Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most common liver disease and it has an increasing prevalence worldwide. NAFLD has been linked to enhanced risk of several diseases such as type 2 diabetes, cardiovascular and chronic kidney diseases. NAFLD is defined by accumulation of excess fat in the liver and its severity includes simple fatty infiltration and non-alcoholic steatohepatitis (NASH). Hepatic fat accumulation results from the excessive delivery of free fatty acids from peripheral tissue and imbalance between lipid synthesis and export in hepatocytes. At the molecular level, several important genes regulate lipid metabolism in the liver. Activation of the sterol regulatory element-binding protein 1 (SREBP1) and its target genes fatty acid synthase (FAS) and acetyl-coenzyme (Co) A carboxylase (ACC) induces lipogenesis in hepatocytes., Peroxisome proliferator–activated receptor (PPAR)α is another transcriptional regulator of lipid metabolism that regulates the expression of genes involved in mitochondrial β-oxidation such as palmitoyl transferase 1 (CPT-1) and long-chain L-3-hydroxyacylcoenzyme A dehydrogenase α (HADHα). Upregulation of the PPARα has been reported to inhibit lipid accumulation in liver cells.
The nuclear factor erythroid 2-related factor 2 (Nrf2) has been recognized as the master transcription factor that upregulates the expression of antioxidant genes such as superoxide dismutase (SOD), hemeoxygenase‐1 (HO‐1) and glutathione peroxidase (GPx)., Nrf2 has been reported to be involved in the development of NAFLD and NASH, because it suppressed the expression of genes related to lipogenesis. In this regard, high-fat diet fed mice lacking the Nrf2 exhibited severe liver injury and accelerated NASH progression, whereas overexpression of Nrf2 via inhibition of Keap1 reduces their sensitivity to NASH. Decreased activation of the Nrf2 signaling has been reported in NAFLD and activation of this transcription factor might be a strategy to prevent oxidative stress and subsequently excessive lipid deposition in the liver. Recently published works studies have shown that small molecule compounds could alleviate lipid accumulation in hepatocytes through activation of the Nrf2 signaling pathway.,,
Kaempferol (3,4/,5,1-tetrahydroxyflavoune) (KMF), a natural flavonoid compound exists in many kinds of vegetables and fruits. Kaempferol has been demonstrated to have several beneficial biological activities, such as anti-inflammatory, antioxidant, and antihyperglycemic effects.– In addition, the studies have reported that KMF had lipid lowering effects. In this regard, KMF has been shown to ameliorate hepatic simple steatosis, non-alcoholic steatohepatitis and NASH in mice fed high fat diet., However, the mechanism by which KMF mitigated lipid accumulation in liver cells remains unclear. The goal of this study was to explore the effects and molecular mechanism of KMF on palmitate (PA)-induced lipid accumulation in HepG2 cells. Here, we have focused on the Nrf2 signaling to understand whether the beneficial effects of KMF is mediated through activation of the Nrf2 signaling pathway. In this study, we treated HepG2 cells with palmitate to create an experimental model of NAFLD. Palmitate, as the most abundant saturate fatty acid in circulation, has been implicated in NAFLD initiation and development.
Materials and methods
Materials and reagents
Kaempferol was obtained by Sigma-Aldrich (St. Louis, MO, USA). The bicinchoninic acid assay (BCA) assay was obtained from Wuhan Boster Bio-engineering Co., Ltd. (Wuhan, China). Antibodies against Nrf2, β-actin, CPT-1, ACC, p-ACC, SREBP1, PPAR-α, and lamin B1 were provided by Cell Signaling Technology. Palmitic acid and common chemicals were purchased from Sigma Aldrich (St. Louis, MO). 2,7-dichlorofluorescein diacetate (DCFHDA) were from Nanjing Jiancheng Bioengineering Institute, China.
Cell culture and treatment
Human hepatoma HepG2 cell line was obtained from Cancer Institute of the Fourth Military Medical University (Xi’an, China). HepG2 cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Meilun Biotechnology, Dalian, China) in a humidified 5%CO2 at 37°C. The cells were treated with 0.5 mM PA for 24 h. HepG2 cells were also pretreated with various concentrations of KMF (5, 10, 20, 40 and 80 μM) 2 h before treatment with PA. The range of KMF concentrations was based on previous reports., Palmitate-BSA complex solution was prepared as previously described. In brief, 100 mM PA was dissolved in 0.1 N NaOH. To make a stock solution of 5 mM, 100 mM PA was diluted with 10% free fatty acid-free BSA-DMEM. The final concentration of PA for cell treatment was 0.5 mM. Selected dose of PA was based on previous reports.,
Small interfering RNA transfection
Nrf2-siRNA was obtained from Suzhou GenePharma LLC, China. A siRNA (scramble) with no homology to any known mammalian gene was used as a negative control. HepG2 cells were seeded in a 6-well cell-culture plate in growth medium until they reach a confluence of 50%. Next, Nrf2-siRNA or negative control siRNA (20 nM) were transiently transfected into the cells using lipofectamine™ 2000 reagent (Invitrogen, Waltham, USA). After 24 h, the transfected cells were treated with PA and KMF for another 24 h and then the cell lysate was used for western blot analysis.
