine residues of VEGFR3 that are required for activation of intracellular signalling pathways are located in the juxtamembrane domain, the kinase domain, and C-terminal tail. Trans-autophosphorylation results in recruitment of adaptor proteins such as CRK (CT10 regulator of kinase), SHC (SRC homology domain containing), and GRB2 (growth factor receptor-bonus protein 2), which in conjunction with phosphatidylinositol-3-kinase (PI3K) activate downstream signalling pathways that include the conserved PI3K/MAPK (mitogen-activated protein kinase)-associated family members AKT, ERK1/2, and JNK (c-Jun N-terminal kinase).,
VEGFC is required for lymphatic development and the VEGFR3/VEGFC signalling axis is particularly important during the expansion of the lymphatic system when LECs start budding off from the cardinal vein. Paracrine secretion of VEGFC provides a chemogradient in areas of active lymphangiogenesis and lymphatic vessel development can therefore be controlled spatially and temporally. VEGFC is only active after proteolytic processing and CCBE1 (collagen and calcium binding extracellular growth factor domain 1) is crucial for the immobilization of pro-VEGFC enabling CCBE1’s cofactor ADAMTS3 (a disintegrin and metalloproteinase with thrombospondin motifs 3) to proteolytically activate the ligand. Due to their essential role in VEGFC processing, mutations in human CCBE1, ADAMTS3, and VEGFC have been shown to cause various forms of primary lymphoedema.
In order to maintain their endothelial cell identity during embryogenesis, LECs employ a positive feedback loop whereby VEGFR3 signalling maintains the expression of the transcription factor PROX1 (homeobox prospero-like 1), which in turn regulates expression of the receptor., Both PROX1 and the transcription factor FOXC2 (forkhead box protein C2) play a role in the proper formation, location and function of the lymphatic valves in a process requiring VEGFR3 activation and its subsequent degradation through an EPSIN-mediated mechanism.
The transcription factor ETV2 is known to have key roles in early developmental processes and is required for lymphangiogenesis through direct regulation of VEGFR3 expression. Similarly, integrin-linked kinase (ILK) is known to play a role in regulating VEGFR3 signalling during lymphatic vascular growth. Signalling downstream of VEGFR3 in LECs can be inhibited by vascular endothelial phosphatase; the only evidenced regulatory VEGFR3 phosphatase identified thus far. VEGFR3 expression has also been shown in nonvascular cell types including osteoblasts, neural progenitor cells and macrophages.
2.3. Signalling via VEGFR3 heterodimers
As previously mentioned, VEGFR3 is able to function as a heterodimer with VEGFR2 as a receptor for VEGFC ligand binding during angiogenesis and haematopoiesis (Figure 3A). In a culture of murine aortic mesoderm explants from Vegfr3 knockout embryos, vascular bed formation was enhanced compared to wild type and heterozygous embryos, and haematopoiesis severely diminished. It has been postulated that in the absence of VEGFR3, a higher abundance of VEGFC is free to signal through VEGFR2, leading to disruption of angiogenesis and blood cell formation during embryogenesis. VEGFR2-positive cells derived from embryonic stem cells serve as vascular progenitors that differentiate into endothelial cells upon VEGFA stimulation. Likewise, VEGFC is also able to stimulate endothelial differentiation into LECs when VEGFR2 and VEGFR3 act in a heterodimeric manner.
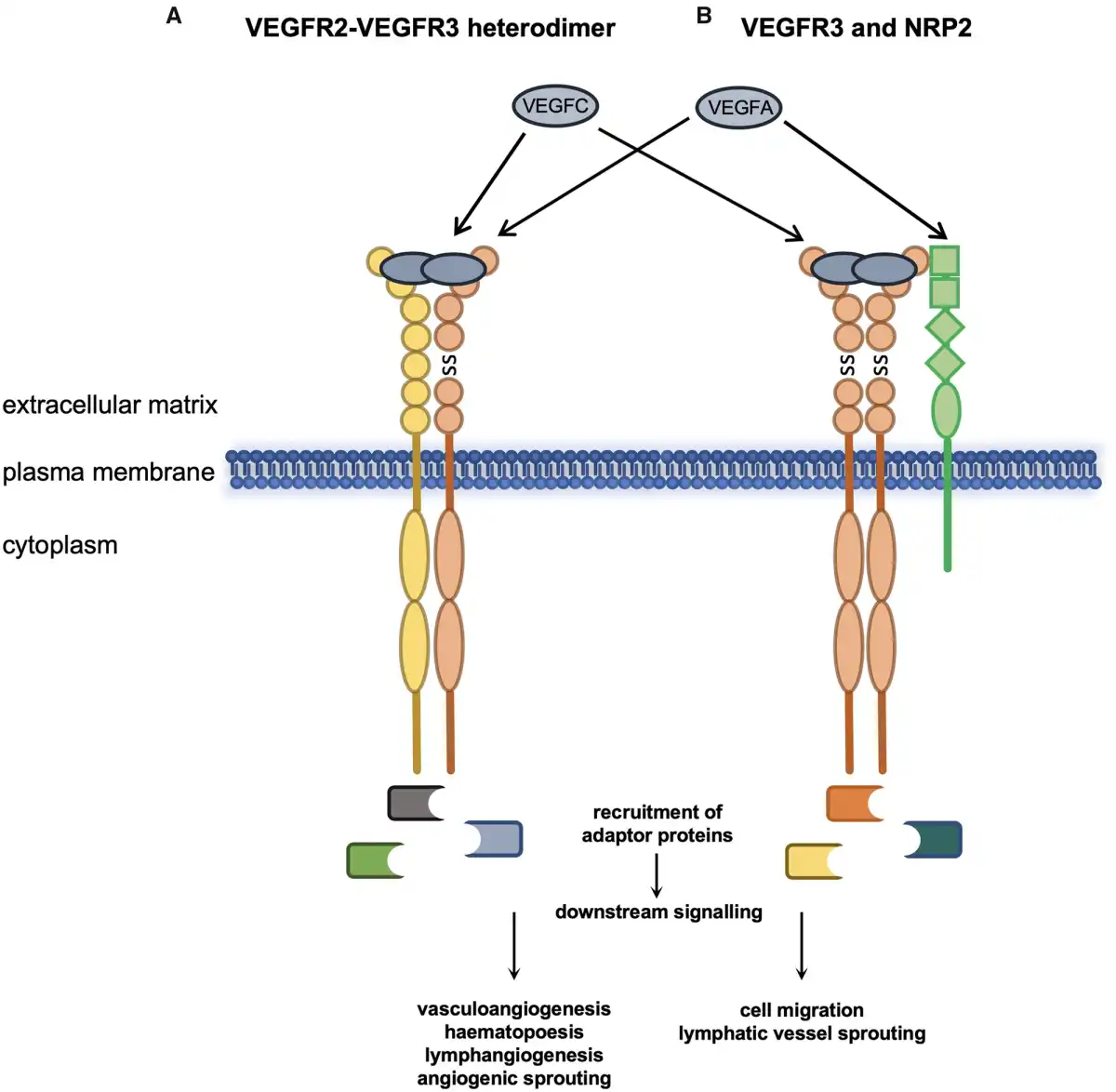
Figure 3
VEGFR3 functions with VEGFR2 and NRP2. Heterodimerization of VEGFR3 and VEGFR2 and activation by VEGFA or VEGFC regulates multiple biological processes in endothelial and endothelial-derived cell lines (A). Association of VEGFR3 homodimers with NRP2 and activation of VEGFR3 by its cognate ligands and NRP2 by VEGFA modulates VEGFR3 function (B). SS, disulphide bond.
Neuropilin 2 (NRP2) has also been shown to be a coreceptor for both VEGFR2 and VEGFR3 and can act in response to VEGFA and VEGFC (Figure 3B). Expression of NRP2 promotes survival of endothelial cells in response to both ligands and promotes migration of human microvascular endothelial cells. Lymphatic system vascular sprouting can be mediated by VEGFR3 and NRP2 interacting at the plasma membrane in response to VEGFC. This process is independent of Vegfr2 since Vegfr2+/−/Nrp2+/− but not Vegfr3+/−/Nrp2+/− double heterozygous mice have normal lymphatic vessel sprouting during development and lymph vessel branching later in life. The hypoxia-regulated transmembrane protein CLP24 (claudin-like protein 24 kDa) is also required for lymphatic vessel sprouting through interaction with VEGFR2 and VEGFR3 promoting downstream phosphorylation of the transcription factor CREB (cyclic adenosine monophosphate response element-binding protein).
As mentioned previously, there is some evidence that dimerization of the VEGFRs can occur in the absence of ligand with reduced downstream signalling. However, it has been shown that both VEGFA and VEGFC strongly promote heterodimerization of VEGFR2 and VEGFR3 in both developing blood vessels and in early lymphatic progenitor cells., The presence of ligand also leads to spatial aggregation of dimers in the leading tip of growing angiogenic sprouts.,,
During angiogenesis, endothelial cells undergo specification to tip or stalk cells of newly developing blood vessels. Endothelial-specific knockout of Vegfr3 in mice led postnatally to excessive angiogenic sprouting and branching whilst decreasing the level of Notch signalling. VEGFR3 regulates angiogenic sprouting even in the presence of VEGFR2 inhibitors, suggesting this function can be independent of VEGFR2-VEGFR3 cooperation., VEGFR3 regulation by the NOTCH pathway has also been shown to facilitate angiogenesis without the requirement of VEGFR2 signalling, however, in the retina VEGFR2 is explicitly required for this process.
