Introduction
The recent success of clinical studies with high-dose omega-3 polyunsaturated fatty acids (ω3-PUFAs), eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and specifically EPA, has reignited interest in the potential mechanism of action of ω3-PUFA therapy in cardiovascular disease (CVD). Reporting in 2018, the Reduction of Cardiovascular Events with Icosapent Ethyl-Intervention Trial (REDUCE-IT) was the largest randomized trial to date to employ high-dose EPA (4 g/day). Results from REDUCE-IT indicated that EPA significantly reduced risk of ischaemic events, including death from cardiovascular causes, in patients with high triglycerides (TG) despite statin use. In addition, OMEGA-REMODEL, a smaller randomized trial that examined high-dose ω3-PUFAs (EPA + DHA, 4 g/day) on remodelling for 6 months post-myocardial infarction (MI), reported significant reductions in ventricular remodelling and inflammation. In stark contrast, the Vitamin D and Omega-3 Trial (VITAL), also reporting in 2018, found no improvement in primary cardiovascular outcomes with low-dose ω3-therapy (1 g/day). However, despite the positive outcomes from REDUCE-IT and OMEGA-REMODEL, there remains controversy regarding the efficacy of ω3-PUFAs in CVD. This controversy stems from a history of clinical trials with low-dose ω3-PUFAs that have failed to show efficacy. More recently, the STRENGTH trial using high-dose mixed ω3-PUFAs was stopped for futility. Additionally, there is ongoing debate regarding the mechanistic basis for the cardioprotective effects of ω3-PUFAs, which has also hampered their wider use in CVD. While there has long been interest in how the effects of ω3-PUFAs differ from ω6-PUFAs, recent results have highlighted the potential that EPA has unique benefits. Therefore, the goals of this review are to outline the mechanistic basis for EPA cardioprotective effects in CVD, and finally to suggest new directions in understanding EPA in CVD. ω3-polyunsaturated fatty acids have multiple biological activities including: (i) distinct physiochemical properties; (ii) as a precursor to oxylipins and other lipid mediators; and as direct receptor agonists at (iii) free fatty acid receptors (Ffars) or (iv) peroxisome proliferator-activated receptors (PPARs).
Distinct effects of eicosapentaenoic acid on membrane structure, lipid oxidation, biomarkers, plaque composition, and endothelial function
ω3-polyunsaturated fatty acids modify lipid composition of caveolae and cellular localization of nitric oxide synthase
Rather than diffusing in an unrestricted fashion, membrane constituents are organized into a complex ‘mosaic’ of specific lipids that create distinct environments or microdomains for various proteins involved in signal transduction and receptor binding., Such microdomains or ‘lipid rafts’ are typically detergent-resistant and enriched with unesterified cholesterol and sphingolipids. These sphingolipids contain saturated fatty acids (SFAs) that provide a highly ordered structure and sequester specific proteins. Proteins that preferentially localize to these domains have a certain tertiary structure that supports their function as an integral membrane protein. Lipid rafts move or ‘float’ as a coherent structural unit within the surrounding liquid-disordered lipid bilayer and can also cluster with other rafts to form larger platforms. The basis for differences in fluidity between rafts and the surrounding membrane is the differences in the degree of hydrocarbon chain saturation of constituent sphingolipids that associate strongly with cholesterol. Treatment of cells with ω3-PUFAs like EPA modifies the acyl chain composition of membrane phospholipids and thereby alters the properties and protein function of various lipid domains.
Caveolae are a lipid raft subtype that appear as microscopic, flask-shaped invaginations along the membrane surface and are commonly found in endothelial cells, adipocytes, and smooth muscle cells. The principal protein component of caveolae is caveolin, a scaffolding protein that binds cholesterol efficiently and interacts with various signalling macromolecules, including G proteins and calcium regulating proteins. Caveolin modulates endothelial nitric oxide synthase (eNOS) activity as it binds directly to the enzyme, thereby blocking access of the co-factor, calcium/calmodulin. The expression of caveolin is markedly elevated under conditions of hypercholesterolaemia because of enrichment of plasma membrane cholesterol levels, resulting in reduced eNOS activation in a manner that can be reversed with statin treatment. In cultured human endothelial cells, EPA treatment modifies the subcellular distribution of eNOS and dramatically changes the lipid composition of caveolae. As a result of its incorporation into phospholipid acyl chains, EPA changes the fluid dynamics of the caveolae by increasing acyl chain unsaturation. Eicosapentaenoic acid treatment also displaces caveolin-1 from caveolae and causes the translocation of eNOS from caveolae fractions to soluble fractions where it can be activated. Finally, EPA changes the overall distribution of caveolin-1 and eNOS in plasma membranes.
Distinct membrane interactions of eicosapentaenoic acid and docosahexaenoic acid
ω3-polyunsaturated fatty acids and their bioactive lipid metabolites incorporate into cellular membranes and lipoproteins associated with vascular tissues, along with the atherosclerotic plaque, where they interfere with inflammation, oxidative stress, and endothelial dysfunction. Treatment with EPA, at pharmacologic doses, affects lipid oxidation rates, membrane organization, and signal transduction. Eicosapentaenoic acid and DHA have disparate effects on membrane lipid dynamics due to differences in their carbon length and number of double bonds. Eicosapentaenoic acid inserts into lipoprotein particles and cellular membranes where it scavenges free radicals through electron stabilization mechanisms associated with its multiple double bonds (Figure 1). The antioxidant effects of EPA were not reproduced with DHA over time or other FDA-approved TG-lowering agents such as gemfibrozil, as well as fenofibric and nicotinic acids.,, The antioxidant effects of EPA are also observed in membranes under conditions of hyperglycaemia. Unlike EPA, other TG-lowering agents like the fibrates are relatively hydrophilic and do not possess chemical moieties capable of stabilizing unpaired free electrons in the membrane hydrocarbon core, while EPA has an extended and stable orientation in the membrane as determined by small-angle X-ray diffraction approaches. In contrast, DHA interacts with the phospholipid head group region with concomitant changes in hydrocarbon core electron density consistent with increased membrane disorder. These differences in membrane interactions with DHA are consistent with the rapid isomerization or conformational changes over a nanoscale time frame.,,,, In a more recent study, the antioxidant effects of EPA in small dense low density lipoprotein (sdLDL) and membranes were compared to other long-chain fatty acids. The results showed that hydrocarbon length and number of double bonds for fatty acids are important predictors of antioxidant activity in lipoproteins and membranes, along with membrane cholesterol domain formation. Eicosapentaenoic acid appears to have the optimal combination of chain length and unsaturation for these antioxidant properties, followed by other ω3-PUFAs. Fatty acids with two or fewer double bonds and the ω6-PUFA arachidonic acid (AA) failed to exhibit antioxidant activity as compared to ω3-PUFAs.
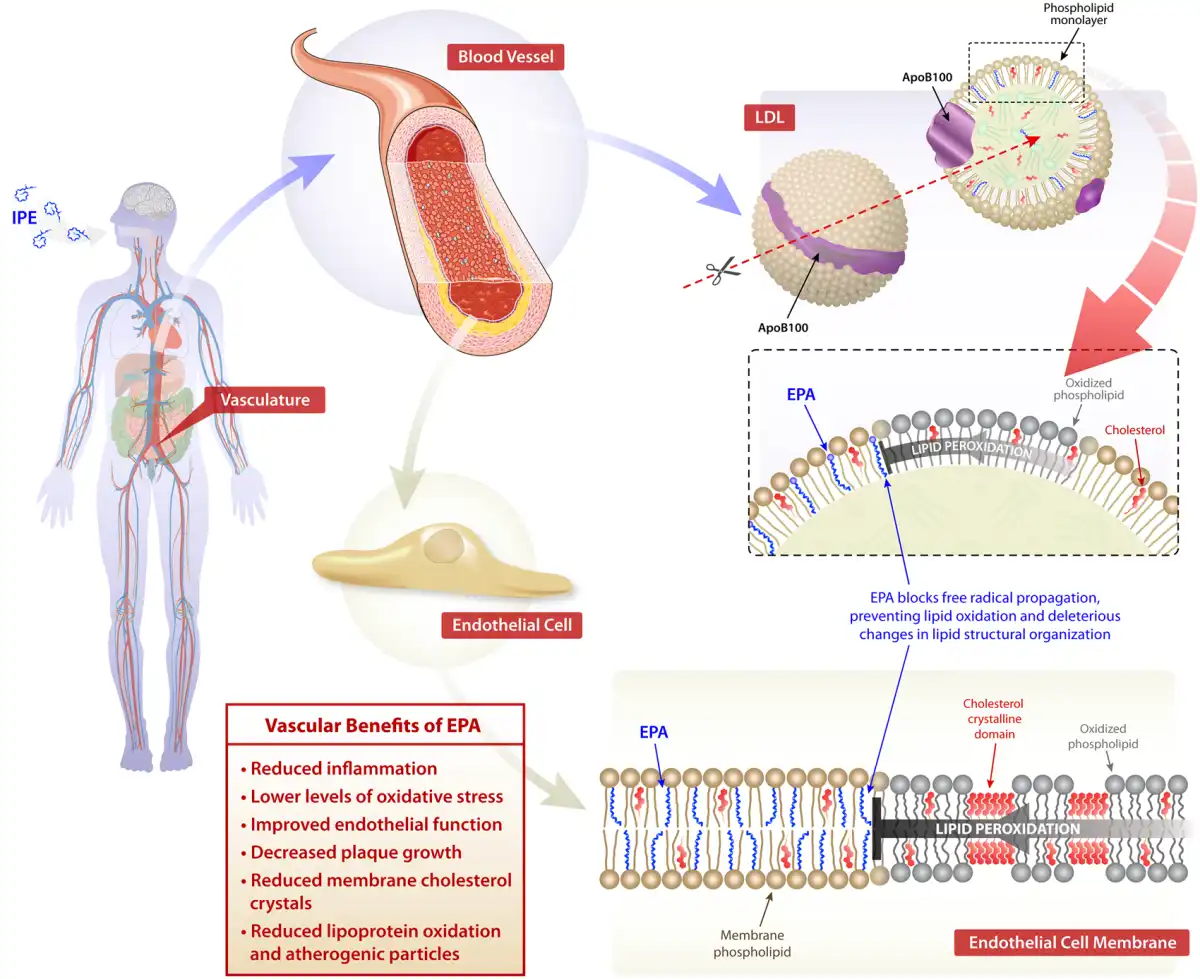
Figure 1
Molecular lipid interactions of eicosapentaenoic acid. Schematic illustration of the proposed location of phospholipid-linked eicosapentaenoic acid in cellular membranes and lipoproteins. Eicosapentaenoic acid is rapidly esterified and incorporated into lipoproteins and membrane phospholipids. Biophysical studies indicate eicosapentaenoic acid has an extended orientation that preserves membrane fluidity and inhibition of lipid oxidation and membrane cholesterol domain formation.
Effects of ω3-polyunsaturated fatty acids on membrane cholesterol crystal formation
A characteristic feature of an atherosclerotic plaque is the formation of cholesterol crystals that originate from cholesterol-enriched membranes of various cells, including macrophages and smooth muscle cells., Over time, these toxic cholesterol crystals destabilize the plaque as they continue to form or precipitate from membrane domains. Using various imaging approaches, cholesterol crystals have been observed in vascular tissue where they associated with the plaque’s fibrous cap. In patients that have experienced myocardial infarction, there is abundant evidence of cholesterol crystals in the diseased plaque in a proportional manner. In addition to excess cholesterol accumulation, membrane cholesterol domains are also stimulated by oxidative stress and high glucose. Along with oxidized LDL, cholesterol crystals activate nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3) inflammasomes, which regulates the processing of pro-interleukin 1 beta (IL-1β) into an active cytokine.