Cell viability
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (Wolsen Biotechnology, Xian, China) assay was applied to investigate the cytotoxicity of PA and KMF in HepG2 cells. HepG2 cells were seeded at a density of 10 × 103 cells/well in 96-ell plates. The cells were then treated with PA for 24 h in the presence or absence of various concentrations of KMF. After washing of the cells with PBS, a volume of 100 μL of MTT (1 mg/mL in PBS) was added to each well of plate. Then the plate was incubated for 4 h at 37°C. Finally, after adding 100 μL DMSO, the absorbance was assessed at 510 nm.
Oil red O staining
Oil red O was used to stain lipids. Briefly, 5 × 105 HepG2 cells were seeded in 6-well plates. After pretreatment for 2 h with various concentrations of KMF, the cells were treated 0.5 mM palmitate and KMF for 24 h. The cells were washed and fixed with 4% paraformaldehyde for 30 min at 37°C. After washing with isopropanol, the cells were stained with Oil Red O solution for 1 h in a 60°C water bath. Lipid droplets in the cells were observed and imaged using a light microscope. To quantify lipid content, lipids were extracted using 100% isopropanol, and then the absorbance was read at 570 nm.
Measurement of triglyceride level
Intracellular triglyceride (TG) level was measured using triglyceride assay kit (Shensuo UNF, China) after lysis of the cells.
Measurement of malondialdehyde levels and superoxide dismutase, and glutathione peroxidase activities
5 × 105 HepG2 cells were cultured at 6-well plates. After 24 h, the cells were treated with PA and KMF for 24 h. The cells were then harvested and the levels of MDA, and the activities of SOD and GPx were measured by commercial kits according to manufacturer’s instructions (Nanjing Jiancheng Bioengineering Institute). MDA reacted with thiobarbituric acid (TBA) to form a red-complex compound featuring absorbance at 532 nm. MDA results were expressed in nmol/mg protein of the cells. In SOD measurements, superoxide ions reacted with 2-(4-iodophenyl)-3- (4-nitrophenol)-5-phenyltetrazolium chloride to form a red formazan dye featuring absorbance at 550 nm. SOD activity was expressed in U/mg protein of the cells. GPx activity was measured by a coupled assay using H2O2 and dithio-bis-nitrobenzoic acid (DTNB). GPx activity was assessed by dinitrobenzoic acid colorimetry and read at a wavelength of 412 nm. GPx activity was expressed in U/mg protein of the cells.
Measurement of intracellular reactive oxygen species level
Intracellular ROS levels were evaluated using a DCFH-DA. HepG2 cells (10 × l03 cells/well) were seeded in 96-well plates for 24 h incubation. After treatment with PA and KMF for 24 h and then washing with PBS buffer, the cells were incubated with DCFH-DA for 30 min. Fluorescence was determined using a microplate reader with excitation/emission wavelength at 485/525 nm.
Real-time quantitative PCR
Total RNA was extracted from the cells using a RNeasy Kit (Qiagen, Valencia, CA, USA). A cDNA Synthesis Kit (Vazyme, China) was used to synthesize first-stranded cDNA. Real-time Polymerase Chain Reaction (PCR) was conducted to evaluate gene expression using SYBR Green I (TaKaRa, Japan). Each experiment was repeated three times. The relative changes in mRNA expression were calculated by the 2 −DDCt method. The data were normalized to GAPDH. The sequences of the primers are shown in Table 1 of the supplementary file.