VEGFR2 and VEGFR3 have also been identified as components of a complex that mediates the response of vascular endothelial cells to fluid sheer stress during development and angiogenesis. In this role, VE-cadherin acts as an adaptor by binding directly to the transmembrane domain of both VEGFR2 and VEGFR3. In vivo Vegfr3 was directly shown to contribute to the response of flow in the aortic endothelium. Interestingly, evidence suggests the role of Vegfr3 as part of a mechanosensitive complex in blood vessel formation is ligand-independent, this is thought to be due to its ability to modulate Vegfr2 signalling. VEGFR3 signalling can also be mediated through integrin/SRC; this however induces a different phosphorylation pattern to that induced by VEGFC and VEGFD. This indicates ligand-independent functions for VEGFR3 and adds to the complexity of the VEGFR3 signalling system.,
3. VEGFR3 signalling and congenital lymphovascular malformation
3.1. Normal development of the lymphatic system
The lymphatic vasculature is an essential part of the body’s circulatory systems with roles in immune surveillance, fat absorption and fluid homeostasis. Lymphangiogenesis refers to the appropriate production and maintenance of a functioning lymphatic system in the vascularized tissues of the body throughout life.,
In brief, the first appearance of the lymphatic system in mice is approximately E9.5 when endothelial cells of the cardinal vein differentiate to the LEC lineage., At around E10.5 these cells bud and emerge from the cardinal and intersomitic veins and, following migration, form a primitive lymphatic plexus and lymph sacs. Afferent and efferent lymph vessels then proceed to emerge throughout the tissues of the developing embryo forming distinct lymph nodes and producing lymphovenous valves for regulation of fluid movement between the lymphatic and cardiovascular systems. At approximately E15 the lymphatic system undergoes maturation and remodelling at which point lymphatic valves are formed. Their function is to control unidirectional movement of lymph.
An important role of the lymphatic system is to maintain body fluid homeostasis by draining plasma filtrate from the interstitium, but it is also intrinsically linked to immune cell function since LECs can secrete chemokines and thereby recruit immune cells and transport them into lymph nodes during inflammatory immune responses. Lymphatic vessels in the gut (also known as lacteals) are responsible for the absorption of dietary fat which is then transported as chyle up through the thoracic duct into the venous circulation., Lymphatic development is a very complex process and the list of genes and gene products involved is rapidly expanding. For a more comprehensive overview of the known cellular and molecular processes controlling the development of the lymphatic system during embryogenesis the readier is directed to some excellent reviews.,,,,
3.2. Dominant negative VEGFR3 mutations in Milroy disease
Lymphoedema is caused by impaired drainage or transport of interstitial fluid which leads to a build-up of lymph, resulting in chronic swelling. It typically affects the limbs but may involve any body site also within the inner body cavities, for example pleural and pericardial effusions or ascites. Lymphoedema can be discomforting and associated with high morbidity from loss of mobility and recurrent infections. There are two main types of lymphoedema: primary lymphoedema, which is the result of an underlying genetic abnormality, and secondary lymphoedema, which arises due to damage to the lymphatic system as a result of trauma, infection or following surgery or radiotherapy.
Primary lymphoedema is a highly heterogeneous condition with many different genetic causes, some as autosomal dominant traits, such as Milroy disease or lymphoedema distichiasis syndrome or as autosomal recessive traits, for example Hennekam syndrome. Although Milroy disease (OMIM: 153100) is a rare condition, it is one of the most frequent causes of congenital primary lymphoedema. It is characterized by symmetrical lymphoedema of the lower limbs, particularly the dorsum of the feet and ankles but may reach the knees. It typically presents at or shortly after birth, although in some cases lymphoedema does not manifest until later in life. In addition, the affected areas are prone to decreased rates of healing even following minor trauma and the affected skin may become brawny and fibrotic. The impaired lymph drainage also predisposes to infection, for example cellulitis, which is also a frequent complication in individuals with Milroy disease.
VEGFR3 was first implicated in Milroy disease when a region of chromosome five was linked to inherited lymphoedema cases and the FLT4 locus was chosen as the best candidate gene in this region for further investigation. A FLT4 missense mutation, p.H1035A, was identified in a Milroy patient and in vitro assessment of the mutation found that, compared to wild type protein, receptor trans-autophosphorylation was inhibited. Subsequently, further mutations in FLT4 have been identified and approximately seventy percent of Milroy cases have been given a molecular diagnosis. Importantly, the 57 reported FLT4 mutations identified in Milroy disease to date are either missense (n = 54) or small in frame deletions (n = 3) within the kinase domain coding region.
Full-length VEGFR3 harbouring a kinase domain mutation is expressed and translocates to the plasma membrane where it can interact with wild type VEGFR3. All mutations identified in Milroy patients thus far have demonstrated decreased catalytic activity and their downstream signalling is also reduced. For example, in response to VEGFC ligand binding, the MAPK pathways normally activated display decreased phosphorylation of sites required for their downstream signalling (Figure 2B)., Karkkainen et al. also showed that the mutant receptors had greater stability and were internalized and degraded at a slower rate compared to wild type receptor. This altered turnover of mutant VEGFR3 receptors would lead to a greater number of mutant receptors available for dimerization at the plasma membrane; thereby reducing the relative amount of wild type VEGFR3 for VEGFC to bind to. Thus, evidence suggests that FLT4 mutations in Milroy disease have a dominant negative effect as they antagonize the activity of wild type protein.,
4. VEGFR3 and congenital heart disease
4.1. Normal development of the heart and great vessels
The cardiovascular system develops early during embryogenesis shortly after gastrulation with the beginning of cardiogenesis. Figure 4 compares the developmental timings of both cardiovascular and lymphovascular systems following fertilization during human and mouse embryogenesis aligned with the established Carnegie stages of human embryonic development.
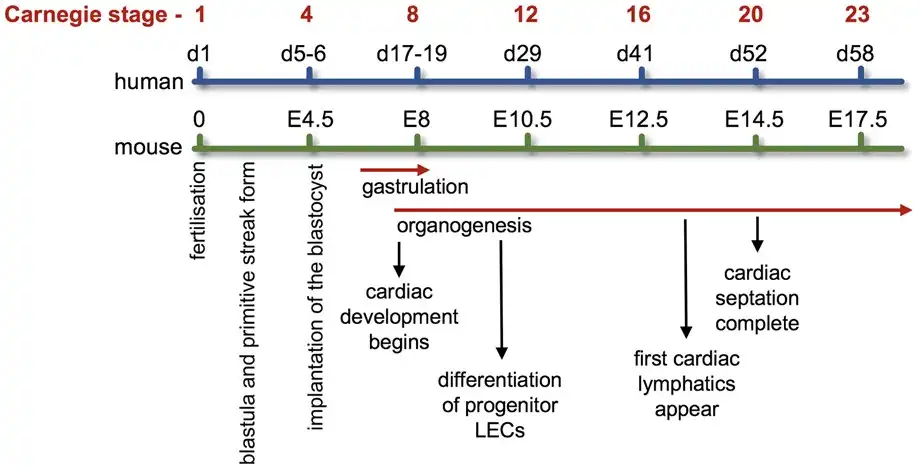
Figure 4
Developmental timing of cardiovascular and lymphovascular system development. Developmental timings in both human and mouse of cardiovascular and lymphovascular development, aligned with the Carnegie stages of embryonic development. For references, see main text.
Heart development begins when progenitor cardiac crescent cells, termed the first heart field (FHF), develop from the early mesoderm and form the primary heart tube. Distinct cells from the second heart field (SHF) are then added to both the rostral and caudal poles before looping of the tube and the occurrence of further complex morphological changes occur to convert the initial in-series circuit to an in-parallel arrangement. Beating of cardiac cells commences at three weeks post-fertilization in humans, well before heart development is complete, followed by the initiation of blood flow approximately a week later.
The completion of cardiogenesis occurs when the four chambers of the heart are defined at the end of septation., Cells originating from both heart fields contribute to the atria, whilst FHF-derived cells form the majority of the left ventricle, and SHF-derived cells the right ventricle and the outflow tract (OFT), which connects the cardiac ventricles to the great vessels. Cardiac neural crest cells are migratory mesenchymal cells originating from the ectoderm of the dorsal ridge of the neural tube, migrating through chiefly the 3rd, 4th, and 6th branchial arches towards the heart. They are essential, together with mesodermal cardiac cells, for OFT formation, and they also contribute significantly to the smooth muscle tunics of the great arteries. The formation of arteries, veins and capillaries connecting the heart to the tissues and organs of the developing embryo occurs throughout development and life via angiogenesis.
It is also worth noting that the heart requires its own extensive lymphatic network in order to maintain myocardial fluid homeostasis and provide immune surveillance. Cardiac lymphatics are established shortly after the development of the heart vasculature during embryogenesis. In mice, this is around E12 before coronary circulation commences and heart development is complete (Figure 4). The heart’s lymphatic network is found in both atria and ventricles extending to at least the mitral valve in humans and enriched around the OFT. In humans, the aortic valve does not have any associated lymphatics. The specific role of VEGFR3 in cardiac lymphatic anatomy and physiology has been reported.