Due to its potent antioxidant activity and lipophilicity, EPA significantly inhibits glucose-induced cholesterol crystalline domain formation in membrane lipid vesicles at pharmacologically relevant concentrations., The inhibition of cholesterol domain formation by EPA is not reproduced with other TG-lowering agents or vitamin E due to differences in membrane distribution and antioxidant activities. These findings suggest that EPA preferentially intercalates into the hydrocarbon core of the membrane bilayer where it can trap free radicals with its multiple double bonds, leading to reduced cholesterol domain formation. The effects of EPA on cholesterol domain formation was not observed with DHA as it has different interactions with the highly planar steroid ring structure of cholesterol that is oriented parallel to surrounding phospholipid acyl chains.,,
Effects of ω3-polyunsaturated fatty acids on cardiovascular biomarkers, plaque biology, and high density lipoprotein (HDL) function
Eicosapentaenoic acid and other ω3-PUFAs have been shown to favourably influence circulating levels of biomarkers associated with cardiovascular risk in patients with dyslipidaemia. Prescription EPA (2–4 g/day) in patients with high or very high triglyceride (TG) levels reduces high-sensitivity C-reactive protein (hsCRP), lipoprotein-associated phospholipase A2 (Lp-PLA2), and oxidized LDL-cholesterol (oxLDL) levels, as well as the arachidonic acid (AA)-to-EPA ratio, as compared to placebo. The ability of EPA to reduce hsCRP is actually enhanced in combination with a statin in proportion to its intensity. Similar effects on hsCRP were not observed with DHA in a similar patient population even with 4 g/day but, like EPA, treatment was associated with reduced Apolipoprotein CIII. Eicosapentaenoic acid treatment also down-regulates cAMP responsive element protein 1 (CREB1) and hypoxia inducible factor 1 (HIF1) gene expression. In cellular studies, EPA reduces production of inflammatory mediators in macrophages, including TNF-α and IL-1β, compared to DHA.
Eicosapentaenoic acid also has direct vascular effects that impact the progression of atherosclerosis as compared to other ω3-PUFAs. In mice that lack apolipoprotein E (Apoe−/−) fed a Western diet supplemented with EPA (1%, w/w) or DHA (1%, w/w) for 3 weeks, EPA treatment reduces plaque volume as compared to DHA. Eicosapentaenoic acid and its metabolites, especially 12-hydroxyeicospentaenoic acid (12-HEPE), preferentially associate with thin-cap plaques and accumulation of anti-inflammatory M2 macrophages, while DHA associates with plaques of various sizes. In the aortic root, total EPA and 12-HEPE levels follow a concentration gradient from the vascular endothelium to the media. In patients with coronary artery disease (CAD), treatment with EPA improves high-density lipoprotein mediated cholesterol efflux along with antioxidant and anti-inflammatory effects. In human endothelial cells, EPA-enriched HDL increases resolvin E3 production while reducing cytokine-stimulated vascular cell adhesion molecule 1 (VCAM-1) expression. As observed in Apo-B containing particles, the lipophilic properties and molecular dimensions of EPA allow it to insert more efficiently into the HDL particle, with improved antioxidant function, as compared to DHA.
Effects of ω3-polyunsaturated fatty acids and statins on endothelial function
Atherosclerosis is causally related to endothelial cell (EC) dysfunction characterized by a loss of normal nitric oxide (NO) bioavailability that results in abnormal vasodilation and plaque development. In isolated tissue, EPA reverses endothelial dysfunction triggered by either oxLDL or high glucose in a manner that is enhanced in combination with a statin. This improvement in function is evidenced by pronounced increases in the release ratio of NO to peroxynitrite (ONOO–) in the endothelial cells. The combined benefit of EPA with a statin on endothelial function and NO synthase activity was not reproduced with DHA under identical conditions. By reducing LDL oxidation, EPA also attenuated the effects of oxLDL on endothelial function unlike DHA over the same time course. Similar effects of EPA vs. DHA were seen ex vivo in arterial segments from a rodent model.
The endothelial benefits of EPA are not caused by differences in expression of endothelial nitric oxide synthase (eNOS) but are likely due to improved eNOS coupling. In the absence of eNOS coupling, excessive rather than NO is generated by eNOS, which leads to increased levels of highly reactive ONOO– molecules that cause cellular and lipoprotein oxidation (oxLDL). EPA treatment of human ECs, along with a statin, reduces the effects of oxLDL on endothelial dysfunction and preserves NO bioavailability. The favourable effects of EPA and the statin may be due to shared antioxidant properties and a common molecular distribution in cellular membranes. There are also benefits of EPA in an ex vivo rodent system. The combined treatment of EPA and a statin produces an additional improvement with respect to NO bioavailability following exposure to hyperglycaemia.
Summary
Based on its unique molecular structure, EPA has several significant effects on cellular and lipoprotein membrane structure and dynamics, including:
Eicosapentaenoic acid incorporated in phospholipid acyl chains modifies membrane dynamics of caveolae (lipid rafts), displaces caveolin-1, the structural backbone of caveolae, and displaces signalling proteins localized to caveolae, like eNOS, allowing for increased activation.
Eicosapentaenoic acid inserts into lipoprotein particles and cellular membranes where it scavenges free radicals through electron stabilization due to its multiple double bonds. Eicosapentaenoic acid, as opposed to DHA, appears to have the optimal combination of chain length and unsaturation for these antioxidant properties.
Eicosapentaenoic acid, as opposed to DHA, significantly inhibits glucose-induced cholesterol crystalline domain formation in membrane lipid vesicles, which is not reproduced with other TG-lowering agents.
Eicosapentaenoic acid and other ω3-PUFAs reduce circulating levels of biomarkers associated with cardiovascular risk in patients with dyslipidaemia.
Eicosapentaenoic acid, in combination with statins, reverses endothelial dysfunction as evidenced by increased nitric oxide bioavailability due to shared antioxidant properties and a common molecular distribution in cellular membranes.
Oxylipins as signalling metabolites derived from eicosapentaenoic acid
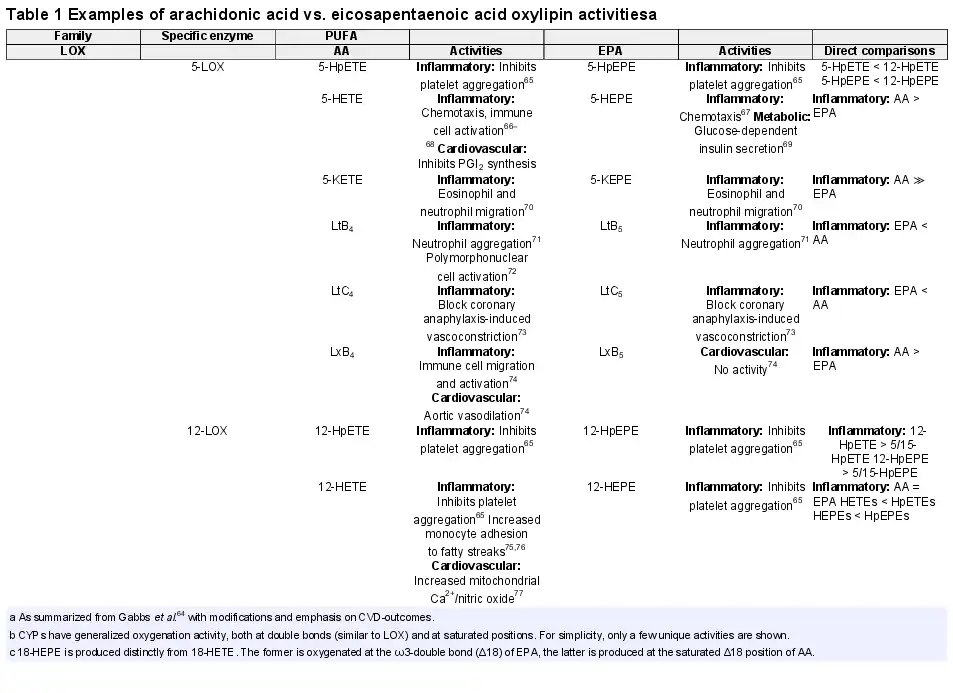
Oxylipins are the subclass of lipid mediators derived by enzymatic oxygenation of PUFAs that have specific biological activities. At the molecular level, PUFAs serve as a reservoir of precursors from which a broad array of lipid mediators are synthesized through oxygenation, amidation, nitrosylation, and more. Oxylipins are produced by enzymes in multiple signalling pathways, including cyclooxygenase (COX) and lipoxygenase families (5-LOX, 12-LOX, and 15-LOX), as well as a broad cross section of cytochrome p450s with epoxygenase (CYPepox), hydroxylase (CYPhx), or ω/ω-1 hydroxylase (CYPω-hx) activities. In many cases, CYPs exhibit these activities simultaneously. While COX enzymes are selective for arachidonic acid (AA), LOX, and CYP have activity on multiple PUFAs including AA and EPA and the pleiotropic activity forms complex webs of overlapping signalling pathways. Table 1 summarizes the most common AA and EPA oxylipins in LOX and CYP signalling. The summary exemplifies the complexity of signalling matrices generated because: (i) the same signalling molecule can be produced from distinct enzymes—e.g. 12-HEPE is produced by 12-LOX and by CYPhx; and (ii) the same enzymes can produce oxylipins from multiple PUFAs—e.g. 12-LOX produces 12-hydroxyeicotetraenoic (12-HETE) from AA and 12-HEPE from EPA.
Our understanding of the initiation of oxylipin synthesis and generation of signalling activity is guided by the common model depicted in Figure 2. Precursor PUFAs stored in membrane phospholipids are made available to enzymes by the activity of phospholipase A2 (PLA2), which releases PUFAs from the sn-2 position into the cytosol where COX, LOX, and CYP divert them into each pathway. Over 70 metabolites of AA are known to exist, and since EPA has an additional double bond (five compared to four in AA), even more metabolites of EPA are likely to exist, and almost 50 have been described to date.
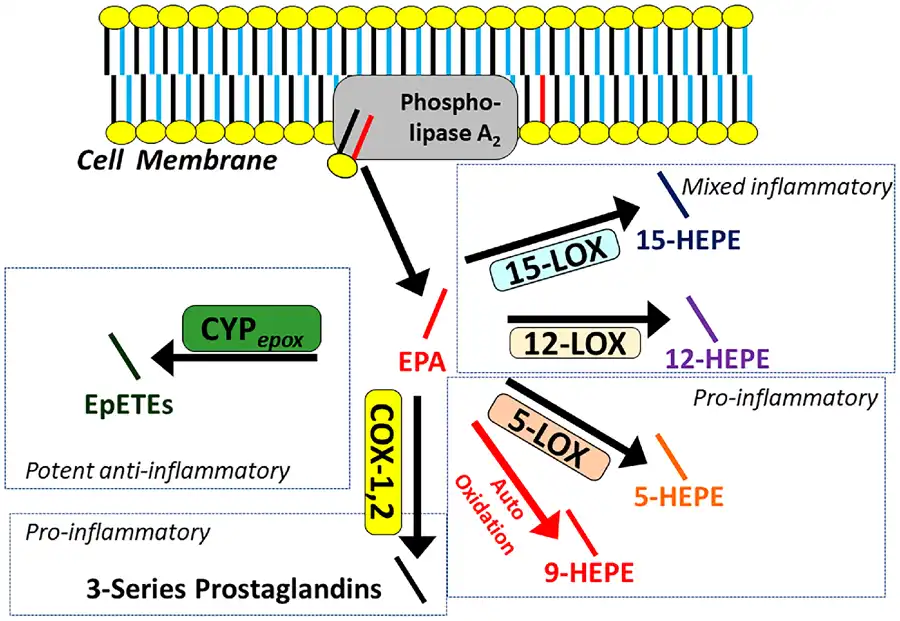
Figure 2
The common model of oxylipin production by oxylipin producing enzymes, including various lipoxygenase, cyclooxygenases, and cytochrome p450 epoxygenases. Polyunsaturated fatty acids are released by phospholipase A2 from the membrane, making them available to enzymes for downstream signalling by oxylipins. Eicosapentaenoic acid is depicted in red, other polyunsaturated fatty acids are depicted in blue. The relative production of eicosapentaenoic acid-derived oxylipins is a function of the relative provision of eicosapentaenoic acid to enzymes by phospholipase A2, which in turn, reflects their relative abundance in the phospholipid membrane. Generalized pathway activities are summarized. For details, see Table 1. COX, cyclooxygenase; CYPepox, cytochrome P450 expoxygenases; LOX, lipoxygenase.