Western blot
Cytoplasmic and nuclear proteins from the cells were isolated as previously described. Protein concentration was measured by the bicinchoninic acid assay. After treatments with PA and KMF for 24 h, HepG2 cells were washed with PBS and then lysed using RIPA buffer supplemented with protease and phosphatase inhibitor cocktail (ThermoFisher Scientific, USA). 30 μg of proteins were separated by SDS-PAGE and transferred onto a polyvinylidene difluoride membrane. The blots were probed using the primary antibodies at 4°C overnight. All primary antibodies were diluted in 1% bovine serum albumin at a dilution of 1:1000. Antibodies against Nrf2, β-actin, CPT-1, ACC, p-ACC, SREBP1, PPAR-α, and lamin B1 were provided by Cell Signaling Technology. After that, the membranes were incubated with goat anti-rabbit (or anti-mouse) HRP-conjugated secondary antibodies for 1 h at room temperature. An ECL Plus detection reagent (Amersham Biosciences) was used to visualize protein bands and the band density was determined by Image J software (NIH, USA). All western blot experiments were conducted at least three times.
Luciferase reporter assay
HepG2 cells were seeded at a density of 2 × 105 cells/well 24-well plates to reach a confluence of 50%. ARE-Luc or the control Renilla pRL-TK vectors were transiently co-transfected into the cells using Lipofectamine 2000 transfection reagent. At 24 h after transfection, the cells were treated with PA and KMF for another 24 h. Luciferase and Renilla activities were assessed using a DualLuciferase® Reporter Assay System (Promega). Renilla activity was used to normalize the relative luciferase activity.
Statistical analysis
The results are expressed as mean ± standard error of the mean (SEM). One-way analysis of variance with Tukey’s test for post hoc analysis were used for statistical analysis. p < 0.05 was considered statistically significant.
Results
Kaempferol ameliorates palmitate-induced lipid accumulation in HepG2 cells
Previous works have suggested that PA can induce hepatocyte lipid accumulation in vitro and in vivo., To investigate the effects of KMF on lipid accumulation, we first evaluated the cytotoxicity effects of KMF on HepG2 cells. The data of MTT assay revealed that KMF (5–40 μM) had no cytotoxic effects on the cells. A slightly decrease of cell viability was observed in treatment of the cells with KMF 80 μM (p < 0.01) (Figure 1(a)). We also assessed the influence of KMF on cell viability of the cells in the presence of 0.5 mM PA for 24 h. As illustrated in Figure 1(b), PA treatment had cytotoxic effects, whereas KMF pretreatment dose-dependently reversed PA-induced decrease in HepG2 cell viability (p < 0.01) (Figure 1(b)). We then evaluated the impact of KMF on lipid metabolism. We first evaluated total lipid content in HepG2 cells by Oil red O staining. The cells were treated with 0.5 mM PA and various concentrations of KMF for 24 h. As illustrated in Figure 2(a) and (b), PA enhanced total lipid content in comparison with the control (p < 0.0001). Importantly, 10, 20 and 40 μM KMF could reduce total lipid content compared with treatment with PA alone. We verified these findings by measuring intracellular TG levels. Increased TG level following PA treatment was significantly prevented by co-treatment of PA with KMF at concentration of 10–40 μM (p < 0.0001) (Figure 2(c)). According to these findings, and because of no cytotoxic effect of KMF 20 μM, we selected KMF at concentration of 20 μM for the further experiments.
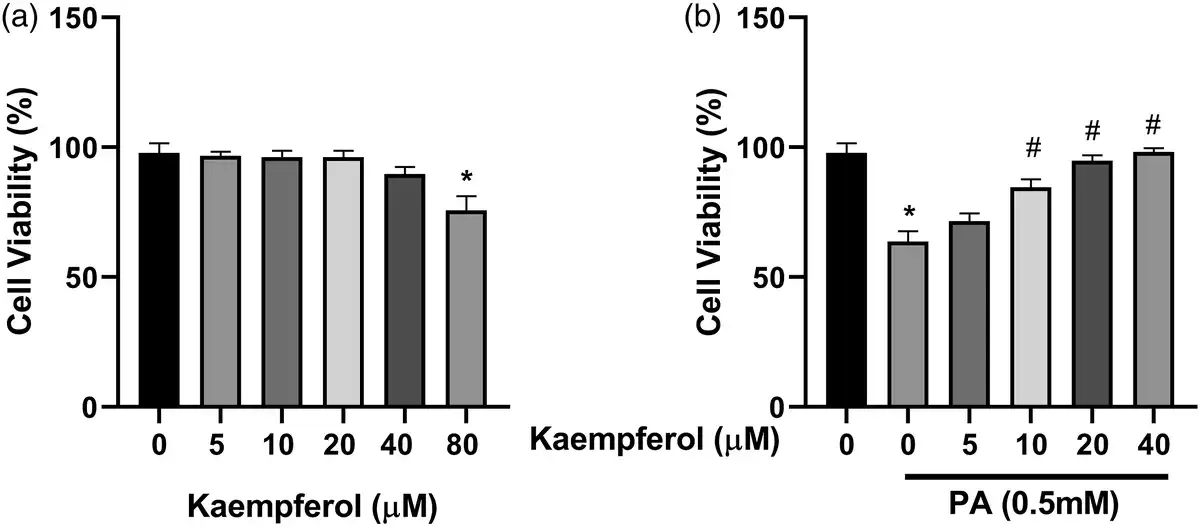
Figure 1
Effects of Kaempferol (KMF) and palmitate (PA) on cell viability. (a) Cell viability after treating with different doses of KMF and (b) combination of KMF and PA (0.5 mM). *p < 0.01 vs. control and #p < 0.01 vs. treatment with PA alone.