Remodelling of cardiac lymphatics has been associated with several diseases including ischaemic heart disease, myocardial infarction and chronic heart failure. Insufficient myocardial drainage via cardiac lymphatics can lead to oedema and inflammatory immune responses in cases of infective endocarditis. The role cardiac lymphatics play in health and disease is an active area of research that is helping develop therapeutic treatments for conditions whose cause or degeneration is due to dysfunction of normal heart lymphangiogenesis.,
4.2. VEGFR3 variants in congenital heart disease
CHDs encompass a spectrum of heterogeneous phenotypes relating to structural defects arising during cardiogenesis. Defects can be singular and localized or a range of complex morphological abnormalities can occur simultaneously. Tetralogy of Fallot (TOF) is the most common, complex cyanotic CHD with a recorded prevalence of approximately 1 in every 2500 live births. TOF is considered a malformation of the OFT during early cardiac development and is defined by four specific structural abnormalities identified postnatally—a ventricular septal defect, anterocephalad deviation of the outflow septum with resultant overriding aorta, variable obstruction of the right ventricular OFT (pulmonary stenosis) and consequent hypertrophy of the right ventricle. Open heart surgery, usually in the first year of life, means up to 90% of TOF patients live to the age of 30. However, event-free survival to age 40 is just 25% since repercussions of surgery, particularly the development of pulmonary regurgitation, and cardiac arrhythmias still cause significant morbidity in adulthood.
The genetic basis of TOF is still relatively unknown although approximately 20% of cases have been linked with chromosomal anomalies. Most significantly around 15% of these syndromic TOF patients have been diagnosed with DiGeorge syndrome (also called 22q11.2 deletion syndrome), in which the gene responsible for many of the disease manifestations is TBX1 (T box transcription factor 1).,
Through whole-exome sequencing (WES) studies of TOF patients by several groups there is now robust evidence that rare deleterious variants in FLT4 are a predisposing factor for sporadic, non-syndromic TOF. Our work, evaluating the largest non-syndromic TOF cohort studied by WES to date, discovered previously unobserved, predicted pathogenic variants in two genes with exome-wide significance, NOTCH1 and FLT4. The occurrence of FLT4 truncating variants (2.4%) was similar to that identified independently by others., Looking at a range of different CHD probands and parents, FLT4 truncating variants appear to be enriched in TOF cases in particular and can be inherited or de novo in an affected child.,
The majority of FLT4 variants predisposing to TOF result in truncation of the protein coding sequence, either by the introduction of stop codons, frame shift mutations or disruption of the conserved splice site regions that dictate the removal of intronic sequences from transcripts before translation. Missense variants that have never been observed in the general population have also been identified in several cases. Interestingly, these are predominantly located in the first immunoglobulin-like domains of VEGFR3 and are predicted in silico to be highly damaging to protein function. In contrast, the VEGFR3 mutations that cause Milroy disease are markedly distinct in both mutation type and location from those predisposing to TOF (Figure 5). All Milroy FLT4 mutations identified thus far are all missense or in-frame deletions that map to the kinase domain,, compared to those identified in TOF cases, which are predominantly truncating variants or missense mutations in the extracellular domains.
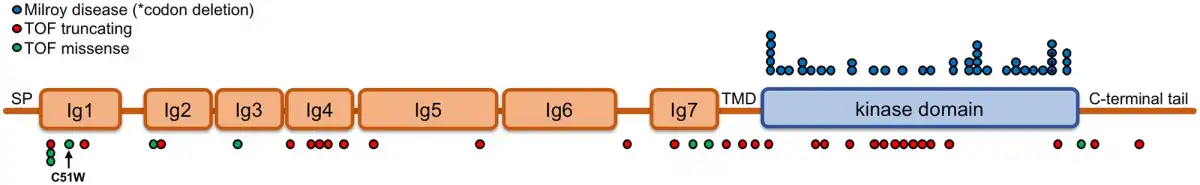
Figure 5
Comparison of VEGFR3 mutations between Milroy disease and TOF. The characterized VEGFR3 mutations known to cause Milroy disease (missense or small in frame deletions, blue dots) are compared to those that predispose to TOF (missense, predicted highly damaging to protein function, scaled combined annotation-dependent depletion score ≥20; previously unobserved in the general population, absent from the gnomAD database, green dots; or truncating, i.e. nonsense, frameshifts and splice donor or acceptor site nucleotide changes, red dots). The location of the de novo point mutant C51W is indicated beneath. References in the main text. Ig, immunoglobulin-like domain; SP, signal peptide; TMD, transmembrane domain.
In addition to the exome sequencing data implicating FLT4 in TOF aetiology, copy number variant (CNV) analysis of CHD patients provides further support. Soemedi et al. identified two TOF cases with CNVs encompassing or adjacent to the FLT4 locus; one with a de novo duplication of FLT4 and several other genes, and another with deletion of the region upstream of the FLT4 locus. A separate study identified a duplication of FLT4 and two proximal genes in a case of aortic arch anomaly, linking the FLT4 locus and potential genetic regulatory sequences with CHD. Intriguingly, two independent TOF cases have been associated with CNVs where only the C-terminal coding region of FLT4 is deleted which, like the truncating variants identified by WES, could result in expression of a C-terminally truncated protein., These observations further connect the region around the FLT4 locus with CHD; however, to date, there is no evidence that deletion of the entire FLT4 gene increases the incidence of TOF. An enrichment of truncating variants but not deleterious CNVs in TOF cases suggests that it is not FLT4 haploinsufficiency that predisposes the condition but rather the expression of truncated or mutated VEGFR3 protein during embryonic development.
Further supporting the role of FLT4 truncating variants in TOF, a family has been recorded where such a mutation has passed through several generations prominently increasing the occurrence of TOF in carriers., Several other cases have been reported where TOF probands have inherited an FLT4 variant from an unaffected parent indicating that the mutant allele has reduced penetrance., However, TOF, like most CHDs, is only very rarely inherited in a Mendelian fashion, therefore susceptibility variants whose penetrance is influenced by other genetic and environmental factors are the expected finding in sporadic cases. FLT4 variants enriched in TOF cases are all extremely rare or absent in the general population; thus far, there has been no evidence from genome-wide association studies that common variants in the region predispose to CHD.
VEGFR2 variants have also been associated with CHDs, including TOF,,, and a meta-analysis of single nucleotide polymorphisms in VEGFA, also a CHD gene, identified a variant that increased the incidence of TOF., Furthermore, mouse embryos modified to solely express Vegfa120 isoform displayed alterations in Vegf and Notch signalling and a cardiac phenotype that mimicked TOF. Gene expression profiling of right ventricular biopsies collected from TOF patients compared to age-matched controls showed changes in transcript levels for VEGF, VEGFR, PDGF, and FGF (fibroblast growth factor) family members, though no change in FLT4 was reported. Immunohistological staining showed that there was an increase in vascularization of the heart vasculature in TOF cases, but the vessels were stunted and could possibly not conduct blood. Hence, while VEGFR2 and VEGFA mutations are associated with multiple CHD phenotypes, evidence strongly supports a role for FLT4 variants primarily in the predisposition to TOF.
5. Future directions
5.1. Vegfr3 animal models of cardiac and lymphatic development
The expression of Vegfr3 protein in early embryonic murine hearts has been observed in the endocardium at E9.5 and throughout the heart at E12.5 with strong staining of specific LECs sprouting proximal to the OFT on the dorsal side., Tbx1, a transcription factor linked to CHDs including TOF and the cardiovascular manifestations of DiGeorge syndrome, is known to regulate Vegfr3 during lymphatic vessel development in mice. The expression of Tbx1 and Vegfr3 is tightly balanced during heart lymphangiogenesis ensuring the morphology, localization and number of cardiac lymphatic vessels is correct.
Genetic lineage tracing in Isl1 (insulin gene enhancer protein 1)-Cre reporter mice indicated that lymphatic cells surrounding the OFT could arise from the pharyngeal mesoderm of the SHF. Isl1 is a key marker of cardiac progenitor cells that form the SHF and, interestingly, tracing back to earlier embryonic stages Isl1+ cells were shown to overlap with Vegfr3+/Prox1+ cells in the pharyngeal core. Endocardial specific ablation of Hand2 (heart and neural crest derivatives-expressed protein 2), a known CHD gene, in mice, results in cardiac malformations resembling the human condition tricuspid atresia and is caused by disruption to Notch-dependent cell-to-cell signalling and dysregulation of Vegfr3 function.
Due to multiple studies pointing to a role for distinct VEGFR3 variants predisposing to a human CHD, it is timely to review the various mouse models that give credence to a function for the receptor in early cardiac development distinct to that in lymphangiogenesis (Table 2).
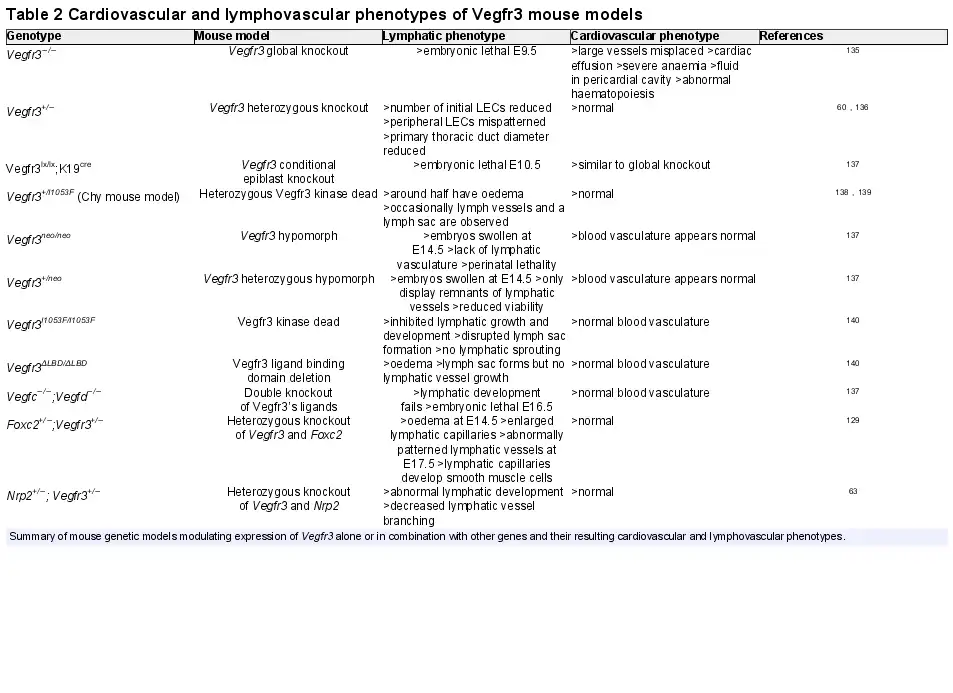
In brief, complete knockout of Vegfr3 results in cardiovascular failure at Day E9.5 with embryos displaying severe anaemia and cardiac effusion. Angiogenesis occurs but the large vessels are disorganized and fluid accumulates in the pericardial cavity resulting in lethality. Considering the well-established role of Vegfr3 in lymphatic development the occurrence of this severe cardiovascular phenotype, before commencement of lymphangiogenesis suggests a distinct role for the receptor in early cardiovascular development.