Eicosapentaenoic acid administration changes the amount and types of oxylipins in plasma
Pharmaceutical EPA as icosapent ethyl at 4 g/day increases membrane EPA content by nearly 8-fold but reduces membrane AA content by 27%. 1.86 g/day EPA as omega-3 acid ethyl esters (in conjunction with 1.5 g/day DHA) not only induces a 540% increase in %EPA but also induces an 11% decrease in %AA. This shift in membrane substrate availability is reflected in treatment-dependent changes in tissue oxylipins, with a general pattern of increasing EPA membrane content, at the expense of AA. Like fatty acids, oxylipins are distributed through plasma and the majority circulate esterified in the glycerolipids of lipoproteins (VLDL, LDL, and HDL). A small portion (<5%) circulate unesterified and bound to albumin in a manner similar to non-esterified fatty acids (often termed ‘free’ oxylipins, which are most often reported). As a general rule, treatments that increase tissue EPA result in concurrent increases in total (esterified + unesterified) plasma alcohols and epoxides derived from EPA: 4 weeks of 3.4 g/day omega-3 acid ethyl esters induced an 8.4-fold increase in plasma %EPA and concurrently increased plasma esterified HEPEs by 5.7-fold, epoxytetraenoic acids (EpETEs) by 4.7-fold, and dihydroxyeicostetraenoic acids (DiHETEs) by 4.1-fold. Treatment reduced %AA to 0.88-fold of baseline and similarly reduced oxylipins derived from AA: HETEs to 0.8-fold of baseline and dihydroxytrienoic acids (DiHETrEs) to 0.81-fold of baseline; epoxyeicosatrienoic acids (EpETrEs) were not significantly reduced, but the data were most consistent with a reduction to 0.9-fold of baseline. For both EPA- and AA-oxylipins, changes in plasma concentration were reflective of the abundance their PUFA precursors. Eicosapentaenoic acid treatment is not only effective at increasing steady-state abundance of EPA oxylipins, but it also decreases the abundance of AA oxylipins in proportion to decreases in AA availability. However, a more detailed examination in the randomized clinical trial (RCT) setting reveals pool-specific changes. In subjects with metabolic syndrome responding to oral EPA (combined with DHA) at 3.4 g/day ω3-acid-ethyl esters, the reduction in AA-oxylipins was restricted to two HETEs, oxoeicosatetraenoic acids (KETEs), and leukotriene B4 (LTB4) derivatives in LDL and HDL respectively, but the same VLDL oxylipins were unchanged. In addition, AA-oxylipins including EpETrEs and DiHETrEs were unchanged for all lipoproteins, indicating that after treatment CYPepox signalling is sustained but LOX and CYPhx signalling are limited by AA.
Eicosapentaenoic acid oxylipins share ligand activity with oxylipins from other polyunsaturated fatty acids
In addition to enzymes that can produce oxylipins from multiple PUFAs, oxylipins also have overlapping activities as receptor ligands, and the classic model of a unique ligand-receptor pair does not hold. This expectation follows from Burr’s original findings that linoleic acid and alpha-linolenic acid are both effective in rescuing rats with essential fatty acid deficiency, which was explained in 1930 as complementary PUFA activities. Unfortunately, few studies systematically compare the bioactivity of ω6 and ω3 homologs. AA and EPA are both effective substrates for CYPepox activity, producing EpETrEs and EpETEs from AA and EPA, respectively. Both series potently induce dilation of rat coronary microvessels by activation of BKCa channels. EpETEs appear to be moderately more potent than their EpETrE homologs, but all homologs bind and activate the channel. Hence, the most likely benefit of EPA is to add the benefit of EpETEs to that of EpETrEs. Additive instances are common. Among 12-LOX pathway metabolites, 12-hydroperoxides (12-HpETE for AA, 12-HpEPE for EPA) equivalently suppress collagen-mediated platelet aggregation and serotonin release.
Instances in which the effect of EPA-oxylipins are different from the AA-homolog are also reported, with examples both where the EPA-oxylipin is more potent than the AA-homolog, and also where it is less potent. Among 5-LOX pathway metabolites, the EPA-derived 5-KEPE is 10-fold less potent than the AA-derived 5-KETE in mobilizing cytosolic Ca2+ in human neutrophils, and it is less potent in stimulating neutrophil or eosinophil migration. Another EPA-oxylipin derived from 5-LOX (5-HEPE) acts on GPR119 to produce an insulinogenic effect but the AA homolog (5-HETE) has only half the effect. Hence, different GPR119-dependent insulinogenic responses among individuals could potentially be explained by availability of cell membrane AA and EPA, independent of genetic polymorphisms. By considering the interactions of PUFAs with each other, a better assessment of how the bioactivities of mixtures are established can be derived.
Finally, ω3-PUFAs can produce distinct oxylipins with no direct homolog. With EPA, this occurs because ω3-PUFAs have one more double bond compared to AA, and hence an additional point for oxygenation not available on AA. For mid-chain alcohols, this metabolite is 18-HEPE, which is a target of much investigative interest, in part because of its role as a precursor to specialized pro-resolving mediators such as Resolvin E1. Interestingly, 18-HEPE protects against pressure overload-induced heart failure. For fatty acid epoxides, the unique metabolite is 17(18)-EpETE, and it is thought to have potent anti-inflammatory activity and to direct macrophage polarization towards the M2 phenotype.
Finally, pathway diversion is a potential mechanism. The phenomenon has been demonstrated in the context of heart failure, where activation of mitochondrial iPLA2γ diverts oxylipin production away from epoxides (EpETrEs) towards mid-chain alcohols (HETEs) which promotes pathology by inducing necrosis and apoptosis, and such a shift could amplify cases where EPA is a neutral antagonist to an oxylipin producing enzyme as is the case with COX.
Summary
There are at least four ways increasing EPA availability can alter the oxylipin signalling pathways and affect downstream physiologic function:
Eicosapentaenoic acid-oxylipins can add to the activity of AA-oxylipins.
Eicosapentaenoic acid-oxylipins can have effects distinct from their AA-homologs.
Eicosapentaenoic acid-oxylipins can neutralize the activity of their AA-homologs.
Eicosapentaenoic acid could divert activity from one oxylipin pathway to another.
As the results from STRENGTH are clarified, if evidence for differences between EPA and DHA emerges, the same molecular rationale that accounts for differences between AA and EPA can similarly explain differences between EPA and DHA oxylipins.
Free fatty acid receptors
While ω3-PUFA incorporation into cellular membranes has profound effects on membrane structure and the organization of membrane-associated signalling complexes, mechanistically, it fails to explain activation of intracellular signalling pathways associated with EPA-mediated cardioprotection. In the early 2000s, a subgroup of orphan receptors, G-protein coupled receptor 40 (GPR40), GPR43, GPR41, and GPR120, were defined as receptors for endogenous free fatty acids and were subsequently renamed free fatty acid receptor 1 (Ffar1), Ffar2, Ffar3, and Ffar4, respectively. Ffar2 and Ffar3 are receptors for short chain FA (≤8 carbons) and will not be discussed further, whereas Ffar1 and Ffar4 are receptors for medium and long-chain FA (≥10 carbons), including, but not limited to, ω3-PUFAs. The discovery of GPRs that bind to and are activated by endogenous free fatty acids establishes and entirely novel paradigm whereby fatty acids function as signalling molecules, not simply as an energy source.
Free fatty acid receptor 1 expression and physiology
In 2003, Ffar1 was the first identified receptor for free FAs and is activated by medium and long-chain FAs. It is located at chromosome 19q13.1 in humans and is expressed as a single-gene product. Ffar1 is highly expressed in the pancreas, GI-tract, brain, and lung, with lower levels of expression in skeletal muscle, heart, liver, bone, and brain. In the pancreas, Ffar1 is highly expressed in the pancreatic islets, including β-cells, where long-chain PUFA stimulation enhances glucose-stimulated insulin secretion,, and α-cells, where it modulates glucagon release. In the gastrointestinal (GI) tract, Ffar1 is expressed in enteroendocrine cells and regulates the release of glucagon like peptide-1 (GLP-1) and peptide hormone YY (PYY) from intestinal L-cells, cholecystokinin (CCK) from intestinal I cells, and gastric inhibitory peptide (GIP) from intestinal K-cells. Additionally, Ffar1 and other Ffars play an important role producing anti-inflammatory signals in the gut to regulate metabolism. Ffar1 is also expressed in several brain regions including the hypothalamus, hippocampus, medulla oblongata, olfactory bulb, cerebellum, striatum, and cerebral cortex, and might have a role in neurodevelopment, inflammation and pain, and neurodegenerative diseases. Finally, Ffar1 is expressed at low levels in mouse heart, 30-fold lower than Ffar4, although relative expression levels of Ffar1 in human heart might be higher (unpublished observation). However, there are no studies of which we are aware that address Ffar1 function in the heart.
Free fatty acid receptor 4 expression and physiology
In 2005, Ffar4 was identified as another receptor for long-chain FAs. Ffar4, which shows little structural similarity to Ffar1, is located at chromosome 10q23.33 in humans, and two splice variants are expressed in humans, a short isoform (Ffar4S, analogous to rodent Ffar4) and long isoform (Ffar4L) that is unique to humans and differs from Ffar4S in a 16 amino acid insertion in the third intracellular loop., Ffar4 expression is detected in several tissues with similar expression patterns in mouse and human, with high levels of expression in lung and GI-tract, and lower levels of expression in other tissues including brain, pancreas, small intestine, adipose, taste buds, muscle, heart, and liver.,, Like Ffar1, Ffar4 is expressed in intestinal L-cells and regulates the release of glucagon like peptide-1 (GLP-1) in STC-1 mouse enteroendocrine cells in vitro and using α-linolenic acid infusion in rats in vivo. However, this was not confirmed in Ffar4 knockout mice, suggesting this effect might be mediated by Ffar1. In the pancreas, Ffar4 is expressed in pancreatic islet α cells where activation induces glucagon release, and ∂ cells where activation induces somatostatin release. Ffar4 might also be expressed in pancreatic, β cells, where Ffar4 was shown to induce insulin release BRINBD11 cells, isolated islets, and in vivo, although this was not observed in Ffar4 knockout mice. Ffar4 is also expressed in adipose tissue, and increases surface expression of GLUT4 to attenuate insulin resistance. Ffar4 is expressed in macrophages and induces a more anti-inflammatory phenotype, characterized by increased expression of anti-inflammatory genes including arginase-1, IL-10, MCL1, Ym-1, Clcc7a, and MMR, with decreased expression of pro-inflammatory genes including IL-6, TNFα, MCP-1, IL-1β, iNOS, adiponectin, and CD11c. Furthermore, activation of Ffar4 in macrophages reduced inflammation in adipose tissue and improved insulin sensitivity in mice. The combined insulin-sensitizing and anti-inflammatory effects of Ffar4 in pancreatic islet cells, adipocytes, and macrophages suggest Ffar4 as a potential therapeutic target in the management of type-II diabetes. Finally, Ffar4 is expressed in mouse heart at 30-fold higher levels than Ffar1, while Ffar2/3 were significantly lower levels. Further, Ffar4 is expressed in isolated mouse cardiac myocytes and fibroblasts. Because Ffar4 is the main Ffar expressed in heart, the remainder of the section will focus on Ffar4 in CVD.
Free fatty acid receptor 4 signalling
Although commonly referred to as a receptor for ω3-PUFAs, Ffar4 binds to and is activated by a wide array of saturated, monounsaturated, and polyunsaturated, medium and long-chain FAs with potencies in the low µM range, including ω3-PUFAs, α-linolenic acid (αLA), EPA, and DHA. However, SFAs tend to show lower efficacy for activation of downstream signalling pathways, suggesting a potential functional bias towards PUFAs that likely affects biological activity in vivo. Additionally, a novel class of branched chain fatty acids, fatty acid esters of hydroxy fatty acids (FAHFAs), were identified as potential ligands for Ffar4 and have anti-diabetic and anti-inflammatory properties.
Ffar4 signals through both Gαq and β-arrestin-2 (not β-arrestin-1), although there are cell-type specific effects and differences between the Ffar4S and Ffar4L isoforms.,, The original description of Ffar4 indicated that activation of the receptor increased intracellular Ca2+ and activation of ERK in STC-1 enteroendocrine tumour cells suggesting signalling through Gq/11, which was confirmed in subsequent studies., Alternatively, Ffar4 signals through βarrestin2 to activate TAB1 which inhibits TAK1, preventing NFκB and JNK pro-inflammatory signalling in macrophages. Interestingly, Ffar4 signals through both Gq and βarrestin2 to activate cytoplasmic phospholipase A2 (cPLA2), resulting in activation of cyclooxygenase 2 (COX2) and production of prostaglandin E2 (PGE2) in macrophages. However, in adipocytes, Ffar4 signals through Gq to increase cell surface expression of Glut4. Recent studies also suggest Ffar4 might signal through Gi to induce ghrelin release in the stomach and somatostatin release in pancreatic δ-cells. Adding to the complexity of Ffar4 signalling, the human Ffar4S and Ffar4L isoforms differentially activate Gq and β-arrestin, with the Ffar4S activating both, whereas the Ffar4L only signals through β-arrestin. However, Ffar4L expression appears rather limited, and the physiological significance of different isoform signalling is not entirely clear., Collectively, these studies indicate the importance of deciphering Ffar4 cell-specific signalling.