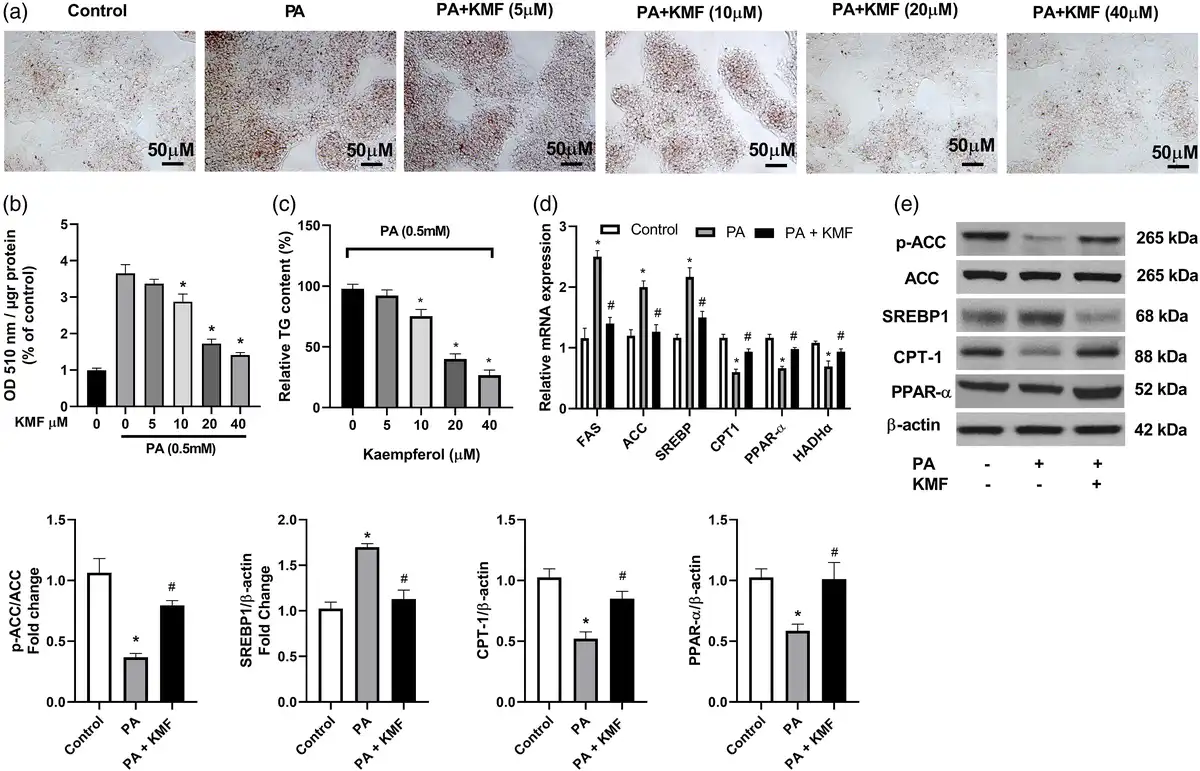
Figure 2
Effects of Kaempferol on PA-induced lipid deposition in HepG2 cells. (a, b) intracellular lipid content in HepG2 cells after treatment with different doses of KMF and PA (0.5 mM). (c) TG level following incubation with various doses of KMF and 0.5 mM PA for 24 h. (d) The relative mRNA expression of genes involved in lipid metabolism following treatment with KMF (20 μM) and 0.5 mM PA for 24 h. (e) The levels of p-ACC, CPT-1, PPAR-α and SREBP1 in the cells treated with KMF (20 μM) and 0.5 mM PA for 24 h. *p < 0.01 vs. control and #p < 0.01 vs. treatment with PA alone.