A similar phenotype was observed when a conditional K19 (keratin 19 promoter)-Cre model was employed to knockout Vegfr3 in the epiblast indicating that the cardiovascular phenotype is dependent on Vegfr3 functioning in the embryo proper and not due to defects in placental development. Intriguingly, mice heterozygous for Vegfr3, when compared with homozygous knockouts, do not display abnormal heart development or haematopoiesis suggesting one functional copy of Vegfr3 is sufficient for normal cardiovascular system development. Although there is no obvious lymphatic phenotype in these mice and they appear healthy it has been shown that the initial number of LECs produced is reduced and peripheral LECs are mispatterned.
Hypomorphic homozygous or heterozygous Vegfr3 mice, in which a neomycin cassette has been inserted between the first two exons of the gene causing dysregulation of expression but not necessarily altered function, displayed disrupted lymphatic system but not cardiovascular system development indicating lymphangiogenesis is more sensitive to the relative abundance of Vegfr3 than cardiogenesis.
Early cardiovascular system development is also independent of Vegfr3’s characterized ligands that are known to be required during lymphangiogenesis. Vegfc/Vegfd double knockout mice display abnormal lymphangiogenesis, but normal blood vasculature development. In addition, evidence suggests that Vegfc and Vegfd are functionally redundant during lymphangiogenesis, supporting the notion that VEGFR3 has a ligand-independent function in embryonic cardiovascular development.
Double heterozygous models of Vegfr3 with either Nrp2 or Foxc2 have abnormal lymphatic system development but no embryonic lethality due to cardiovascular failure, further indicating that the function of Vegfr3 in heart development is distinct to that which mediates LEC maturation and lymphangiogenesis.,
Finally, mice engineered to express versions of Vegfr3 that either could not bind ligand (ligand binding domain knockout) or were kinase dead (inactivating missense mutation in the kinase domain) also had normal cardiovascular development before lymphatic system dysfunction.
A role for Vegfr3 in valvulogenesis has recently been shown by Fontana et al., during cardiac development in zebrafish. Fluid shear stress acting on endocardial cells lining the atrioventricular valve leaflet independently activates Notch or Klf2a (Krüppel-like Factor 2) signalling and spatial antagonism between the Notch and Vegfr3 pathways defines atrioventricular valve morphology.
5.2. Defining the distinct functions of VEGFR3 during development and disease
The identification of FLT4 as the causal or predisposing genetic factor for two unrelated human conditions, Milroy disease and TOF, respectively, highlights the distinct roles VEGFR3 plays in development. Since CHD is not considered to be part of the Milroy phenotype, and congenital lymphoedema is not considered to be a constituent of the TOF phenotype, how different mutations contribute to disease pathology is an intriguing research proposition.
The early embryonic lethality due to cardiac failure of homozygous knockout, or conditional epiblast knockout, of Vegfr3 in mice demonstrates the receptor has a crucial function in cardiogenesis during early embryonic development. The lack of a cardiovascular phenotype in Vegfc/Vegfd double homozygous knockout mice suggests this function of Vegfr3 in heart development is different to that during lymphangiogenesis since it is not dependent on its activation by such ligands. This is supported further by the homozygous mouse models in which Vegfr3’s ligand binding domain has been deleted or kinase domain has been inactivated by mutation of an amino acid residue that is critical for the receptor’s enzymatic activity. Therefore, we can conclude that ligand binding, activation and trans-autophosphorylation of Vegfr3 that is essential during lymphatic system development is not required for cardiogenesis.
Heterozygous Vegfr3 knockout mice have normal cardiovascular system development suggesting the function of the receptor during cardiogenesis is not sensitive to the level of Vegfr3 protein. This also contrasts with its role in lymphatic system development which is sensitive to changes in the level of Vegfr3 expression, as highlighted by the hypomorphic mouse model. This reemphasizes that haploinsufficiency as a disease mechanism of the FLT4 variants that greatly increase the risk of TOF is unlikely, and that the disease pathogenesis is instead related to the expression of mutated VEGFR3 protein. Further supporting this is the extraordinarily low occurrence of truncating FLT4 variants observed in exome and genome sequences from the Genome Aggregation Database (gnomAD) of over 100 000 people. Indeed, it has been calculated from the gnomAD database that FLT4 is very intolerant to such loss of function variants.
Vegfr3 mouse genetic models that do not have a cardiovascular phenotype all still express the receptor with an N-terminal targeting sequence and C-terminal tail. In contrast, the truncations seen in TOF all result in coding sequences that are shorter from the C-terminus whilst retaining their N-terminal signal sequence. Therefore, it is tempting to suggest that in TOF it is expression and dysfunction of C-terminally truncated mutant VEGFR3 proteins that inappropriately modulates cellular functions early in development, leading to disrupted cardiogenesis. The truncated VEGFR3 proteins could act through aberrant interaction with other proteins such as wild type VEGFR3, proteins of the exocytotic pathway, coreceptors, ligands or an as yet uncharacterized or pathological binding partner. Such an interaction could occur in endocardial cells, vascular endothelial cells or cardiac LECs and only be disruptive to heart development in particular physiological environments or genetic backgrounds.
VEGFR3 can modulate vascular permeability in blood vessel endothelial cells, where, even though it is weakly expressed, it plays a physiological role controlling the expression of the major angiogenesis regulator VEGFR2. VEGFR2 has a role in cardiomyocyte hypertrophy through paracrine signalling between endothelial cells and cardiomyocytes during physiological myocardial growth. This is mediated by the VEGFA-VEGFR2-DLL4 (delta-like protein 4)-NOTCH signalling axis. If the FLT4 truncating mutations observed in TOF cases disrupted this function of VEGFR2 in cells of the SHF, where FLT4 expression has been reported, then that could lead to abnormal cardiac development. A role for VEGFR2 in formation of the arterial pole of the heart from the early pharyngeal mesoderm should also be considered in TOF aetiology.
The FLT4 gene also shows intolerance to missense variants in the gnomAD database (FLT4 Z-score = 3.73). The FLT4 missense mutations identified in a small number of TOF cases that are previously unobserved and predicted to be highly damaging to protein function are almost all located in the receptor’s extracellular domain and could be acting in a similar manner to the truncating mutations. Of note, the de novo mutation, C51W, identified in a TOF proband (Figure 5), would disrupt the C51-C111 disulfide bond that is important for the structure of the first immunoglobulin-like domain of VEGFR3 and would possibly disrupt proper ligand binding.
Though it is now clear that VEGFR3 has a role in both Milroy disease and TOF, there are undoubtedly significant gaps in our knowledge regarding the different functions of this multifaceted receptor. Identifying the cell types important for Vegfr3’s role in cardiogenesis could be done by employing conditional mouse models that could also be used to assess when during development Vegfr3 is required, for example, in early or late OFT progenitor cells. If generation of a knock-in mouse harbouring a truncated version of Vegfr3 had a phenotype mimicking TOF it would be an extremely powerful tool for delineating the mechanism by which heart malformations occur in disease. Another approach would be to generate human embryonic stem cells harbouring different FLT4 TOF variants using the most up to date genome editing tools followed by experiments assessing for changes in their differentiation to relevant developmental cell types such as cardiomyocytes, neural crest cells or an endothelial lineage.
6. Summary
The functions of VEGFR3 in lymphangiogenesis, angiogenesis, and cardiogenesis and the link to human conditions of distinct genetic variants of the gene make it an enticing avenue for future research. However, due to the complicated nature of these different processes and the difficulty separating them experimentally, researchers must carefully plan the techniques they adopt to elucidate both the physiological functions and disease mechanisms associated with VEGFR3.
Conflict of interest: none declared.
Funding
This work was supported by the British Heart Foundation [Programme Grant RG/15/12/31616 to B.D.K., Personal Chair to B.D.K.].
References
- 1. Levick JR, Michel CC. Microvascular fluid exchange and the revised starling principle. Cardiovasc Res2010;87:198–210.
- 2. Breslin JW, Yang Y, Scallan JP, Sweat RS, Adderley SP, Murfee WL. Lymphatic vessel network structure and physiology. Compr Physiol2018;9:207–299.
- 3. McAloon CJ, Boylan LM, Hamborg T, Stallard N, Osman F, Lim PB, Hayat SA. The changing face of cardiovascular disease 2000-2012: an analysis of the world health organisation global health estimates data. Int J Cardiol2016;224:256–264.
- 4. Green A. Outcomes of congenital heart disease: a review. Pediatr Nurs2004;30:280–284.