Free fatty acid receptor 4 as a mechanistic basis for ω3-polyunsaturated fatty acid-mediated cardioprotection
There are very few studies that directly evaluate Ffar4 as a mechanistic basis for EPA-mediated cardioprotection, but evidence suggests EPA-Ffar4 signalling might explain at least some of the protective effects of EPA in heart failure (HF) and atherosclerosis. In the heart, EPA supplementation prevents interstitial fibrosis and contractile dysfunction in a mouse model of pressure overload heart failure (transverse aortic constriction, TAC)., In fact, EPA-mediated prevention of fibrosis has now been replicated in many models including: mice,,, rats, dogs,, rabbits, and humans. However, dietary supplementation with EPA fails to increase EPA incorporation into cardiac myocytes or fibroblasts, suggesting a mechanism independent of membrane incorporation accounts for EPA-mediated anti-fibrotic and cardioprotective effects., Interestingly, Ffar4 is expressed in the mouse heart, at 30-fold higher levels of expression than Ffar1, and is expressed in both cardiac myocytes and fibroblasts. Ffar4 is also detected in human hearts, although Ffar1 is expressed at proportionally higher levels in human vs. mouse heart (unpublished observation). More importantly, in primary cultures of mouse cardiac fibroblasts, Ffar4 is both sufficient and necessary to prevent TGFβ1-induced fibrosis, but whether Ffar4 mediates EPA-mediated anti-fibrotic and cardioprotective effects in vivo remains to be determined. In a model of vascular injury induced by FeCl3, endogenously produced ω3-PUFAs in fat-1 mice prevents neointimal hyperplasia and vascular inflammation in the common carotid artery, which required Ffar4, as protection is lost in fat-1/Ffar4 knockout mice. Interestingly, Ffar4 is anti-inflammatory, reducing macrophage infiltration into adipose tissue in mice fed a high-fat diet and inducing macrophage phenotype switching from pro-inflammatory M1 to anti-inflammatory M2. Given the important role of inflammation in the pathology of CVD, it will be important to understand the interaction between Ffar4-mediated anti-inflammatory signals and direct Ffar4 actions in the cardiovascular system.
Summary
Ffar1 and Ffar4 signalling is the newest mechanism proposed to explain EPA-cardioprotective signalling. In summary:
Ffar1 and Ffar4 are G-protein-coupled receptors for medium and long-chain FAs (C10–C24), which include but are not limited to ω3-PUFAs.
Ffar1 regulates metabolism and is highly expressed in pancreatic islets, regulating insulin release in β cells and glucagon release from α cells, and the GI tract, regulating gut hormone release including, GLP-1, PPY, CCK, and GIP, as well as inflammatory signalling in the gut.
Ffar4 also regulates metabolism and is expressed in the pancreatic islets, regulating glucagon release from α cells and somatostatin release from ∂ cells, and in the GI tract, regulating GLP-1 release.
Ffar4 regulates inflammation, and in macrophages, ω3-PUFAs induce an M2-like anti-inflammatory phenotype. Ffar4 anti-inflammatory signalling in macrophages reduces inflammation in adipose tissue in obesity.
Ffar4 is expressed in the mouse heart, in both cardiac myocytes and fibroblasts, at much higher levels than Ffar1. Ffar4 attenuates pro-fibrotic signalling in cultured fibroblasts, and might mediate at least some of the cardioprotective functions of EPA, but this remains to be tested.
Peroxisome proliferator-activated receptors
Peroxisome proliferator-activated receptor structure, expression, and pharmacology
The PPARs are a subfamily of ligand-activated nuclear receptors/transcription factors that belong to the superfamily of nuclear receptors., Structurally, PPARs contain an N-terminal transactivating domain, a highly conserved DNA-binding domain that recognizes PPAR-specific DNA binding domains, or PPAR-response elements (PPREs), and a C-terminal ligand-binding domain., Ligand binding to PPARs facilitates heterodimerization with the retinoid-X-receptor (RXR), binding to PPREs, a six nucleotide dimeric repeat separated by a single nucleotide (AGGTCA-N-AGGTCA), and activation of transcription., Of note, the binding pocket of PPARs is relatively large compared to other nuclear receptors, facilitating binding of endogenous FAs and greater overall flexibility in ligand binding.
There are three PPAR isoforms PPARα, PPARβ/∂, and PPARγ that differ in tissue distribution, ligand binding profiles, and regulation of distinct target genes/physiologic functions., Peroxisome proliferator-activated receptor α, the first cloned PPAR, is expressed in metabolically active tissues including, liver, heart, skeletal muscle, and kidney, and regulates genes associated with fatty acid uptake, intracellular transport, and β-oxidation. Peroxisome proliferator-activated receptor γ is highly expressed in adipocytes and is a master regulator of adipogenesis, energy balance, and lipid biosynthesis; however, PPARγ is also expressed in several other tissues including skeletal and cardiac muscle where it regulates fatty acid oxidation. PPARβ/δ is widely expressed, with higher expression in the large and small intestine, heart, brain, and adipose and regulates fatty acid oxidation, improves lipid profiles, and attenuates adiposity.
While several synthetic PPAR agonists are in clinical use, including thiazolidinediones (TZDs, PPARγ agonists) and fibrates (PPARα agonists),, all three PPARs also bind to and are activated by endogenous FAs, typically in the low µM range, although with differing specificity for each PPAR isoform. While all PPARs bind PUFAs with similar affinity, PPARα also shows high affinity for MUFAs and SFAs, PPARβ/δ shows lower affinity for MUFAs and SFAs, and PPARγ is the most selective for PUFAs showing little or no affinity for MUFAs and SFAs. Of note, EPA is one of the most potent PUFAs at all three PPARs. Additionally, PPARs can also bind to certain oxylipins derived from AA,, and it seems likely oxylipins derived from other FAs would likely also bind to PPARs, although this has not been rigorously examined.
Peroxisome proliferator-activated receptors in atherosclerosis
Peroxisome proliferator-activated receptors have received significant attention as potential therapeutic targets for the management of atherosclerosis. As discussed above, clinical trials indicate that EPA, one of the more potent endogenous ligands for PPARs, lowers plasma TG without raising LDL cholesterol and can reduce atherosclerotic plaques. Furthermore, REDUCE-IT demonstrated that EPA (icosapent ethyl) reduced residual atherosclerotic cardiovascular disease (ASCVD) risk beyond statins in patients with ASCVD but also in diabetic patients with multiple risk factors. However, most of the current mechanistic understanding of PPARs in vascular disease is based on studies using high-affinity synthetic PPAR agonists, including fibrates (PPARα) and TZDs (PPARγ).
Functionally, all three PPARs isoforms are expressed in vascular endothelial cells, smooth muscle cells, and multiple inflammatory cells including macrophages and foam cells in atherosclerotic plaques., In vascular endothelial cells, PPARα activation inhibits NFκB activation and suppresses inflammation, reducing expression of VCAM-1 and leucocyte adhesion, whereas PPARγ and PPARδ may increase nitric oxide (NO) and reduce oxidative stress in diabetes. In vascular smooth muscle cells, PPARα attenuates inflammation, whereas PPARγ down-regulates matrix metalloproteinase (MMP) expression, such as MMP9 that might be involved in plaque rupture. In macrophages and foam cells within atherosclerotic lesions, PPARγ generally attenuates inflammation by regulating inflammatory gene expression.,,
However, mouse models with global deletion of PPARα (PPARαKO) display slower development of atherosclerosis, conversely PPARα deletion in macrophages increases atherosclerosis, suggesting cell-type specific effects of PPARα signalling are crucial to atherogenesis. Furthermore, endothelial cell-specific deletion of PPARγ induces dyslipidaemia, suggesting that endothelial PPARγ is also a potentially important regulator of atherogenesis. In total, PPARs in the vascular system are important in atherogenesis, and while synthetic PPAR agonists show some promise in therapeutic treatment of atherosclerosis, far fewer studies have examined the benefits of endogenous FAs, such as EPA. However, EPA activation of PPARγ attenuates pro-inflammatory signalling and inhibits endothelial lipase expression in macrophages. Interestingly, DHA signalling through PPARα increases phagocytosis and induces an M2-like phenotype in macrophages, while maresins, a class of oxylipins derived from DHA, signalling through PPARα inhibit inflammation in endothelial cells. In summary, mechanistic studies designed to explain a potential for a clinical benefit of EPA acting through PPARs in atherosclerosis are somewhat lacking.
Peroxisome proliferator-activated receptors in the liver
In the liver, PPARα activation modulates the expression of multiple target genes involved in TG metabolism. Clinically, PPARα agonists (gemfibrozil, clofibrate, fenofibrate, and fenofibric acid) decrease plasma TG levels and increase HDL levels. Despite their ability to effectively lower TGs, clinical trials have failed to show that their use was associated with reduced risk for cardiovascular events when added to statins as compared to statin therapy alone. These findings suggest that TG-lowering alone through this mechanism does not lead to reduced cardiovascular risk. As an endogenous ligand for PPARs, EPA may have additional effects through this pathway that limit atherosclerosis and vascular inflammation. Differences between natural and synthetic PPARα ligands are currently being explored.
Peroxisome proliferator-activated receptors in heart failure
Peroxisome proliferator-activated receptor regulation of metabolism in the heart is another potential therapeutic target for the management of HF. Under basal conditions, mitochondrial FA oxidation is the major source of energy production in the myocardium, but in HF, mitochondrial FA oxidation often declines and glycolytic pathways become more predominant, ultimately contributing to contractile dysfunction. Mechanistically, PPARα-RXRα complex formation is decreased in hypertrophic cardiac myocytes, while PPARα in turn might complex with silent information regulator 1 (Sirt1) in HF, contributing to the down-regulation of genes regulating FA oxidation. In PPARαKO mice, chronic pressure overload reduces FA oxidation and worsens pathologic remodelling., Conversely, cardiac myocyte-specific overexpression of PPARα induces hypertrophy, cardiac lipotoxicity, and insulin resistance similar to diabetic cardiomyopathy. Cardiac myocyte-specific knockout of PPARγ induces cardiac hypertrophy,, while cardiac myocyte-specific overexpression of PPARγ and rosiglitazone (PPARγ agonist) also induce hypertrophy., Cardiac myocyte-specific deletion of PPARδ disrupts FA oxidation and induces cardiomyopathy. In summary, the data from PPAR knockout and transgenic mice is sometimes conflicting, but suggests that fine regulation of PPAR signalling in cardiac myocytes is essential to maintain cardiac homeostasis.
Eicosapentaenoic acid and mixed ω3-PUFAs demonstrated improved HF outcomes in GISSI-HF and OMEGA-REMODEL., However, much less is known about EPA-PPAR signalling in cardiac myocytes. In cultured neonatal cardiac myocytes, EPA mediated activation of PPARα attenuates endothelin-1 induced hypertrophy. In rats fed a standard or high-fat diet, the PPARα agonist WY14643 increased monounsaturated FA and ω3-PUFA replacement with ω6-PUFA. In models of cardiac allografts, EPA reduced allograft rejection, which was abrogated by a PPPAγ antagonist., In humans, ω3-PUFA supplementation (3.4 g/day) 2–3 weeks before elective cardiac surgery increased nuclear transactivation of PPARγ, increased FA metabolic, anti-inflammatory, and antioxidant gene expression, and increased mitochondrial respiration rate in atrial biopsies. These studies do suggest that EPA signalling through PPARs have significant biological effects in the heart, but whether EPA signalling through PPARs prevents HF is still an open question.
Summary
Peroxisome proliferator-activated receptors are a second and distinct receptor-mediated mechanism proposed to explain EPA-cardioprotective signalling. In summary:
Peroxisome proliferator-activated receptors are nuclear receptors/transcription factors, and upon ligand binding, PPARs heterodimerize with RXR, bind to PPAR-response elements, and activate transcription.
There are three PPAR isoforms, PPARα, PPARβ/δ, and PPARγ, and synthetic PPAR agonists are in clinical use, including thiazolidinediones (TZDs, PPARγ agonists) and fibrates (PPARα agonists), to regulate diabetes.
Clinical trials suggest that EPA attenuates atherosclerosis. Further, EPA is one of the most potent FA activators of PPARs, suggesting a potential mechanistic explanation for EPA-mediated protection in atherosclerosis, but this remains to be tested.