Next, we focused on molecular mechanisms by which KMF ameliorates TG accumulation in HepG2 cells by evaluating the key molecules involved in fatty acid synthesis and oxidation. We found that compared with the control cells, the mRNA and protein levels of ACC, FAS, and SREBP-1 were significantly increased following treatment with PA (p < 0.001). Kaempferol suppressed the mRNA and protein levels of ACC, FAS, and SREBP-1, (Figure 2(d) and (e)) in PA-treated cells, suggesting that KMF reduces de novo fatty acid synthesis in hepatocytes. To further validate the effect of KMF on lipid metabolism, we focused our efforts on genes responsible for driving fatty acid oxidation. HepG2 cells treated with PA showed lower mRNA and protein levels of CPT-1 and HADHα (p < 0.01) compared to untreated cells (Figure 2(d)), whereas the mRNA and protein levels of these genes were significantly up-regulated by KMF. It has been suggested that PPAR-α regulates lipid metabolism through activation of the expression of CPT1, and HADHα. Because these genes are up-regulated by KMF, we examined whether the expression of PPARα was also increased by KMF. KMF was able to enhance the mRNA and protein levels of PPARα in HepG2 cells treated with PA (p < 0.01), suggesting that KMF might improve lipid catabolism through regulating the expression of PPARα (Figure 2(d) and (e)).
Kaempferol alleviated PA-induced oxidative stress in HepG2 cells
Previous reports have shown that PA induces hepatocyte oxidative stress in vitro.,– To explore the role of KMF in PA-induced oxidative stress in hepatocytes, we evaluated several markers of oxidative stress. The results revealed that KMF reversed elevated ROS generation by PA in HepG2 cells (p < 0.0001) (Figure 3(a)). The activities of SOD and GPx were significantly elevated by stimulation with KMF (p < 0.01). Kaempferol significantly increased the activities of SOD and GPx in the cells treated with PA (p < 0.01) (Figure 3(b) and (c)). In comparison with the control group, the cells treated with PA had higher levels of MDA, and KMF significantly lowered the level of MDA induced by PA in HepG2 cells (Figure 3(d)).
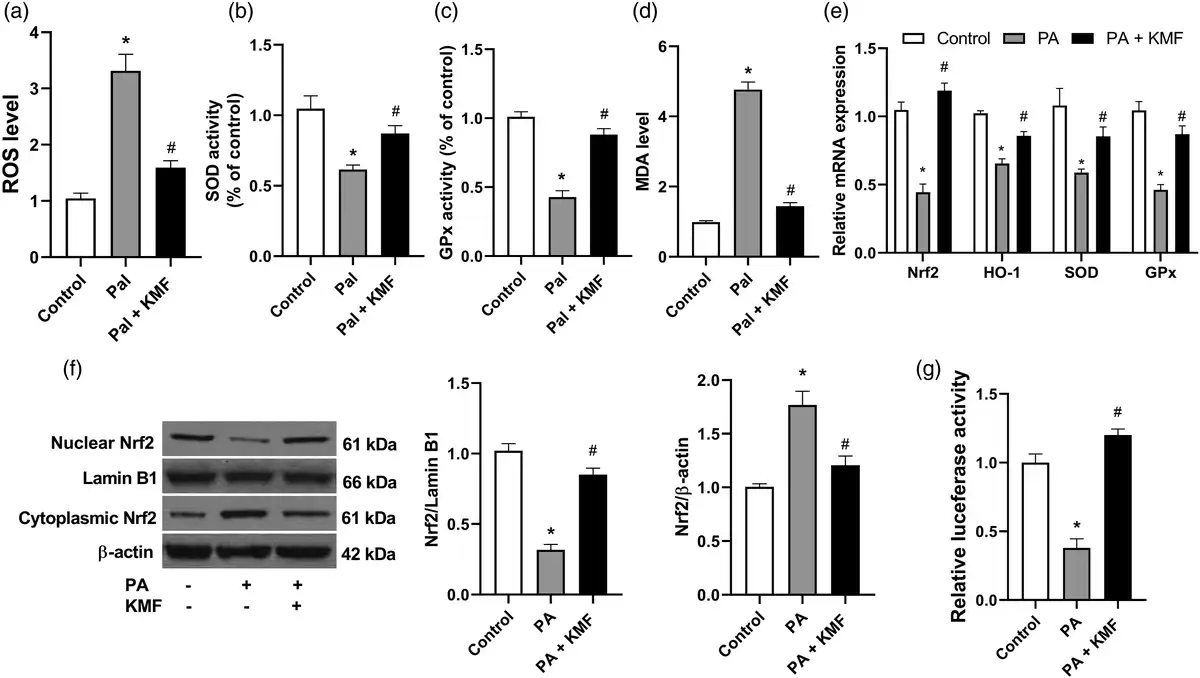
Figure 3
The impact of Kaempferol on the oxidative stress markers. (a) The levels of ROS, (b) SOD, (c) GPx and (d) MDA following treatment of the cells with KMF (20 μM) and PA (0.5 mM) for 24 h. (d) The relative mRNA expression of Nrf2, HO-1, SOD and GPx following treatment with KMF and PA for 24 h. (e) The levels of nuclear and cytoplasmic Nrf2 in the cells treated with KMF and PA for 24 h. (f) Luciferase activity in the cells treated with KMF and PA. *p < 0.01 vs. control and #p < 0.01 vs. treatment with PA alone.