- 5. Griffiths C, Barker J, Bleiker T, Chalmers R, Creamer D. Rook's Textbook of Dermatology. Chichester, West Sussex; Hoboken, NJ: John Wiley & Sons Inc.; 2016.
- 6. Lacal PM, Graziani G. Therapeutic implication of vascular endothelial growth factor receptor-1 (VEGFR-1) targeting in cancer cells and tumor microenvironment by competitive and non-competitive inhibitors. Pharmacol Res2018;136:97–107.
- 7. Harris R, Miners JS, Allen S, Love S. VEGFR1 and VEGFR2 in Alzheimer's disease. J Alzheimers Dis2017;61:741–752.
- 8. Moe K, Heidecke H, Dechend R, Staff AC. Dysregulation of circulating autoantibodies against VEGF-A, VEGFR-1 and PLGF in preeclampsia—a role in placental and vascular health? Pregnancy Hypertens 2017;10:83–89.
- 9. Rapisarda A, Melillo G. Role of the VEGF/VEGFR axis in cancer biology and therapy. Adv Cancer Res2012;114:237–267.
- 10. Liu D, Song J, Ji X, Liu Z, Cong M, Hu B. Association of genetic polymorphisms on VEGFA AND VEGFR2 with risk of coronary heart disease. Medicine (Baltimore)2016;95:e3413. [CVOCROSSCVO]
- 11. Ji Y, Chen S, Li K, Li L, Xu C, Xiang B. Signaling pathways in the development of infantile hemangioma. J Hematol Oncol2014;7:13.
- 12. Su JL, Yen CJ, Chen PS, Chuang SE, Hong CC, Kuo IH, Chen HY, Hung MC, Kuo ML. The role of the VEGF-C/VEGFR-3 axis in cancer progression. Br J Cancer2007;96:541–545.
- 13. Irrthum A, Karkkainen MJ, Devriendt K, Alitalo K, Vikkula M. Congenital hereditary lymphedema caused by a mutation that inactivates VEGFR3 tyrosine kinase. Am J Hum Genet2000;67:295–301.
- 14. Page DJ, Miossec MJ, Williams SG, Monaghan RM, Fotiou E, Cordell HJ, Sutcliffe L, Topf A, Bourgey M, Bourque G, Eveleigh R, Dunwoodie SL, Winlaw DS, Bhattacharya S, Breckpot J, Devriendt K, Gewillig M, Brook JD, Setchfield KJ, Bu’Lock FA, O’Sullivan J, Stuart G, Bezzina CR, Mulder BJM, Postma AV, Bentham JR, Baron M, Bhaskar SS, Black GC, Newman WG, Hentges KE, Lathrop GM, Santibanez-Koref M, Keavney BD. Whole exome sequencing reveals the major genetic contributors to nonsyndromic tetralogy of fallot. Circ Res2019;124:553–563.
- 15. Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science1989;246:1306–1309.
- 16. Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling? In control of vascular function. Nat Rev Mol Cell Biol2006;7:359–371.
- 17. Karaman S, Leppanen VM, Alitalo K. Vascular endothelial growth factor signaling in development and disease. Development2018;145:dev151019.
- 18. Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J2011;437:169–183.
- 19. Sarabipour S, Ballmer-Hofer K, Hristova K. VEGFR-2 conformational switch in response to ligand binding. Elife2016;5:e13876.
- 20. Jin ZG, Ueba H, Tanimoto T, Lungu AO, Frame MD, Berk BC. Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ Res2003;93:354–363.
- 21. Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res2006;312:549–560.
- 22. Douglas NC, Zimmermann RC, Tan QK, Sullivan-Pyke CS, Sauer MV, Kitajewski JK, Shawber CJ. VEGFR-1 blockade disrupts peri-implantation decidual angiogenesis and macrophage recruitment. Vasc Cell2014;6:16.
- 23. Cudmore MJ, Hewett PW, Ahmad S, Wang KQ, Cai M, Al-Ani B, Fujisawa T, Ma B, Sissaoui S, Ramma W, Miller MR, Newby DE, Gu Y, Barleon B, Weich H, Ahmed A. The role of heterodimerization between VEGFR-1 and VEGFR-2 in the regulation of endothelial cell homeostasis. Nat Commun2012;3:972.
- 24. Nilsson I, Bahram F, Li X, Gualandi L, Koch S, Jarvius M, Soderberg O, Anisimov A, Kholova I, Pytowski B, Baldwin M, Yla-Herttuala S, Alitalo K, Kreuger J, Claesson-Welsh LV. Receptor 2/-3 heterodimers detected in situ by proximity ligation on angiogenic sprouts. EMBO J2010;29:1377–1388.
- 25. Mamer SB, Chen S, Weddell JC, Palasz A, Wittenkeller A, Kumar M, Imoukhuede PI. Discovery of high-affinity PDGF-VEGFR interactions: redefining RTK dynamics. Sci Rep2017;7:16439.
- 26. Ball SG, Shuttleworth CA, Kielty CM. Vascular endothelial growth factor can signal through platelet-derived growth factor receptors. J Cell Biol2007;177:489–500.
- 27. Simons M. An inside view: VEGF receptor trafficking and signaling. Physiology (Bethesda)2012;27:213–222.
- 28. Otrock ZK, Makarem JA, Shamseddine AI. Vascular endothelial growth factor family of ligands and receptors. Blood Cells Mol Dis2007;38:258–268.
- 29. Park SA, Jeong MS, Ha KT, Jang SB. Structure and function of vascular endothelial growth factor and its receptor system. BMB Rep2018;51:73–78.
- 30. Uccelli A, Wolff T, Valente P, Maggio Pellegrino DN, Gurke M, Banfi L, Gianni-Barrera AR. Vascular endothelial growth factor biology for regenerative angiogenesis. Swiss Med Wkly2019;149:w20011.
- 31. Bruce D, Tan PH. Vascular endothelial growth factor receptors and the therapeutic targeting of angiogenesis in cancer: where do we go from here? Cell Commun Adhes 2011;18:85–103.
- 32. Shibuya M. VEGF-VEGFR system as a target for suppressing inflammation and other diseases. Endocr Metab Immune Disord Drug Targets2015;15:135–144.
- 33. Oszajca K, Szemraj J, Wyrzykowski D, Chrzanowska B, Salamon A, Przewratil P. Single-nucleotide polymorphisms of VEGF-A and VEGFR-2 genes and risk of infantile hemangioma. Int J Dermatol2018;57:1201–1207.
- 34. Leppanen VM, Tvorogov D, Kisko K, Prota AE, Jeltsch M, Anisimov A, Markovic-Mueller S, Stuttfeld E, Goldie KN, Ballmer-Hofer K, Alitalo K. Structural and mechanistic insights into VEGF receptor 3 ligand binding and activation. Proc Natl Acad Sci USA2013;110:12960–12965.
- 35. Jeltsch M, Karpanen T, Strandin T, Aho K, Lankinen H, Alitalo K. Vascular endothelial growth factor (VEGF)/VEGF-C mosaic molecules reveal specificity determinants and feature novel receptor binding patterns. J Biol Chem2006;281:12187–12195.
- 36. Hughes DC. Alternative splicing of the human VEGFGR-3/FLT4 gene as a consequence of an integrated human endogenous retrovirus. J Mol Evol2001;53:77–79.
- 37. Dixelius J, Makinen T, Wirzenius M, Karkkainen MJ, Wernstedt C, Alitalo K, Claesson-Welsh L. Ligand-induced vascular endothelial growth factor receptor-3 (VEGFR-3) heterodimerization with VEGFR-2 in primary lymphatic endothelial cells regulates tyrosine phosphorylation sites. J Biol Chem2003;278:40973–40979.
- 38. Singh N, Tiem M, Watkins R, Cho YK, Wang Y, Olsen T, Uehara H, Mamalis C, Luo L, Oakey Z, Ambati BK. Soluble vascular endothelial growth factor receptor 3 is essential for corneal alymphaticity. Blood2013;121:4242–4249.
- 39. Joukov V, Pajusola K, Kaipainen A, Chilov D, Lahtinen I, Kukk E, Saksela O, Kalkkinen N, Alitalo K. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the FLT4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J1996;15:1751.
- 40. Achen MG, Jeltsch M, Kukk E, Makinen T, Vitali A, Wilks AF, Alitalo K, Stacker SA. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (FLK1) and VEGF receptor 3 (FLT4). Proc Natl Acad Sci USA1998;95:548–553.
- 41. Salameh A, Galvagni F, Bardelli M, Bussolino F, Oliviero S. Direct recruitment of CRK and GRB2 to VEGFR-3 induces proliferation, migration, and survival of endothelial cells through the activation of ERK, AKT, and JNK pathways. Blood2005;106:3423–3431.
- 42. Coso S, Zeng Y, Opeskin K, Williams ED. Vascular endothelial growth factor receptor-3 directly interacts with phosphatidylinositol 3-kinase to regulate lymphangiogenesis. PLoS One2012;7:e39558.
- 43. Jha SK, Rauniyar K, Jeltsch M. Key molecules in lymphatic development, function, and identification. Ann Anat2018;219:25–34.
- 44. Vaahtomeri K, Karaman S, Makinen T, Alitalo K. Lymphangiogenesis guidance by paracrine and pericellular factors. Genes Dev2017;31:1615–1634.
- 45. Le Guen L, Karpanen T, Schulte D, Harris NC, Koltowska K, Roukens G, Bower NI, van Impel A, Stacker SA, Achen MG, Schulte-Merker S, Hogan BM. Ccbe1 regulates VEGFC-mediated induction of vegfr3 signaling during embryonic lymphangiogenesis. Development2014;141:1239–1249.