PPARs are expressed in the heart and regulate FA oxidation. While EPA and mixed ω3-PUFAs demonstrated improved HF outcomes, whether this is mediated by PPARs remains to be tested.
Final summary and future directions
The recent success of high-dose ω3-PUFAs, particularly EPA administered as icosapent ethyl, in a clinical outcome trial has brought its role in CVD back into the spotlight. However, questions still remain about ω3-PUFAs, as the benefit of mixed and low-dose ω3-PUFAs has been equivocal at best. Undoubtably, understanding the underlying mechanisms of ω3-PUFAs in the cardiovascular system will lead to improved formulations that produce greater efficacy in those patient populations most likely to benefit from such an intervention. Potential mechanisms to explain the cardioprotective benefit of EPA, in particular, include its unique physiochemical interactions in membranes and lipoproteins, the production of EPA-derived cardioprotective oxylipins, activation of Ffar4 signalling, and induction of favourable PPAR-mediated gene expression. A major challenge going forward is how to systematically elucidate the key biological mechanism(s) underpinning the emerging cardioprotective effects of EPA. In light of multiple potential mechanisms to explain such cardioprotection, traditional reductionist experimental approaches might prove inadequate to address what is essentially a systems biological challenge where multiple mechanisms are involved that vary among different cell and tissue types. New experimental paradigms will likely be needed to address this important question.
Funding
This work was supported by National Institutes of Health and National Heart Lung Blood Institutes [R01 HL130099 and R01 HL152215 to T.D.O. and G.C.S., K01 HL133416 to A.M.N.]. This paper was published as part of a supplement supported by an unrestricted educational grant from Amarin Pharma, Inc.
Conflict of interest: T.D.O., R.P.M., M.J.B., A.M.N., and G.C.S. have received honoraria for consultation and instruction, and T.D.O., R.P.M., A.M.N., and M.J.B. have received grant/research support from Amarin Pharma Inc. R.P.M. has also received support from Pfizer Inc., Amgen Inc., ARCA Biopharma, and Novartis AG. R.P.M. is also a paid speaker and consultant for Amarin Pharma Inc., Pfizer Inc., and Novartis AG. A.M.N. has received funding for research to her institution from Amgen, Janssen, Amarin, Sanofi, Regeneron, and honoraria and consulting fees from Amgen, AstraZeneca, Janssen, Esperion, Amarin, Sanofi, Regeneron, NovoNordisk, Novartis, The Medicines Company, New Amsterdam, and Pfizer.
References
- 1. Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, Doyle RT Jr, Juliano RA, Jiao L, Granowitz C, Tardif JC, Ballantyne CM; REDUCE-IT Investigators. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med2019;380:11–22.
- 2. Heydari B, Abdullah S, Pottala JV, Shah R, Abbasi S, Mandry D, Francis SA, Lumish H, Ghoshhajra BB, Hoffmann U, Appelbaum E, Feng JH, Blankstein R, Steigner M, McConnell JP, Harris W, Antman EM, Jerosch-Herold M, Kwong RY. Effect of omega-3 acid ethyl esters on left ventricular remodeling after acute myocardial infarction: the OMEGA-REMODEL randomized clinical trial. Circulation2016;134:378–391.
- 3. Manson JE, Cook NR, Lee IM, Christen W, Bassuk SS, Mora S, Gibson H, Albert CM, Gordon D, Copeland T, D'Agostino D, Friedenberg G, Ridge C, Bubes V, Giovannucci EL, Willett WC, Buring JE; VITAL Research Group. Marine n-3 fatty acids and prevention of cardiovascular disease and cancer. N Engl J Med2019;380:23–32.
- 4. Burr ML, Ashfield-Watt PA, Dunstan FD, Fehily AM, Breay P, Ashton T, Zotos PC, Haboubi NA, Elwood PC. Lack of benefit of dietary advice to men with angina: results of a controlled trial. Eur J Clin Nutr2003;57:193–200.
- 5. Kromhout D, Giltay EJ, Geleijnse JM; Alpha Omega Trial Group. n-3 fatty acids and cardiovascular events after myocardial infarction. N Engl J Med2010;363:2015–2026.
- 6. Rauch B, Schiele R, Schneider S, Diller F, Victor N, Gohlke H, Gottwik M, Steinbeck G, Del Castillo U, Sack R, Worth H, Katus H, Spitzer W, Sabin G, Senges J; for the OMEGA Study Group. OMEGA, a randomized, placebo-controlled trial to test the effect of highly purified omega-3 fatty acids on top of modern guideline-adjusted therapy after myocardial infarction. Circulation2010;122:2152–2159.
- 7. Galan P, Kesse-Guyot E, Czernichow S, Briancon S, Blacher J, Hercberg S; for the SU.FOL.OM3 Collaborative Group. Effects of B vitamins and omega 3 fatty acids on cardiovascular diseases: a randomised placebo controlled trial. BMJ2010;341:c6273.
- 8. Group ASC, Bowman L, Mafham M, Wallendszus K, Stevens W, Buck G, Barton J, Murphy K, Aung T, Haynes R, Cox J, Murawska A, Young A, Lay M, Chen F, Sammons E, Waters E, Adler A, Bodansky J, Farmer A, McPherson R, Neil A, Simpson D, Peto R, Baigent C, Collins R, Parish S, Armitage J. Effects of n-3 fatty acid supplements in diabetes mellitus. N Engl J Med2018;379:1540–1550.
- 9. Risk Prevention Study Collaborative Group. n-3 fatty acids in patients with multiple cardiovascular risk factors. N Engl J Med2013;368:1800–1808.
- 10. Ferrieres J. The return of triglycerides and revival of omega-3 fatty acids! Arch Cardiovasc Dis 2020;113:369–373.
- 11. Brown DA. Seeing is believing: visualization of rafts in model membranes. Proc Natl Acad Sci USA2001;98:10517–10518.
- 12. Mason RP, Jacob RF. Membrane microdomains and vascular biology: emerging role in atherogenesis. Circulation2003;107:2270–2273.
- 13. Brown DA, London E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J Biol Chem2000;275:17221–17224.
- 14. Li Q, Zhang Q, Wang M, Zhao S, Ma J, Luo N, Li N, Li Y, Xu G, Li J. Eicosapentaenoic acid modifies lipid composition in caveolae and induces translocation of endothelial nitric oxide synthase. Biochimie2007;89:169–177.
- 15. Smart EJ, Graf GA, McNiven MA, Sessa WC, Engelman JA, Scherer PE, Okamoto T, Lisanti MP. Caveolins, liquid-ordered domains, and signal transduction. Mol Cell Biol1999;19:7289–7304.
- 16. Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA, Michel T. Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J Biol Chem1996;271:22810–22814.
- 17. Feron O, Dessy C, Desager JP, Balligand JL. Hydroxy-methylgluataryl-coenzyme A reductase inhibition promotes endothelial nitric oxide synthase activation through a decrease in caveolin abundance. Circulation2001;103:113–118.
- 18. Mason RP, Jacob RF, Shrivastava S, Sherratt SCR, Chattopadhyay A. Eicosapentaenoic acid reduces membrane fluidity, inhibits cholesterol domain formation, and normalizes bilayer width in atherosclerotic-like model membranes. Biochim Biophys Acta2016;1858:3131–3140.
- 19. Shaikh SR. Biophysical and biochemical mechanisms by which dietary N-3 polyunsaturated fatty acids from fish oil disrupt membrane lipid rafts. J Nutr Biochem2012;23:101–105.
- 20. Shaikh SR, Wassall SR, Brown DA, Kosaraju R. n-3 polyunsaturated fatty acids, lipid microclusters, and vitamin E. Curr Top Membr2015;75:209–231.
- 21. Williams JA, Batten SE, Harris M, Rockett BD, Shaikh SR, Stillwell W, Wassall SR. Docosahexaenoic and eicosapentaenoic acids segregate differently between raft and nonraft domains. Biophys J2012;103:228–237.
- 22. Mason RP, Libby P, Bhatt DL. Emerging mechanisms of cardiovascular protection for the omega-3 fatty acid eicosapentaenoic acid. Arterioscler Thromb Vasc Biol2020;40:1135–1147.
- 23. Mason RP, Jacob RF. Eicosapentaenoic acid inhibits glucose-induced membrane cholesterol crystalline domain formation through a potent antioxidant mechanism. Biochim Biophys Acta2015;1848:502–509.
- 24. Mason RP, Sherratt SCR, Jacob RF. Eicosapentaenoic acid inhibits oxidation of ApoB-containing lipoprotein particles of different size in vitro when administered alone or in combination with atorvastatin active metabolite compared with other triglyceride-lowering agents. J Cardiovasc Pharmacol2016;68:33–40.
- 25. Sherratt SCR, Mason RP. Eicosapentaenoic acid and docosahexaenoic acid have distinct membrane locations and lipid interactions as determined by X-ray diffraction. Chem Phys Lipids2018;212:73–79.
- 26. Shaikh SR, Kinnun JJ, Leng X, Williams JA, Wassall SR. How polyunsaturated fatty acids modify molecular organization in membranes: insight from NMR studies of model systems. Biochim Biophys Acta2015;1848:211–219.
- 27.
- 28. Mason RP, Walter MF, Day CA, Jacob RF. Active metabolite of atorvastatin inhibits membrane cholesterol domain formation by an antioxidant mechanism. J Biol Chem2006;281:9337–9345.
- 29. Tulenko TN, Chen M, Mason PE, Mason RP. Physical effects of cholesterol on arterial smooth muscle membranes: evidence of immiscible cholesterol domains and alterations in bilayer width during atherogenesis. J Lipid Res1998;39:947–956.
- 30. Grebe A, Latz E. Cholesterol crystals and inflammation. Curr Rheumatol Rep2013;15:313.
- 31. Kellner-Weibel G, Yancey PG, Jerome WG, Walser T, Mason RP, Phillips MC, Rothblat GH. Crystallization of free cholesterol in model macrophage foam cells. Arterioscler Thromb Vasc Biol1999;19:1891–1898.
- 32. Abela GS, Aziz K. Cholesterol crystals cause mechanical damage to biological membranes: a proposed mechanism of plaque rupture and erosion leading to arterial thrombosis. Clin Cardiol2005;28:413–420.
- 33. Dai J, Tian J, Hou J, Xing L, Liu S, Ma L, Yu H, Ren X, Dong N, Yu B. Association between cholesterol crystals and culprit lesion vulnerability in patients with acute coronary syndrome: an optical coherence tomography study. Atherosclerosis2016;247:111–117.
- 34. Self-Medlin Y, Byun J, Jacob RF, Mizuno Y, Mason RP. Glucose promotes membrane cholesterol crystalline domain formation by lipid peroxidation. Biochim Biophys Acta2009;1788:1398–1403.
- 35. Karasawa T, Takahashi M. Role of NLRP3 inflammasomes in atherosclerosis. J Atheroscler Thromb2017;24:443–451.
- 36. Braeckman RA, Stirtan WG, Soni PN. Pharmacokinetics of eicosapentaenoic acid in plasma and red blood cells after multiple oral dosing with icosapent ethyl in healthy subjects. Clin Pharmacol Drug Develop2014;3:101–108.
- 37. Soni SP, LoCascio DS, Liu Y, Williams JA, Bittman R, Stillwell W, Wassall SR. Docosahexaenoic acid enhances segregation of lipids between: 2H-NMR study. Biophys J2008;95:203–214.
- 38. Wassall SR, Stillwell W. Docosahexaenoic acid domains: the ultimate non-raft membrane domain. Chem Phys Lipids2008;153:57–63.
- 39. Ballantyne CM, Bays HE, Kastelein JJ, Stein E, Isaacsohn JL, Braeckman RA, Soni PN. Efficacy and safety of eicosapentaenoic acid ethyl ester (AMR101) therapy in statin-treated patients with persistent high triglycerides (from the ANCHOR study). Am J Cardiol2012;110:984–992.
- 40. Bays HE, Ballantyne CM, Braeckman RA, Stirtan WG, Soni PN. Icosapent ethyl, a pure ethyl ester of eicosapentaenoic acid: effects on circulating markers of inflammation from the MARINE and ANCHOR studies. Am J Cardiovasc Drugs2013;13:37–46.
- 41. Bays HE, Ballantyne CM, Kastelein JJ, Isaacsohn JL, Braeckman RA, Soni PN. Eicosapentaenoic acid ethyl ester (AMR101) therapy in patients with very high triglyceride levels (from the Multi-center, plAcebo-controlled, Randomized, double-blINd, 12-week study with an open-label Extension [MARINE] trial). Am J Cardiol2011;108:682–690.