The Nrf2 signaling pathway has been suggested to play an important role in the cellular defense against oxidative stress through upregulating the expression of antioxidant genes., In this study we evaluated the effects of KMF on Nrf2 activation and its downstream signaling. As shown in Figure 3(e), KMF prevented suppression of the expression of Nrf2 and its target genes including HO-1, SOD and GPx in the cells treated with PA (p < 0.01). We confirmed these findings by protein evaluation of Nrf2 by western blotting. Analysis of nuclear and cytosol fractions revealed that while PA decreased Nrf2 nuclear translocation, a significant increase of Nrf2 nuclear translocation was observed in the combined treatment of PA and KMF (p < 0.01) (Figure 3(f)). Moreover, our data demonstrated that PA decreased luciferase reporter activity (p < 0.001), whereas KMF prevented this reduction, indicating the transcriptional activation of Nrf2 in the cells treated with KMF (p < 0.01) (Figure 3(g)). These data suggest that KMF ameliorates PA-induced oxidative stress by activating the Nrf2 signaling pathway.
Nrf2 is critical for KMF-mediated protection against PA-induced oxidative stress
To evaluate whether Nrf2 activation had a significant role in KMF protection against PA-induced oxidative stress, we silenced Nrf2 using specific Nrf2 targeted siRNA. As shown in Figure 4(a), Nrf2 expression was significantly decreased after Nrf2 siRNA transfection. The ability of KMF to activate Nrf2 signaling was abolished by Nrf2 knockdown in HepG2 cells. The results showed that transfecting scramble siRNA led to increased expression of antioxidant genes such as HO-1, SOD and GPx in the cells treated with KMF (p < 0.01) (Figure 4(b)–(d)). However, KMF-mediated activation of the Nrf2 and its target genes was no found in the knockdown cells treated with PA. Furthermore, Nrf2 siRNA abolished the KMF-induced reduction in ROS and MDA levels in PA treated cells (Figure 4(e) and (f)).
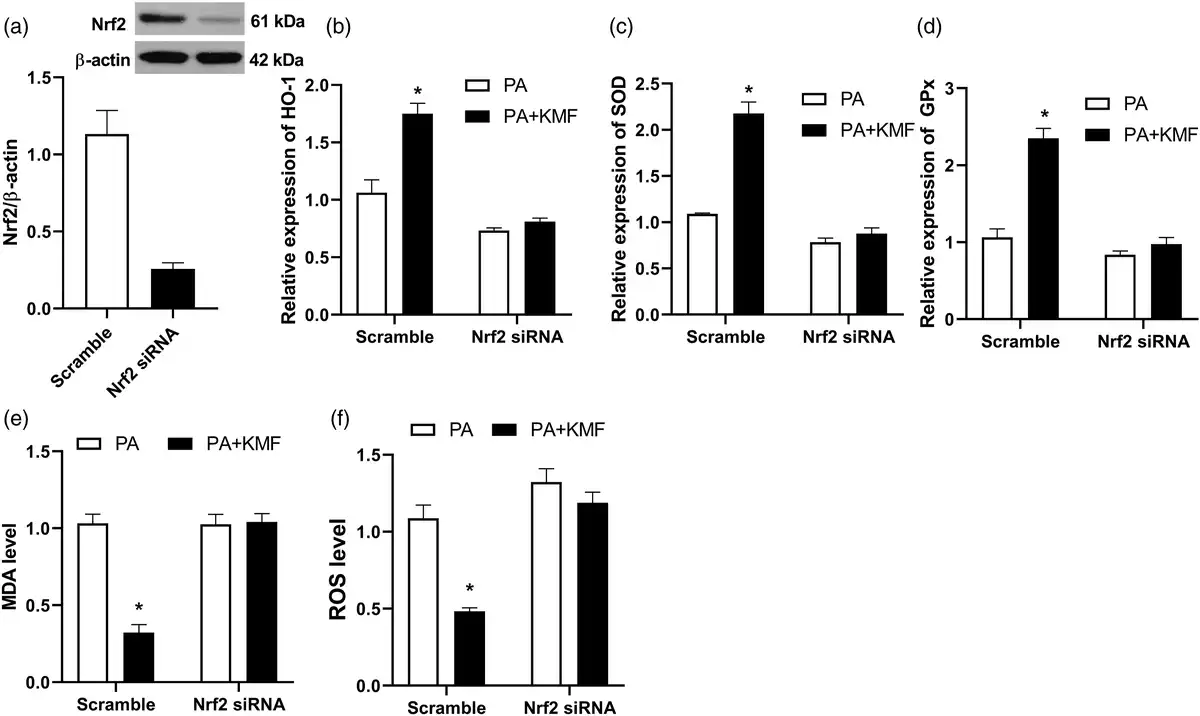
Figure 4
The effect of Nrf2 knockdown on PA-induced oxidative stress. (a) Nrf2 knockdown using siRNA. (b, c, d) After 24 h of transfection with Nrf2 or scrambled (control) siRNA, the relative expression of HO-1, SOD and GPx were analyzed using real-time PCR. The level of MDA (e) and ROS (f) in the scramble and Nrf2 knockdown cells. *p < 0.01 vs. PA treated cells.