- 46. Gordon K, Varney R, Keeley V, Riches K, Jeffery S, Van Zanten M, Mortimer P, Ostergaard P, Mansour S. Update and audit of the st george's classification algorithm of primary lymphatic anomalies: a clinical and molecular approach to diagnosis. J Med Genet2020;57:653–659.
- 47. Brouillard P, Dupont L, Helaers R, Coulie R, Tiller GE, Peeden J, Colige A, Vikkula M. Loss of ADAMTS3 activity causes hennekam lymphangiectasia-lymphedema syndrome 3. Hum Mol Genet2017;26:4095–4104.
- 48. Alders M, Hogan BM, Gjini E, Salehi F, Al-Gazali L, Hennekam EA, Holmberg EE, Mannens MM, Mulder MF, Offerhaus GJ, Prescott TE, Schroor EJ, Verheij JB, Witte M, Zwijnenburg PJ, Vikkula M, Schulte-Merker S, Hennekam RC. Mutations in CCBE1 cause generalized lymph vessel dysplasia in humans. Nat Genet2009;41:1272–1274.
- 49. Srinivasan RS, Escobedo N, Yang Y, Interiano A, Dillard ME, Finkelstein D, Mukatira S, Gil HJ, Nurmi H, Alitalo K, Oliver G. The Prox1-Vegfr3 feedback loop maintains the identity and the number of lymphatic endothelial cell progenitors. Genes Dev2014;28:2175–2187.
- 50. Pan MR, Chang TM, Chang HC, Su JL, Wang HW, Hung WC. Sumoylation of Prox1 controls its ability to induce VEGFR3 expression and lymphatic phenotypes in endothelial cells. J Cell Sci2009;122:3358–3364.
- 51. Liu X, Pasula S, Song H, Tessneer KL, Dong Y, Hahn S, Yago T, Brophy ML, Chang B, Cai X, Wu H, McManus J, Ichise H, Georgescu C, Wren JD, Griffin C, Xia L, Srinivasan RS, Chen H. Temporal and spatial regulation of epsin abundance and VEGFR3 signaling are required for lymphatic valve formation and function. Sci Signal2014;7:ra97.
- 52. Wu H, Rahman HNA, Dong Y, Liu X, Lee Y, Wen A, To KHT, Xiao L, Birsner AE, Bazinet L, Wong S, Song K, Brophy ML, Mahamud MR, Chang B, Cai X, Pasula S, Kwak S, Yang W, Bischoff J, Xu J, Bielenberg DR, Dixon JB, D’Amato RJ, Srinivasan RS, Chen H. Epsin deficiency promotes lymphangiogenesis through regulation of VEGFR3 degradation in diabetes. J Clin Invest2018;128:4025–4043.
- 53. Gauvrit S, Villasenor A, Strilic B, Kitchen P, Collins MM, Marin-Juez R, Guenther S, Maischein HM, Fukuda N, Canham MA, Brickman JM, Bogue CW, Jayaraman PS, Stainier DYR. HHEX is a transcriptional regulator of the VEGFC/FLT4/PROX1 signaling axis during vascular development. Nat Commun2018;9:2704.
- 54. Davis JA, Koenig AL, Lubert A, Chestnut B, Liu F, Palencia Desai S, Winkler T, Pociute K, Choi K, Sumanas S. ETS transcription factor Etsrp/Etv2 is required for lymphangiogenesis and directly regulates vegfr3/flt4 expression. Dev Biol2018;440:40–52.
- 55. Urner S, Planas‐Paz L, Hilger LS, Henning C, Branopolski A, Kelly‐Goss M, Stanczuk L, Pitter B, Montanez E, Peirce SM, Mäkinen T, Lammert E. Identification of ILK as a critical regulator of VEGFR3 signalling and lymphatic vascular growth. EMBO J2019;38:e99322.
- 56. Deng Y, Zhang X, Simons M. Molecular controls of lymphatic VEGFR3 signaling. Arterioscler Thromb Vasc Biol2015;35:421–429.
- 57. Orlandini M, Spreafico A, Bardelli M, Rocchigiani M, Salameh A, Nucciotti S, Capperucci C, Frediani B, Oliviero S. Vascular endothelial growth factor-D activates VEGFR-3 expressed in osteoblasts inducing their differentiation. J Biol Chem2006;281:17961–17967.
- 58. Le Bras B, Barallobre MJ, Homman-Ludiye J, Ny A, Wyns S, Tammela T, Haiko P, Karkkainen MJ, Yuan L, Muriel MP, Chatzopoulou E, Breant C, Zalc B, Carmeliet P, Alitalo K, Eichmann A, Thomas JL. VEGF-C is a trophic factor for neural progenitors in the vertebrate embryonic brain. Nat Neurosci2006;9:340–348.
- 59. Schmeisser A, Christoph M, Augstein A, Marquetant R, Kasper M, Braun-Dullaeus RC, Strasser RH. Apoptosis of human macrophages by FLT-4 signaling: implications for atherosclerotic plaque pathology. Cardiovasc Res2006;71:774–784.
- 60. Hamada K, Oike Y, Takakura N, Ito Y, Jussila L, Dumont DJ, Alitalo K, Suda T. VEGF-C signaling pathways through VEGFR-2 AND VEGFR-3 in vasculoangiogenesis and hematopoiesis. Blood2000;96:3793–3800.
- 61. Suzuki H, Watabe T, Kato M, Miyazawa K, Miyazono K. Roles of vascular endothelial growth factor receptor 3 signaling in differentiation of mouse embryonic stem cell-derived vascular progenitor cells into endothelial cells. Blood2005;105:2372–2379.
- 62. Favier B, Alam A, Barron P, Bonnin J, Laboudie P, Fons P, Mandron M, Herault JP, Neufeld G, Savi P, Herbert JM, Bono F. Neuropilin-2 interacts with VEGFR-2 AND VEGFR-3 and promotes human endothelial cell survival and migration. Blood2006;108:1243–1250.
- 63. Xu Y, Yuan L, Mak J, Pardanaud L, Caunt M, Kasman I, Larrivee B, Del Toro R, Suchting S, Medvinsky A, Silva J, Yang J, Thomas JL, Koch AW, Alitalo K, Eichmann A, Bagri A. Neuropilin-2 mediates VEGF-C-induced lymphatic sprouting together with VEGFR3. J Cell Biol2010;188:115–130.
- 64. Saharinen P, Helotera H, Miettinen J, Norrmen C, D'Amico G, Jeltsch M, Langenberg T, Vandevelde W, Ny A, Dewerchin M, Carmeliet P, Alitalo K. Claudin-like protein 24 interacts with the VEGFR-2 and VEGFR-3 pathways and regulates lymphatic vessel development. Genes Dev2010;24:875–880.
- 65. Alam A, Herault JP, Barron P, Favier B, Fons P, Delesque-Touchard N, Senegas I, Laboudie P, Bonnin J, Cassan C, Savi P, Ruggeri B, Carmeliet P, Bono F, Herbert JM. Heterodimerization with vascular endothelial growth factor receptor-2 (VEGFR-2) is necessary for VEGFR-3 activity. Biochem Biophys Res Commun2004;324:909–915.
- 66. Tvorogov D, Anisimov A, Zheng W, Leppanen VM, Tammela T, Laurinavicius S, Holnthoner W, Helotera H, Holopainen T, Jeltsch M, Kalkkinen N, Lankinen H, Ojala PM, Alitalo K. Effective suppression of vascular network formation by combination of antibodies blocking VEGFR ligand binding and receptor dimerization. Cancer Cell2010;18:630–640.
- 67. Takahashi H, Kato K, Ueyama K, Kobayashi M, Baik G, Yukawa Y, Suehiro JI, Matsunaga YT. Visualizing dynamics of angiogenic sprouting from a three-dimensional microvasculature model using stage-top optical coherence tomography. Sci Rep2017;7:42426.
- 68. Tammela T, Zarkada G, Nurmi H, Jakobsson L, Heinolainen K, Tvorogov D, Zheng W, Franco CA, Murtomaki A, Aranda E, Miura N, Yla-Herttuala S, Fruttiger M, Makinen T, Eichmann A, Pollard JW, Gerhardt H, Alitalo K. VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing notch signalling. Nat Cell Biol2011;13:1202–1213.
- 69. Tammela T, Zarkada G, Wallgard E, Murtomaki A, Suchting S, Wirzenius M, Waltari M, Hellstrom M, Schomber T, Peltonen R, Freitas C, Duarte A, Isoniemi H, Laakkonen P, Christofori G, Yla-Herttuala S, Shibuya M, Pytowski B, Eichmann A, Betsholtz C, Alitalo K. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature2008;454:656–660.
- 70. Benedito R, Rocha SF, Woeste M, Zamykal M, Radtke F, Casanovas O, Duarte A, Pytowski B, Adams RH. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature2012;484:110–114.
- 71. Zarkada G, Heinolainen K, Makinen T, Kubota Y, Alitalo K. VEGFR3 does not sustain retinal angiogenesis without VEGFR2. Proc Natl Acad Sci USA2015;112:761–766.
- 72. Baeyens N, Nicoli S, Coon BG, Ross TD, Van den Dries K, Han J, Lauridsen HM, Mejean CO, Eichmann A, Thomas JL, Humphrey JD, Schwartz MA. Vascular remodeling is governed by a VEGFR3-dependent fluid shear stress set point. Elife2015;4:e04645.
- 73. Park YG, Choi J, Jung HK, Song IK, Shin Y, Park SY, Seol JW. Fluid shear stress regulates vascular remodeling via VEGFR-3 activation, although independently of its ligand, VEGF-C, in the uterus during pregnancy. Int J Mol Med2017;40:1210–1216.