- 42. Braeckman RA, Manku MS, Bays HE, Stirtan WG, Soni PN. Icosapent ethyl, a pure EPA omega-3 fatty acid: effects on plasma and red blood cell fatty acids in patients with very high triglyceride levels (results from the MARINE study). Prostag Leukotr Ess2013;89:195–201.
- 43. Satoh N, Shimatsu A, Kotani K, Sakane N, Yamada K, Suganami T, Kuzuya H, Ogawa Y. Purified eicosapentaenoic acid reduces small dense LDL, remnant lipoprotein particles, and C-reactive protein in metabolic syndrome. Diabetes Care2007;30:144–146.
- 44. Satoh-Asahara N, Shimatsu A, Sasaki Y, Nakaoka H, Himeno A, Tochiya M, Kono S, Takaya T, Ono K, Wada H, Suganami T, Hasegawa K, Ogawa Y. Highly purified eicosapentaenoic acid increases interleukin-10 levels of peripheral blood monocytes in obese patients with dyslipidemia. Diabetes Care2012;35:2631–2639.
- 45. Dunbar RL, Nicholls SJ, Maki KC, Roth EM, Orloff DG, Curcio D, Johnson J, Kling D, Davidson MH. Effects of omega-3 carboxylic acids on lipoprotein particles and other cardiovascular risk markers in high-risk statin-treated patients with residual hypertriglyceridemia: a randomized, controlled, double-blind trial. Lipids Health Dis2015;14:98.
- 46. Tsunoda F, Lamon-Fava S, Asztalos BF, Iyer LK, Richardson K, Schaefer EJ. Effects of oral eicosapentaenoic acid versus docosahexaenoic acid on human peripheral blood mononuclear cell gene expression. Atherosclerosis2015;241:400–408.
- 47. Mickleborough TD, Tecklenburg SL, Montgomery GS, Lindley MR. Eicosapentaenoic acid is more effective than docosahexaenoic acid in inhibiting proinflammatory mediator production and transcription from LPS-induced human asthmatic alveolar macrophage cells. Clin Nutr2009;28:71–77.
- 48. Sato T, Horikawa M, Takei S, Yamazaki F, Ito TK, Kondo T, Sakurai T, Kahyo T, Ikegami K, Sato S, Sato R, Jinno Y, Kawano H, Naoe S, Arita M, Kashiwagi Y, Setou M. Preferential incorporation of administered eicosapentaenoic acid into thin-cap atherosclerotic plaques. Arterioscler Thromb Vasc Biol2019;39:1802–1816.
- 49. Tanaka N, Ishida T, Nagao M, Mori T, Monguchi T, Sasaki M, Mori K, Kondo K, Nakajima H, Honjo T, Irino Y, Toh R, Shinohara M, Hirata K. Administration of high dose eicosapentaenoic acid enhances anti-inflammatory properties of high-density lipoprotein in Japanese patients with dyslipidemia. Atherosclerosis2014;237:577–583.
- 50. Tanaka N, Irino Y, Shinohara M, Tsuda S, Mori T, Nagao M, Oshita T, Mori K, Hara T, Toh R, Ishida T, Hirata KI. Eicosapentaenoic acid-enriched high-density lipoproteins exhibit anti-atherogenic properties. Circ J2018;82:596–601.
- 51. Sherratt SCR, Mason RP. Eicosapentaenoic acid inhibits oxidation of high density lipoprotein particles in a manner distinct from docosahexaenoic acid. Biochem Biophys Res Commun2018;496:335–338.
- 52. Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci USA1987;84:9265–9269.
- 53. Kojda G, Harrison D. Interactions between NO and reactive oxygen species: pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovasc Res1999;43:562–571.
- 54. Rees DD, Palmer RM, Moncada S. Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proc Natl Acad Sci USA1989;86:3375–3378.
- 55. Mason RP, Dawoud H, Jacob RF, Sherratt SCR, Malinski T. Eicosapentaenoic acid improves endothelial function and nitric oxide bioavailability in a manner that is enhanced in combination with a statin. Biomed Pharmacother2018;103:1231–1237.
- 56. Shearer GC, Newman JW. Impact of circulating esterified eicosanoids and other oxylipins on endothelial function. Curr Atheroscler Rep2009;11:403–410.
- 57. Capdevila J, Pramanik B, Napoli JL, Manna S, Falck JR. Arachidonic acid epoxidation: epoxyeicosatrienoic acids are endogenous constituents of rat liver. Arch Biochem Biophys1984;231:511–517.
- 58. Fowler CJ, Doherty P, Alexander SPH. Endocannabinoid turnover. Adv Pharmacol2017;80:31–66.
- 59. Trostchansky A, Bonilla L, Gonzalez-Perilli L, Rubbo H. Nitro-fatty acids: formation, redox signaling, and therapeutic potential. Antioxid Redox Signal2013;19:1257–1265.
- 60. Caro AA, Cederbaum AI. Role of cytochrome P450 in phospholipase A2- and arachidonic acid-mediated cytotoxicity. Free Radic Biol Med2006;40:364–375.
- 61. Spector AA. Arachidonic acid cytochrome P450 epoxygenase pathway. J Lipid Res2009;50:S52–6.
- 62. Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res2004;43:55–90.
- 63. Spector AA, Kim HY. Cytochrome P epoxygenase pathway of polyunsaturated fatty acid metabolism. Biochim Biophys Acta2015;1851:356–365.
- 64. Gabbs M, Leng S, Devassy JG, Monirujjaman M, Aukema HM. Advances in our understanding of oxylipins derived from dietary PUFAs. Adv Nutr2015;6:513–540.
- 65. Takenaga M, Hirai A, Terano T, Tamura Y, Kitagawa H, Yoshida S. Comparison of the in vitro effect of eicosapentaenoic acid (EPA)-derived lipoxygenase metabolites on human platelet function with those of arachidonic acid. Thromb Res1986;41:373–384.
- 66. Goetzl EJ, Brash AR, Tauber AI, Oates JA, Hubbard WC. Modulation of human neutrophil function by monohydroxy-eicosatetraenoic acids. Immunology1980;39:491–501.
- 67. Heidel JR, Taylor SM, Laegreid WW, Silflow RM, Liggitt HD, Leid RW. In vivo chemotaxis of bovine neutrophils induced by 5-lipoxygenase metabolites of arachidonic and eicosapentaenoic acid. Am J Pathol1989;134:671–676.
- 68. Valone FH, Franklin M, Sun FF, Goetzl EJ. Alveolar macrophage lipoxygenase products of arachidonic acid: isolation and recognition as the predominant constituents of the neutrophil chemotactic activity elaborated by alveolar macrophages. Cell Immunol1980;54:390–401.
- 69. Kogure R, Toyama K, Hiyamuta S, Kojima I, Takeda S. 5-Hydroxy-eicosapentaenoic acid is an endogenous GPR119 agonist and enhances glucose-dependent insulin secretion. Biochem Biophys Res Commun2011;416:58–63.
- 70. Powell WS, Gravel S, Gravelle F. Formation of a 5-oxo metabolite of 5,8,11,14,17-eicosapentaenoic acid and its effects on human neutrophils and eosinophils. J Lipid Res1995;36:2590–2598.
- 71. Terano T, Salmon JA, Moncada S. Biosynthesis and biological activity of leukotriene B5. Prostaglandins1984;27:217–232.
- 72. Hafstrom I, Palmblad J, Malmsten CL, Radmark O, Samuelsson B. Leukotriene B4—a stereospecific stimulator for release of lysosomal enzymes from neutrophils. FEBS Lett1981;130:146–148.
- 73. Juan H, Peskar BA, Simmet T. Effect of exogenous 5,8,11,14,17-eicosapentaenoic acid on cardiac anaphylaxis. Br J Pharmacol1987;90:315–325.
- 74. Maddox JF, Serhan CN. Lipoxin A4 and B4 are potent stimuli for human monocyte migration and adhesion: selective inactivation by dehydrogenation and reduction. J Exp Med1996;183:137–146.
- 75. Patricia MK, Kim JA, Harper CM, Shih PT, Berliner JA, Natarajan R, Nadler JL, Hedrick CC. Lipoxygenase products increase monocyte adhesion to human aortic endothelial cells. Arterioscler Thromb Vasc Biol1999;19:2615–2622.
- 76. Reilly KB, Srinivasan S, Hatley ME, Patricia MK, Lannigan J, Bolick DT, Vandenhoff G, Pei H, Natarajan R, Nadler JL, Hedrick CC. 12/15-Lipoxygenase activity mediates inflammatory monocyte/endothelial interactions and atherosclerosis in vivo. J Biol Chem2004;279:9440–9450.
- 77. Nazarewicz RR, Zenebe WJ, Parihar A, Parihar MS, Vaccaro M, Rink C, Sen CK, Ghafourifar P. 12(S)-hydroperoxyeicosatetraenoic acid (12-HETE) increases mitochondrial nitric oxide by increasing intramitochondrial calcium. Arch Biochem Biophys2007;468:114–120.
- 78. Thollon C, Iliou JP, Cambarrat C, Robin F, Vilaine JP. Nature of the cardiomyocyte injury induced by lipid hydroperoxides. Cardiovasc Res1995;30:648–655.
- 79. Matsuda H, Miyatake K, Dahlen SE. Pharmacodynamics of 15(S)-hydroperoxyeicosatetraenoic (15-HPETE) and 15(S)-hydroxyeicosatetraenoic acid (15-HETE) in isolated arteries from guinea pig, rabbit, rat and human. J Pharmacol Exp Ther1995;273:1182–1189.
- 80. Hersberger M. Potential role of the lipoxygenase derived lipid mediators in atherosclerosis: leukotrienes, lipoxins and resolvins. Clin Chem Lab Med2010;48:1063–1073.
- 81. Weylandt KH, Krause LF, Gomolka B, Chiu CY, Bilal S, Nadolny A, Waechter SF, Fischer A, Rothe M, Kang JX. Suppressed liver tumorigenesis in fat-1 mice with elevated omega-3 fatty acids is associated with increased omega-3 derived lipid mediators and reduced TNF-alpha. Carcinogenesis2011;32:897–903.
- 82. Endo J, Sano M, Isobe Y, Fukuda K, Kang JX, Arai H, Arita M. 18-HEPE, an n-3 fatty acid metabolite released by macrophages, prevents pressure overload-induced maladaptive cardiac remodeling. J Exp Med2014;211:1673–1687.
- 83. Hercule HC, Schunck WH, Gross V, Seringer J, Leung FP, Weldon SM, da Costa Goncalves A, Huang Y, Luft FC, Gollasch M. Interaction between P450 eicosanoids and nitric oxide in the control of arterial tone in mice. Arterioscler Thromb Vasc Biol2009;29:54–60.
- 84. Oltman CL, Weintraub NL, VanRollins M, Dellsperger KC. Epoxyeicosatrienoic acids and dihydroxyeicosatrienoic acids are potent vasodilators in the canine coronary microcirculation. Circ Res1998;83:932–939.
- 85. Proctor KG, Falck JR, Capdevila J. Intestinal vasodilation by epoxyeicosatrienoic acids: arachidonic acid metabolites produced by a cytochrome P450 monooxygenase. Circ Res1987;60:50–59.
- 86. Dhanasekaran A, Gruenloh SK, Buonaccorsi JN, Zhang R, Gross GJ, Falck JR, Patel PK, Jacobs ER, Medhora M. Multiple antiapoptotic targets of the PI3K/Akt survival pathway are activated by epoxyeicosatrienoic acids to protect cardiomyocytes from hypoxia/anoxia. Am J Physiol Heart Circ Physiol2008;294:H724–H735.
- 87. VanRollins M. Epoxygenase metabolites of docosahexaenoic and eicosapentaenoic acids inhibit platelet aggregation at concentrations below those affecting thromboxane synthesis. J Pharmacol Exp Ther1995;274:798–804.
- 88. Zhang Y, Oltman CL, Lu T, Lee HC, Dellsperger KC, VanRollins M. EET homologs potently dilate coronary microvessels and activate BK(Ca) channels. Am J Physiol Heart Circul Physiol2001;280:H2430–40.
- 89. Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science1999;285:1276–1279.
- 90. Campbell WB, Deeter C, Gauthier KM, Ingraham RH, Falck JR, Li P1. 15-Dihydroxyeicosatrienoic acid relaxes bovine coronary arteries by activation of K(Ca) channels. Am J Physiol Heart Circ Physiol2002;282:H1656–H1664.