Nrf2 is important for KMF-mediated protection against PA-induced lipid accumulation
Next, we studied the function of Nrf2 activation in PA-treated cells. Importantly, we observed an increase in TG level in Nrf2 knockdown cells following PA treatment or even in the cells co-treated with PA and KMF (p < 0.01) (Figure 5(a)). In addition, the inhibitory effect of KMF on TG level in PA treated cells was significantly abolished by Nrf2 inhibition (Figure 4(a)). At the molecular level, the KMF-mediated inhibition of the expression of FAS, ACC and SREPB-1 was not observed in the knockdown of Nrf2 (Figure 5(b)–(d)). These findings were confirmed at the protein level of p-ACC and SREBP1 (Figure 4(h)). We also observed that Nrf2 inhibition using siRNA suppressed the KMF-induced activation of genes involved in β oxidation (CPT-1, HADHα and PPAR-α) (Figure 5(e), (f) and (h)). Taken together, above findings suggest that that KMF-mediated the inhibition of lipid accumulation in hepatocytes might be mediated through activation of the Nrf2 signaling pathway.
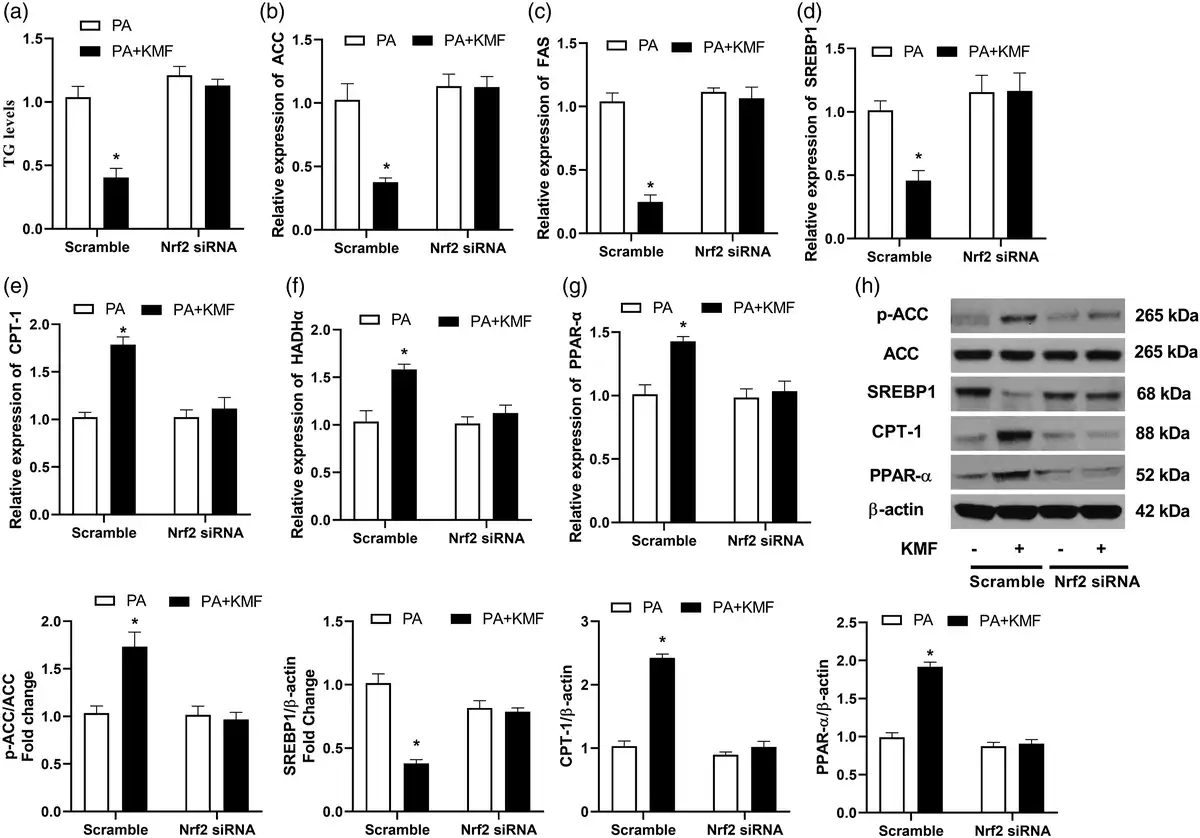
Figure 5
The effect of Nrf2 knockdown on PA-induced lipid accumulation. After 24 h of transfection with Nrf2 or scrambled (control) siRNA, the level of TG (a), the relative expression of ACC (b), FAS (c), SREBP (d), CPT-1 (e), HADHα (f) and PPAR-α (g) were analyzed using real-time PCR. The protein levels p-ACC, SREBP1, CPT1 and PPAR-α were evaluated using western blotting (h) in the scramble and Nrf2 silenced cells. *p < 0.01 vs. PA treated cells.
Discussion
The development of NAFLD and NASH has been associated to increased lipids deposition. Previous investigations have suggested that free fatty acids specifically statured fatty acids such as laurate and PA enhance lipid accumulation and induce pro-inflammatory responses in hepatocytes., In this study, we induced lipid accumulation in HepG2 cells by exposing them to pathophysiologically concentration of PA (0.5 mM) to simulate the excessive influx of fatty acids into hepatocytes. Our results suggested that treatment of HepG2 cells with PA results in increased TG level, which was evident from Oil Red O staining. Lipid accumulation following PA treatment was closely linked with activation of de novo lipogenesis and inhibition of β-oxidation. These data are in agreement with previous reports.,