- 74. Galvagni F, Pennacchini S, Salameh A, Rocchigiani M, Neri F, Orlandini M, Petraglia F, Gotta S, Sardone GL, Matteucci G, Terstappen GC, Oliviero S. Endothelial cell adhesion to the extracellular matrix induces c-Src-dependent VEGFR-3 phosphorylation without the activation of the receptor intrinsic kinase activity. Circ Res2010;106:1839–1848.
- 75. Domigan CK, Ziyad S, Iruela-Arispe ML. Canonical and noncanonical vascular endothelial growth factor pathways: new developments in biology and signal transduction. Arterioscler Thromb Vasc Biol2015;35:30–39.
- 76. Mortimer PS, Rockson SG. New developments in clinical aspects of lymphatic disease. J Clin Invest2014;124:915–921.
- 77. Kazenwadel J, Harvey NL. Morphogenesis of the lymphatic vasculature: a focus on new progenitors and cellular mechanisms important for constructing lymphatic vessels. Dev Dyn2016;245:209–219.
- 78. Wigle JT, Oliver G. Prox1 function is required for the development of the murine lymphatic system. Cell1999;98:769–778.
- 79. Escobedo N, Oliver G. Lymphangiogenesis: origin, specification, and cell fate determination. Annu Rev Cell Dev Biol2016;32:677–691.
- 80. Card CM, Yu SS, Swartz MA. Emerging roles of lymphatic endothelium in regulating adaptive immunity. J Clin Invest2014;124:943–952.
- 81. Hokkanen K, Tirronen A, Yla-Herttuala S. Intestinal lymphatic vessels and their role in chylomicron absorption and lipid homeostasis. Curr Opin Lipidol2019;30:370–376.
- 82. Cifarelli V, Eichmann A. The intestinal lymphatic system: functions and metabolic implications. Cell Mol Gastroenterol Hepatol2019;7:503–513.
- 83. Coso S, Bovay E, Petrova TV. Pressing the right buttons: signaling in lymphangiogenesis. Blood2014;123:2614–2624.
- 84. Wong BW, Zecchin A, Garcia-Caballero M, Carmeliet P. Emerging concepts in organ-specific lymphatic vessels and metabolic regulation of lymphatic development. Dev Cell2018;45:289–301.
- 85. Browse NL, Stewart G. Lymphoedema: pathophysiology and classification. J Cardiovasc Surg (Torino)1985;26:91–106. [CVOCROSSCVO]
- 86. McGuinness CL, Burnand KG. Lymphoedema. Trop Doct2001;31:2–7.
- 87. Connell FC, Gordon K, Brice G, Keeley V, Jeffery S, Mortimer PS, Mansour S, Ostergaard P. The classification and diagnostic algorithm for primary lymphatic dysplasia: an update from 2010 to include molecular findings. Clin Genet2013;84:303–314.
- 88. Brice GW, Mansour S, Ostergaard P, Connell F, Jeffery S, Mortimer P. Milroy disease. In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (eds). Genereviews((r)). Seattle (WA): University of Washington; 1993–2020. http://www.genereviews.org/.
- 89. Ferrell RE, Levinson KL, Esman JH, Kimak MA, Lawrence EC, Barmada MM, Finegold DN. Hereditary lymphedema: evidence for linkage and genetic heterogeneity. Hum Mol Genet1998;7:2073–2078.
- 90. Connell FC, Ostergaard P, Carver C, Brice G, Williams N, Mansour S, Mortimer PS, Jeffery S; Lymphoedema Consortium. Analysis of the coding regions of VEGFR3 and VEGFC in Milroy disease and other primary lymphoedemas. Hum Genet2009;124:625–631.
- 91. Gordon K, Spiden SL, Connell FC, Brice G, Cottrell S, Short J, Taylor R, Jeffery S, Mortimer PS, Mansour S, Ostergaard P. FLT4/VEGFR3 and Milroy disease: novel mutations, a review of published variants and database update. Hum Mutat2013;34:23–31.
- 92. Verstraeten VL, Holnthoner W, van Steensel MA, Veraart JC, Bladergroen RS, Heckman CA, Keskitalo S, Frank J, Alitalo K, van Geel M, Steijlen PM. Functional analysis of FLT4 mutations associated with Nonne-Milroy lymphedema. J Invest Dermatol2009;129:509–512.
- 93. Griffin HR, Hall DH, Topf A, Eden J, Stuart AG, Parsons J, Peart I, Deanfield JE, O'Sullivan J, Babu-Narayan SV, Gatzoulis MA, Bu'lock FA, Bhattacharya S, Bentham J, Farrall M, Granados Riveron J, Brook JD, Burn J, Cordell HJ, Goodship JA, Keavney B. Genetic variation in VEGF does not contribute significantly to the risk of congenital cardiovascular malformation. PLoS One2009;4:e4978.
- 94. Zhou F, Chang Z, Zhang L, Hong YK, Shen B, Wang B, Zhang F, Lu G, Tvorogov D, Alitalo K, Hemmings BA, Yang Z, He Y. Akt/protein kinase B is required for lymphatic network formation, remodeling, and valve development. Am J Pathol2010;177:2124–2133.
- 95. Ichise T, Yoshida N, Ichise H. H-, N- and Kras cooperatively regulate lymphatic vessel growth by modulating VEGFR3 expression in lymphatic endothelial cells in mice. Development2010;137:1003–1013.
- 96. Karkkainen MJ, Ferrell RE, Lawrence EC, Kimak MA, Levinson KL, McTigue MA, Alitalo K, Finegold DN. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat Genet2000;25:153–159.
- 97. Streeter GL. Developmental Horizons in Human Embryos. Washington: Department of Embryology of the Carnegie Institution of Washington; 1948.
- 98. Munoz-Chapuli R, Perez-Pomares JM. Cardiogenesis: an embryological perspective. J Cardiovasc Transl Res2010;3:37–48.
- 99. Gittenberger-de Groot AC, Bartelings MM, Deruiter MC, Poelmann RE. Basics of cardiac development for the understanding of congenital heart malformations. Pediatr Res2005;57:169–176.
- 100. Van Vliet P, Wu SM, Zaffran S, Puceat M. Early cardiac development: a view from stem cells to embryos. Cardiovasc Res2012;96:352–362.
- 101. Brade T, Pane LS, Moretti A, Chien KR, Laugwitz KL. Embryonic heart progenitors and cardiogenesis. Cold Spring Harb Perspect Med2013;3:a013847.
- 102. Brakenhielm E, Alitalo K. Cardiac lymphatics in health and disease. Nat Rev Cardiol2019;16:56–68.
- 103. Bradham RR, Parker EF. The cardiac lymphatics. Ann Thorac Surg1973;15:526–535.
- 104. Ratajska A, Gula G, Flaht-Zabost A, Czarnowska E, Ciszek B, Jankowska-Steifer E, Niderla-Bielinska J, Radomska-Lesniewska D. Comparative and developmental anatomy of cardiac lymphatics. Sci World J2014;2014:183170.
- 105. Karunamuni G, Yang K, Doughman YQ, Wikenheiser J, Bader D, Barnett J, Austin A, Parsons-Wingerter P, Watanabe M. Expression of lymphatic markers during avian and mouse cardiogenesis. Anat Rec2010;293:259–270.
- 106. Flaht-Zabost A, Gula G, Ciszek B, Czarnowska E, Jankowska-Steifer E, Madej M, Niderla-Bielińska J, Radomska-Leśniewska D, Ratajska A. Cardiac mouse lymphatics: developmental and anatomical update. Anat Rec2014;297:1115–1130.
- 107. Vuorio T, Ylä-Herttuala E, Laakkonen JP, Laidinen S, Liimatainen T, Ylä-Herttuala S. Downregulation of VEGFR3 signaling alters cardiac lymphatic vessel organization and leads to a higher mortality after acute myocardial infarction. Sci Rep2018;8:16709.
- 108. Kholová I, Dragneva G, Čermáková P, Laidinen S, Kaskenpää N, Hazes T, Čermáková E, Šteiner I, Ylä-Herttuala S. Lymphatic vasculature is increased in heart valves, ischaemic and inflamed hearts and in cholesterol-rich and calcified atherosclerotic lesions. Eur J Clin Invest2011;41:487–497.
- 109. Telinius N, Hjortdal VE. Role of the lymphatic vasculature in cardiovascular medicine. Heart2019;105:1777–1784.
- 110. Ferencz C, Rubin JD, McCarter RJ, Brenner JI, Neill CA, Perry LW, Hepner SI, Downing JW. Congenital heart disease: prevalence at livebirth. The Baltimore-Washington Infant Study. Am J Epidemiol1985;121:31–36.
- 111. Bailliard F, Anderson RH. Tetralogy of fallot. Orphanet J Rare Dis2009;4:2.
- 112. Shinebourne EA, Babu-Narayan SV, Carvalho JS. Tetralogy of fallot: from fetus to adult. Heart2006;92:1353–1359.
- 113. Cuypers JA, Menting ME, Konings EE, Opic P, Utens EM, Helbing WA, Witsenburg M, van den Bosch AE, Ouhlous M, van Domburg RT, Rizopoulos D, Meijboom FJ, Boersma E, Bogers AJ, Roos-Hesselink JW. Unnatural history of tetralogy of fallot: prospective follow-up of 40 years after surgical correction. Circulation2014;130:1944–1953.
- 114. Baldini A. Digeorge's syndrome: a gene at last. Lancet2003;362:1342–1343.
- 115. Chen L, Fulcoli FG, Tang S, Baldini A. Tbx1 regulates proliferation and differentiation of multipotent heart progenitors. Circ Res2009;105:842–851.