- 91. López-Vicario C, Alcaraz-Quiles J, García-Alonso V, Rius B, Hwang SH, Titos E, Lopategi A, Hammock BD, Arroyo V, Clària J. Inhibition of soluble epoxide hydrolase modulates inflammation and autophagy in obese adipose tissue and liver: role for omega-3 epoxides. Proc Natl Acad Sci U S A2015;112:536–541.
- 92. Hercule HC, Salanova B, Essin K, Honeck H, Falck JR, Sausbier M, Ruth P, Schunck WH, Luft FC, Gollasch M. The vasodilator 17,18-epoxyeicosatetraenoic acid targets the pore-forming BK alpha channel subunit in rodents. Exp Physiol2007;92:1067–1076.
- 93. Lu T, Katakam PV, VanRollins M, Weintraub NL, Spector AA, Lee HC. Dihydroxyeicosatrienoic acids are potent activators of Ca(2+)-activated K(+) channels in isolated rat coronary arterial myocytes. J Physiol2001;534:651–667.
- 94. Ballantyne CM, Manku MS, Bays HE, Philip S, Granowitz C, Doyle RT Jr, Juliano RA. Icosapent ethyl effects on fatty acid profiles in statin-treated patients with high triglycerides: the randomized, placebo-controlled ANCHOR study. Cardiol Ther2019;8:79–90.
- 95. Shearer GC, Borkowski K, Puumala SL, Harris WS, Pedersen TL, Newman JW. Abnormal lipoprotein oxylipins in metabolic syndrome and partial correction by omega-3 fatty acids. Prostagland Leukot Ess2018;128:1–10.
- 96. Shearer GC, Harris WS, Pedersen TL, Newman JW. Detection of omega-3 oxylipins in human plasma and response to treatment with omega-3 acid ethyl esters. J Lipid Res2010;51:2074–2081.
- 97. Burr GO, Burr MM. On the nature and role of the fatty acids essential in nutrition. J Biol Chem1930;86:587.
- 98. Burr GO, Burr MM, Miller ES. On the fatty acids essential in nutrition. III. J Biol Chem1932;97:1–9.
- 99. Spector AA, Kim HY. Discovery of essential fatty acids. J Lipid Res2015;56:11–21.
- 100. Moon SH, Liu X, Cedars AM, Yang K, Kiebish MA, Joseph SM, Kelley J, Jenkins CM, Gross RW. Heart failure-induced activation of phospholipase iPLA2gamma generates hydroxyeicosatetraenoic acids opening the mitochondrial permeability transition pore. J Biol Chem2018;293:115–129.
- 101. Laneuville O, Breuer DK, Xu N, Huang ZH, Gage DA, Watson JT, Lagarde M, DeWitt DL, Smith WL. Fatty acid substrate specificities of human prostaglandin-endoperoxide H synthase-1 and -2. Formation of 12-hydroxy-(9Z, 13E/Z, 15Z)- octadecatrienoic acids from alpha-linolenic acid. J Biol Chem1995;270:19330–19336.
- 102. Milligan G, Shimpukade B, Ulven T, Hudson BD. Complex pharmacology of free fatty acid receptors. Chem Rev2017;117:67–110.
- 103. Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, Ellis C, Elshourbagy NA, Goetz AS, Minnick DT, Murdock PR, Sauls HR Jr, Shabon U, Spinage LD, Strum JC, Szekeres PG, Tan KB, Way JM, Ignar DM, Wilson S, Muir AI. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem2003;278:11303–11311.
- 104. Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M, Tanaka Y, Uejima H, Tanaka H, Maruyama M, Satoh R, Okubo S, Kizawa H, Komatsu H, Matsumura F, Noguchi Y, Shinohara T, Hinuma S, Fujisawa Y, Fujino M. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature2003;422:173–176.
- 105. Kotarsky K, Nilsson NE, Flodgren E, Owman C, Olde B. A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochem Biophys Res Commun2003;301:406–410.
- 106. Edfalk S, Steneberg P, Edlund H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes2008;57:2280–2287.
- 107. Liou AP, Lu X, Sei Y, Zhao X, Pechhold S, Carrero RJ, Raybould HE, Wank S. The G-protein-coupled receptor GPR40 directly mediates long-chain fatty acid-induced secretion of cholecystokinin. Gastroenterology2011;140:903–912.
- 108. Parker HE, Habib AM, Rogers GJ, Gribble FM, Reimann F. Nutrient-dependent secretion of glucose-dependent insulinotropic polypeptide from primary murine K cells. Diabetologia2009;52:289–298.
- 109. Tan JK, McKenzie C, Marino E, Macia L, Mackay CR. Metabolite-sensing G protein-coupled receptors-facilitators of diet-related immune regulation. Annu Rev Immunol2017;35:371–402.
- 110. Khan MZ, He L. The role of polyunsaturated fatty acids and GPR40 receptor in brain. Neuropharmacology2017;113:639–651.
- 111. Eclov JA, Qian Q, Redetzke R, Chen Q, Wu SC, Healy CL, Ortmeier SB, Harmon E, Shearer GC, O’Connell TD. EPA, not DHA, prevents fibrosis in pressure overload induced heart failure; potential role of free fatty acid receptor 4. J Lipid Res2015;56:2297–2308.
- 112. Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, Sugimoto Y, Miyazaki S, Tsujimoto G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med2005;11:90–94.
- 113. Burns RN, Moniri NH. Agonism with the omega-3 fatty acids alpha-linolenic acid and docosahexaenoic acid mediates phosphorylation of both the short and long isoforms of the human GPR120 receptor. Biochem Biophys Res Commun2010;396:1030–1035.
- 114. Watson SJ, Brown AJ, Holliday ND. Differential signaling by splice variants of the human free fatty acid receptor GPR120. Mol Pharmacol2012;81:631–642.
- 115. Tanaka T, Yano T, Adachi T, Koshimizu TA, Hirasawa A, Tsujimoto G. Cloning and characterization of the rat free fatty acid receptor GPR120: in vivo effect of the natural ligand on GLP-1 secretion and proliferation of pancreatic beta cells. Naunyn Schmiedebergs Arch Pharmacol2008;377:515–522.
- 116. Cornall LM, Mathai ML, Hryciw DH, McAinch AJ. Diet-induced obesity up-regulates the abundance of GPR43 and GPR120 in a tissue specific manner. Cell Physiol Biochem2011;28:949–958.
- 117. Xiong Y, Swaminath G, Cao Q, Yang L, Guo Q, Salomonis H, Lu J, Houze JB, Dransfield PJ, Wang Y, Liu JJ, Wong S, Schwandner R, Steger F, Baribault H, Liu L, Coberly S, Miao L, Zhang J, Lin DC, Schwarz M. Activation of FFA1 mediates GLP-1 secretion in mice. Evidence for allosterism at FFA1. Mol Cell Endocrinol2013;369:119–129.
- 118. Suckow AT, Polidori D, Yan W, Chon S, Ma JY, Leonard J, Briscoe CP. Alteration of the glucagon axis in GPR120 (FFAR4) knockout mice: a role for GPR120 in glucagon secretion. J Biol Chem2014;289:15751–15763.
- 119. Stone VM, Dhayal S, Brocklehurst KJ, Lenaghan CS, Winzell M, Hammar M, Xu X, Smith DM, Morgan NG. GPR120 (FFAR4) is preferentially expressed in pancreatic delta cells and regulates somatostatin secretion from murine islets of Langerhans. Diabetologia2014;57:1182–1191.
- 120. Moran BM, Abdel-Wahab YH, Flatt PR, McKillop AM. Evaluation of the insulin-releasing and glucose-lowering effects of GPR120 activation in pancreatic beta-cells. Diabetes Obes Metab2014;16:1128–1139.
- 121. Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell2010;142:687–698.
- 122. Christiansen E, Watterson KR, Stocker CJ, Sokol E, Jenkins L, Simon K, Grundmann M, Petersen RK, Wargent ET, Hudson BD, Kostenis E, Ejsing CS, Cawthorne MA, Milligan G, Ulven T. Activity of dietary fatty acids on FFA1 and FFA4 and characterisation of pinolenic acid as a dual FFA1/FFA4 agonist with potential effect against metabolic diseases. Br J Nutr2015;113:1677–1688.
- 123. Yore MM, Syed I, Moraes-Vieira PM, Zhang T, Herman MA, Homan EA, Patel RT, Lee J, Chen S, Peroni OD, Dhaneshwar AS, Hammarstedt A, Smith U, McGraw TE, Saghatelian A, Kahn BB. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell2014;159:318–332.
- 124. Hudson BD, Shimpukade B, Mackenzie AE, Butcher AJ, Pediani JD, Christiansen E, Heathcote H, Tobin AB, Ulven T, Milligan G. The pharmacology of TUG-891, a potent and selective agonist of the free fatty acid receptor 4 (FFA4/GPR120), demonstrates both potential opportunity and possible challenges to therapeutic agonism. Mol Pharmacol2013;84:710–725.
- 125. Liu Y, Chen LY, Sokolowska M, Eberlein M, Alsaaty S, Martinez-Anton A, Logun C, Qi HY, Shelhamer JH. The fish oil ingredient, docosahexaenoic acid, activates cytosolic phospholipase A(2) via GPR120 receptor to produce prostaglandin E(2) and plays an anti-inflammatory role in macrophages. Immunology2014;143:81–95.
- 126. Engelstoft MS, Park WM, Sakata I, Kristensen LV, Husted AS, Osborne-Lawrence S, Piper PK, Walker AK, Pedersen MH, Nohr MK, Pan J, Sinz CJ, Carrington PE, Akiyama TE, Jones RM, Tang C, Ahmed K, Offermanns S, Egerod KL, Zigman JM, Schwartz TW. Seven transmembrane G protein-coupled receptor repertoire of gastric ghrelin cells. Mol Metab2013;2:376–392.
- 127. Galindo MM, Voigt N, Stein J, van Lengerich J, Raguse JD, Hofmann T, Meyerhof W, Behrens M. G protein-coupled receptors in human fat taste perception. Chem Senses2012;37:123–139.
- 128. Milligan G, Alvarez-Curto E, Watterson KR, Ulven T, Hudson BD. Characterizing pharmacological ligands to study the long-chain fatty acid receptors GPR40/FFA1 and GPR120/FFA4. Br J Pharmacol2015;172:3254–3265.
- 129. Chen J, Shearer GC, Chen Q, Healy CL, Beyer AJ, Nareddy VB, Gerdes AM, Harris WS, O'Connell TD, Wang D. Omega-3 fatty acids prevent pressure overload-induced cardiac fibrosis through activation of cyclic GMP/protein kinase G signaling in cardiac fibroblasts. Circulation2011;123:584–593.
- 130. Kain V, Ingle KA, Colas RA, Dalli J, Prabhu SD, Serhan CN, Joshi M, Halade GV. Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J Mol Cell Cardiol2015;84:24–35.
- 131. Nagai T, Anzai T, Mano Y, Kaneko H, Anzai A, Sugano Y, Maekawa Y, Takahashi T, Yoshikawa T, Fukuda K. Eicosapentaenoic acid suppresses adverse effects of C-reactive protein overexpression on pressure overload-induced cardiac remodeling. Heart Vessels2013;28:404–411.
- 132. Ghule AE, Kandhare AD, Jadhav SS, Zanwar AA, Bodhankar SL. Omega-3-fatty acid adds to the protective effect of flax lignan concentrate in pressure overload-induced myocardial hypertrophy in rats via modulation of oxidative stress and apoptosis. Int Immunopharmacol2015;28:751–763.
- 133. Mayyas F, Jaradat R, Alzoubi KH. Cardiac effects of fish oil in a rat model of streptozotocin-induced diabetes. Nutr Metab Cardiovasc Dis2018;28:592–599.
- 134. Poudyal H, Panchal SK, Ward LC, Brown L. Effects of ALA, EPA and DHA in high-carbohydrate, high-fat diet-induced metabolic syndrome in rats. J Nutr Biochem2013;24:1041–1052.
- 135. Ramadeen A, Connelly KA, Leong-Poi H, Hu X, Fujii H, Laurent G, Domenichiello AF, Bazinet RP, Dorian P. Docosahexaenoic acid, but not eicosapentaenoic acid, supplementation reduces vulnerability to atrial fibrillation. Circ Arrhythm Electrophysiol2012;5:978–983.
- 136. Ramadeen A, Laurent G, dos Santos CC, Hu X, Connelly KA, Holub BJ, Mangat I, Dorian P. N- 3 polyunsaturated fatty acids alter expression of fibrotic and hypertrophic genes in a dog model of atrial cardiomyopathy. Heart Rhythm2010;7:520–528.
- 137. Kitamura K, Shibata R, Tsuji Y, Shimano M, Inden Y, Murohara T. Eicosapentaenoic acid prevents atrial fibrillation associated with heart failure in a rabbit model. Am J Physiol Heart Circ Physiol2011;300:H1814–H1821.
- 138. Li X, Ballantyne LL, Mewburn JD,, Kang JX,, Barkley RM,, Murphy RC,, Yu Y,, Funk CD. Endogenously generated omega-3 fatty acids attenuate vascular inflammation and neointimal hyperplasia by interaction with free fatty acid receptor 4 in mice. J Am Heart Assoc2015;4:e001856.
- 139. Han L, Shen WJ, Bittner S, Kraemer FB, Azhar S. PPARs: regulators of metabolism and as therapeutic targets in cardiovascular disease. Part II: PPAR-beta/delta and PPAR-gamma. Future Cardiol2017;13:279–296.
- 140. Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, Grimaldi PA, Kadowaki T, Lazar MA, O'Rahilly S, Palmer CN, Plutzky J, Reddy JK, Spiegelman BM, Staels B, Wahli W. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev2006;58:726–741.
- 141. Glass CK, Ogawa S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat Rev Immunol2006;6:44–55.
- 142. Bervejillo L, Ferreira AM. Understanding peroxisome proliferator-activated receptors: from the structure to the regulatory actions on metabolism. Adv Exp Med Biol2019;1127:39–57.
- 143. Brown JD, Plutzky J. Peroxisome proliferator-activated receptors as transcriptional nodal points and therapeutic targets. Circulation2007;115:518–533.
- 144. Grygiel-Gorniak B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications—a review. Nutr J2014;13: 17.
- 145. Berger JP, Akiyama TE, Meinke PT. PPARs: therapeutic targets for metabolic disease. Trends Pharmacol Sci2005;26:244–251.
- 146. Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med2004;10:355–361.
- 147. Cheang WS, Tian XY, Wong WT, Huang Y. The peroxisome proliferator-activated receptors in cardiovascular diseases: experimental benefits and clinical challenges. Br J Pharmacol2015;172:5512–5522.
- 148. Xu HE, Lambert MH, Montana VG, Parks DJ, Blanchard SG, Brown PJ, Sternbach DD, Lehmann JM, Wisely GB, Willson TM, Kliewer SA, Milburn MV. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell1999;3:397–403.
- 149. Nelson JR, Wani O, May HT, Budoff M. Potential benefits of eicosapentaenoic acid on atherosclerotic plaques. Vasc Pharmacol2017;91:1–9.
- 150. Wong ND, Toth PP, Amsterdam EA; American Society for Preventive Cardiology. Most important advances in preventive cardiology during this past decade: viewpoint from the American Society for Preventive Cardiology. Trends Cardiovasc Med2019;doi: 10.1016/j.tcm.2019.11.013.
- 151. Jackson SM, Parhami F, Xi XP, Berliner JA, Hsueh WA, Law RE, Demer LL. Peroxisome proliferator-activated receptor activators target human endothelial cells to inhibit leukocyte-endothelial cell interaction. Arterioscler Thromb Vasc Biol1999;19:2094–2104.
- 152. Marx N, Sukhova GK, Collins T, Libby P, Plutzky J. PPARalpha activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation1999;99:3125–3131.
- 153. Sethi S, Ziouzenkova O, Ni H, Wagner DD, Plutzky J, Mayadas TN. Oxidized omega-3 fatty acids in fish oil inhibit leukocyte-endothelial interactions through activation of PPAR alpha. Blood2002;100:1340–1346.
- 154. Huang PH, Sata M, Nishimatsu H, Sumi M, Hirata Y, Nagai R. Pioglitazone ameliorates endothelial dysfunction and restores ischemia-induced angiogenesis in diabetic mice. Biomed Pharmacother2008;62:46–52.
- 155. Quintela AM, Jiménez R, Gómez-Guzmán M, Zarzuelo MJ, Galindo P, Sánchez M, Vargas F, Cogolludo Á, Tamargo J, Pérez-Vizcaíno F, Duarte J. Activation of peroxisome proliferator-activated receptor-beta/-delta (PPARbeta/delta) prevents endothelial dysfunction in type 1 diabetic rats. Free Radic Biol Med2012;53:730–741.
- 156. Tian XY, Wong WT, Wang N, Lu Y, Cheang WS, Liu J, Liu L, Liu Y, Lee SS, Chen ZY, Cooke JP, Yao X, Huang Y. PPARdelta activation protects endothelial function in diabetic mice. Diabetes2012;61:3285–3293.
- 157. Wong WT, Tian XY, Xu A, Yu J, Lau CW, Hoo RL, Wang Y, Lee VW, Lam KS, Vanhoutte PM, Huang Y. Adiponectin is required for PPARgamma-mediated improvement of endothelial function in diabetic mice. Cell Metab2011;14:104–115.
- 158. Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP, Delerive P, Fadel A, Chinetti G, Fruchart JC, Najib J, Maclouf J, Tedgui A. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature1998;393:790–793.
- 159. Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature1998;391:79–82.
- 160. Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med2001;7:48–52.
- 161. Tordjman KM, Semenkovich CF, Coleman T, Yudovich R, Bak S, Osher E, Vechoropoulos M, Stern N. Absence of peroxisome proliferator-activated receptor-alpha abolishes hypertension and attenuates atherosclerosis in the Tsukuba hypertensive mouse. Hypertension2007;50:945–951.
- 162. Babaev VR, Ishiguro H, Ding L, Yancey PG, Dove DE, Kovacs WJ, Semenkovich CF, Fazio S, Linton MF. Macrophage expression of peroxisome proliferator-activated receptor-alpha reduces atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation2007;116:1404–1412.
- 163. Kanda T, Brown JD, Orasanu G, Vogel S, Gonzalez FJ, Sartoretto J, Michel T, Plutzky J. PPARgamma in the endothelium regulates metabolic responses to high-fat diet in mice. J Clin Invest2009;119:110–124.
- 164. Jung UJ, Torrejon C, Chang CL, Hamai H, Worgall TS, Deckelbaum RJ. Fatty acids regulate endothelial lipase and inflammatory markers in macrophages and in mouse aorta: a role for PPARgamma. Arterioscler Thromb Vasc Biol2012;32:2929–2937.
- 165. Chang HY, Lee HN, Kim W, Surh YJ. Docosahexaenoic acid induces M2 macrophage polarization through peroxisome proliferator-activated receptor gamma activation. Life Sci2015;120:39–47.
- 166. Jung TW, Park HS, Choi GH, Kim D, Ahn SH, Kim DS, Lee T, Jeong JH. Maresin 1 attenuates pro-inflammatory reactions and ER stress in HUVECs via PPARalpha-mediated pathway. Mol Cell Biochem2018;448:335–347.
- 167. AIM-HIGHInvestigators, Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W.Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med2011;365:2255–2267.
- 168. Davidson MH, Rosenson RS, Maki KC, Nicholls SJ, Ballantyne CM, Mazzone T, Carlson DM, Williams LA, Kelly MT, Camp HS, Lele A, Stolzenbach JC. Effects of fenofibric acid on carotid intima-media thickness in patients with mixed dyslipidemia on atorvastatin therapy: randomized, placebo-controlled study (FIRST). Arterioscler Thromb Vasc Biol2014;34:1298–1306.
- 169. ACCORD Study Group, Ginsberg HN, Elam MB, Lovato LC, Crouse JR, 3rd Leiter LA, Linz P, Friedewald WT, Buse JB, Gerstein HC, Probstfield J, Grimm RH, Ismail-Beigi F, Bigger JT, Goff DC Jr, Cushman WC, Simons-Morton DG, Byington RP.Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med2010;362:1563–1574.
- 170. HPS2-THRIVE Collaborative Group, Landray MJ, Haynes R, Hopewell JC, Parish S, Aung T, Tomson J, Wallendszus K, Craig M, Jiang L, Collins R, Armitage J.Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med2014;371:203–212.
- 171. Karwi QG, Uddin GM, Ho KL, Lopaschuk GD. Loss of metabolic flexibility in the failing heart. Front Cardiovasc Med2018;5:68.
- 172. Kanda H, Nohara R, Hasegawa K, Kishimoto C, Sasayama S. A nuclear complex containing PPARalpha/RXRalpha is markedly downregulated in the hypertrophied rat left ventricular myocardium with normal systolic function. Heart Vessels2000;15:191–196.
- 173. Oka S, Alcendor R, Zhai P, Park JY, Shao D, Cho J, Yamamoto T, Tian B, Sadoshima J. PPARalpha-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway. Cell Metab2011;14:598–611.
- 174. Guellich A, Damy T, Lecarpentier Y, Conti M, Claes V, Samuel JL, Quillard J, Hebert JL, Pineau T, Coirault C. Role of oxidative stress in cardiac dysfunction of PPARalpha-/- mice. Am J Physiol Heart Circ Physiol2007;293:H93–H102.
- 175. Smeets PJ, Teunissen BE, Willemsen PH, van Nieuwenhoven FA, Brouns AE, Janssen BJ, Cleutjens JP, Staels B, van der Vusse GJ, van Bilsen M. Cardiac hypertrophy is enhanced in PPAR alpha-/- mice in response to chronic pressure overload. Cardiovasc Res2008;78:79–89.
- 176. Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest2002;109:121–130.
- 177. Ding G, Fu M, Qin Q, Lewis W, Kim HW, Fukai T, Bacanamwo M, Chen YE, Schneider MD, Mangelsdorf DJ, Evans RM, Yang Q. Cardiac peroxisome proliferator-activated receptor gamma is essential in protecting cardiomyocytes from oxidative damage. Cardiovasc Res2007;76:269–279.
- 178. Duan SZ, Ivashchenko CY, Russell MW, Milstone DS, Mortensen RM. Cardiomyocyte-specific knockout and agonist of peroxisome proliferator-activated receptor-gamma both induce cardiac hypertrophy in mice. Circ Res2005;97:372–379.
- 179. Smeets PJ, Teunissen BE, Planavila A, de Vogel-van den Bosch H, Willemsen PH, van der Vusse GJ, van Bilsen M. Inflammatory pathways are activated during cardiomyocyte hypertrophy and attenuated by peroxisome proliferator-activated receptors PPARalpha and PPARdelta. J Biol Chem2008;283:29109–29118.
- 180. Cheng L, Ding G, Qin Q, Huang Y, Lewis W, He N, Evans RM, Schneider MD, Brako FA, Xiao Y, Chen YE, Yang Q. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med2004;10:1245–1250.
- 181. Tavazzi L, Maggioni AP, Marchioli R, Barlera S, Franzosi MG, Latini R, Lucci D, Nicolosi GL, Porcu M, Tognoni G; Gissi-HF Investigators. Effect of n-3 polyunsaturated fatty acids in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet2008;372:1223–1230.
- 182. Shimojo N, Jesmin S, Sakai S, Maeda S, Miyauchi T, Mizutani T, Aonuma K, Kawano S. Fish oil constituent eicosapentaenoic acid inhibits endothelin-induced cardiomyocyte hypertrophy via PPAR-alpha. Life Sci2014;118:173–178.
- 183. Baranowski M, Blachnio-Zabielska A, Gorski J. Peroxisome proliferator-activated receptor alpha activation induces unfavourable changes in fatty acid composition of myocardial phospholipids. J Physiol Pharmacol2009;60:13–20.
- 184. Iwami D, Zhang Q, Aramaki O, Nonomura K, Shirasugi N, Niimi M. Purified eicosapentaenoic acid induces prolonged survival of cardiac allografts and generates regulatory T cells. Am J Transplant2009;9:1294–1307.
- 185. Yin R, Huang H, Zhang J, Zhu J, Jing H, Li Z. Dietary n-3 fatty acids attenuate cardiac allograft vasculopathy via activating peroxisome proliferator-activated receptor-gamma. Pediatr Transplant2008;12:550–556.
- 186. Anderson EJ, Thayne KA, Harris M, Shaikh SR, Darden TM, Lark DS, Williams JM, Chitwood WR, Kypson AP, Rodriguez E. Do fish oil omega-3 fatty acids enhance antioxidant capacity and mitochondrial fatty acid oxidation in human atrial myocardium via PPARgamma activation? Antioxid Redox Signal 2014;21:1156–1163.