- 116. Homsy J, Zaidi S, Shen YF, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, Jin SC, Deanfield J, Giardini A, Porter GA, Kim R, Bilguvar K, Lopez-Giraldez F, Tikhonova I, Mane S, Romano-Adesman A, Qi HJ, Vardarajan B, Ma LJ, Daly M, Roberts AE, Russell MW, Mital S, Newburger JW, Gaynor JW, Breitbart RE, Iossifov I, Ronemus M, Sanders SJ, Kaltman JR, Seidman JG, Brueckner M, Gelb BD, Goldmuntz E, Lifton RP, Seidman CE, Chung WK. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science2015;350:1262–1266.
- 117. Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, Zeng X, Qi H, Chang W, Sierant MC, Hung WC, Haider S, Zhang J, Knight J, Bjornson RD, Castaldi C, Tikhonoa IR, Bilguvar K, Mane SM, Sanders SJ, Mital S, Russell MW, Gaynor JW, Deanfield J, Giardini A, Porter GA Jr, Srivastava D, Lo CW, Shen Y, Watkins WS, Yandell M, Yost HJ, Tristani-Firouzi M, Newburger JW, Roberts AE, Kim R, Zhao H, Kaltman JR, Goldmuntz E, Chung WK, Seidman JG, Gelb BD, Seidman CE, Lifton RP, Brueckner M. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet2017;49:1593–1601.
- 118. Szot JO, Cuny H, Blue GM, Humphreys DT, Ip E, Harrison K, Sholler GF, Giannoulatou E, Leo P, Duncan EL, Sparrow DB, Ho JWK, Graham RM, Pachter N, Chapman G, Winlaw DS, Dunwoodie SL. A screening approach to identify clinically actionable variants causing congenital heart disease in exome data. Circ Genom Precis Med2018;11:e001978.
- 119. Reuter MS, Jobling R, Chaturvedi RR, Manshaei R, Costain G, Heung T, Curtis M, Hosseini SM, Liston E, Lowther C, Oechslin E, Sticht H, Thiruvahindrapuram B, Mil SV, Wald RM, Walker S, Marshall CR, Silversides CK, Scherer SW, Kim RH, Bassett AS. Haploinsufficiency of vascular endothelial growth factor related signaling genes is associated with tetralogy of fallot. Genet Med2019;21:1001–1007.
- 120. Sevim Bayrak C, Zhang P, Tristani-Firouzi M, Gelb BD, Itan Y. De novo variants in exomes of congenital heart disease patients identify risk genes and pathways. Genome Med2020;12:9.
- 121. Soemedi R, Wilson IJ, Bentham J, Darlay R, Töpf A, Zelenika D, Cosgrove C, Setchfield K, Thornborough C, Granados-Riveron J, Blue GM, Breckpot J, Hellens S, Zwolinkski S, Glen E, Mamasoula C, Rahman TJ, Hall D, Rauch A, Devriendt K, Gewillig M, O’ Sullivan J, Winlaw DS, Bu’Lock F, Brook JD, Bhattacharya S, Lathrop M, Santibanez-Koref M, Cordell HJ, Goodship JA, Keavney BD. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am J Hum Genet2012;91:489–501.
- 122. Xie HM, Werner P, Stambolian D, Bailey-Wilson JE, Hakonarson H, White PS, Taylor DM, Goldmuntz E. Rare copy number variants in patients with congenital conotruncal heart defects. Birth Defects Res2017;109:271–295.
- 123. Pitt DB. A family study of fallots tetrad. Australas Ann Med1962;11:179–183.
- 124. Madonna R, De Caterina RV. Receptor switching in heart development and disease. Cardiovasc Res2009;84:4–6.
- 125. Morgenthau A, Frishman WH. Genetic origins of tetralogy of fallot. Cardiol Rev2018;26:86–92.
- 126. Wang W, Xu A, Xu H. The roles of vascular endothelial growth factor gene polymorphisms in congenital heart diseases: a meta-analysis. Growth Factors2018;36:232–238.
- 127. van den Akker NM, Molin DG, Peters PP, Maas S, Wisse LJ, van Brempt R, van Munsteren CJ, Bartelings MM, Poelmann RE, Carmeliet P, Gittenberger-de Groot AC. Tetralogy of fallot and alterations in vascular endothelial growth factor—a signaling and notch signaling in mouse embryos solely expressing the VEGF120 isoform. Circ Res2007;100:842–849.
- 128. Peters TH, Sharma V, Yilmaz E, Mooi WJ, Bogers AJ, Sharma HS. DNA microarray and quantitative analysis reveal enhanced myocardial VEGF expression with stunted angiogenesis in human tetralogy of fallot. Cell Biochem Biophys2013;67:305–316.
- 129. Petrova TV, Karpanen T, Norrmen C, Mellor R, Tamakoshi T, Finegold D, Ferrell R, Kerjaschki D, Mortimer P, Yla-Herttuala S, Miura N, Alitalo K. Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat Med2004;10:974–981.
- 130. Klotz L, Norman S, Vieira JM, Masters M, Rohling M, Dube KN, Bollini S, Matsuzaki F, Carr CA, Riley PR. Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature2015;522:62–67.
- 131. Chen L, Mupo A, Huynh T, Cioffi S, Woods M, Jin CL, McKeehan W, Thompson-Snipes L, Baldini A, Illingworth E. Tbx1 regulates vegfr3 and is required for lymphatic vessel development. J Cell Biol2010;189:417–424.
- 132. Martucciello S, Turturo MG, Cioffi S, Chen L, Baldini A, Illingworth E. A dual role for Tbx1 in cardiac lymphangiogenesis through genetic interaction with Vegfr3. FASEB J;doi: 10.1096/fj.201902202R.
- 133. Maruyama K, Miyagawa-Tomita S, Mizukami K, Matsuzaki F, Kurihara H. Isl1-expressing non-venous cell lineage contributes to cardiac lymphatic vessel development. Dev Biol2019;452:134–143.
- 134. VanDusen NJ, Casanovas J, Vincentz JW, Firulli BA, Osterwalder M, Lopez-Rios J, Zeller R, Zhou B, Grego-Bessa J, De La Pompa JL, Shou W, Firulli AB. Hand2 is an essential regulator for two notch-dependent functions within the embryonic endocardium. Cell Rep2014;9:2071–2083.
- 135. Dumont DJ, Jussila L, Taipale J, Lymboussaki A, Mustonen T, Pajusola K, Breitman M, Alitalo K. Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science1998;282:946–949.
- 136. Hagerling R, Pollmann C, Andreas M, Schmidt C, Nurmi H, Adams RH, Alitalo K, Andresen V, Schulte-Merker S, Kiefer F. A novel multistep mechanism for initial lymphangiogenesis in mouse embryos based on ultramicroscopy. EMBO J2013;32:629–644.
- 137. Haiko P, Makinen T, Keskitalo S, Taipale J, Karkkainen MJ, Baldwin ME, Stacker SA, Achen MG, Alitalo K. Deletion of vascular endothelial growth factor C (VEGF-C) and VEGF-D is not equivalent to VEGF receptor 3 deletion in mouse embryos. Mol Cell Biol2008;28:4843–4850.
- 138. Lyon MF, Glenister PH, Loutit JF, Evans EP, Peters J. A presumed deletion covering the W and Ph loci of the mouse. Genet Res1984;44:161–168.
- 139. Karkkainen MJ, Saaristo A, Jussila L, Karila KA, Lawrence EC, Pajusola K, Bueler H, Eichmann A, Kauppinen R, Kettunen MI, Yla-Herttuala S, Finegold DN, Ferrell RE, Alitalo KA. Model for gene therapy of human hereditary lymphedema. Proc Natl Acad Sci USA2001;98:12677–12682.
- 140. Zhang L, Zhou F, Han W, Shen B, Luo J, Shibuya M, He Y. VEGFR-3 ligand-binding and kinase activity are required for lymphangiogenesis but not for angiogenesis. Cell Res2010;20:1319–1331.
- 141. Fontana F, Haack T, Reichenbach M, Knaus P, Puceat M, Abdelilah-Seyfried S. Antagonistic activities of Vegfr3/Flt4 and Notch1b Fine-tune mechanosensitive signaling during zebrafish cardiac valvulogenesis. Cell Rep2020;32:107883.
- 142. Ober EA, Olofsson B, Makinen T, Jin SW, Shoji W, Koh GY, Alitalo K, Stainier DY. VEGFC is required for vascular development and endoderm morphogenesis in zebrafish. EMBO Rep2004;5:78–84.
- 143. Apitz C, Webb GD, Redington AN. Tetralogy of fallot. Lancet2009;374:1462–1471.
- 144. Ruderfer DM, Hamamsy T, Lek M, Karczewski KJ, Kavanagh D, Samocha KE, Daly MJ, MacArthur DG, Fromer M, Purcell SM; Exome Aggregation Consortium. Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nat Genet2016;48:1107–1111.
- 145. Heinolainen K, Karaman S, D’Amico G, Tammela T, Sormunen R, Eklund L, Alitalo K, Zarkada G. VEGFR3 modulates vascular permeability by controlling VEGF/VEGFR2 signaling. Circ Res2017;120:1414–1425.
- 146. Kivela R, Hemanthakumar KA, Vaparanta K, Robciuc M, Izumiya Y, Kidoya H, Takakura N, Peng X, Sawyer DB, Elenius K, Walsh K, Alitalo K. Endothelial cells regulate physiological cardiomyocyte growth via VEGFR2-mediated paracrine signaling. Circulation2019;139:2570–2584.