The rate of lipid accumulation is governed by activities of two major pathways involved in lipid metabolism, de novo lipogenesis and β-oxidation. Our findings clearly demonstrate that pretreatment with KMF significantly prevented lipid accumulation in cells exposed to PA. Reduction in lipid accumulation in the cells pretreated with KMF was accompanied with decrease in the mRNA and protein levels of lipogenesis related genes (FAS, ACC and SREBP1) and upregulation of β-oxidation related genes (CPT1, HADHα and PPARα). In the line with above findings, a study demonstrated that KMF prevented lipid accumulation by coordinately suppressing lipid synthesis and up-regulating fatty acid oxidation in adipocytes. Specifically, it was revealed that KMF can bind to and activate PPARα and subsequently induces the expression of genes related to fatty acid oxidation. In another study, KMF administration was able to ameliorate fat accumulation in the liver of HFD-fed rats through PPARα-mediated increased fatty acid oxidation and down-regulation of SREBP expression. Tie et al. also provided evidence regarding the protective role of KMF against lipid accumulation in hepatocytes exposed to oleic acid. They used oleic acid as an inducer of lipid accumulation, whereas we used palmitate. It is on note that the differential effects of oleic and palmitic acids on lipid-droplet accumulation in the hepatic cell line HepG2, has been previously reported. Palmitic acid, as the abundant saturated fatty acid present in the diet and serum is able to trigger lipotoxicity in hepatocytes. Contrarily, unsaturated fatty acids, such as oleic acid are much nontoxic, and are proven able to combat PA-induced lipotoxicity in hepatocytes. Taken together, above findings imply that the inhibitory impact of KMF on lipid accumulation in hepatocytes is probably exerted through modulating the expression of key molecules involved in lipid metabolism. However, the molecular mechanisms underlying KMF-mediated inhibition of lipid accumulation in hepatocytes are unclear.
Oxidative stress has been identified to have a major role in progression from NAFLD to NASH., Evidence has suggested that elevated ROS generation is the main factor driving mitochondrial dysfunction, hepatocyte damage, inflammation and fibrosis in the pathogenesis of NAFLD., Therefore, suppressing hepatic oxidative stress through activating the antioxidant system might be strategy for NAFLD prevention and treatment. In this regard, increasing the activities of antioxidant enzymes is an important approach for alleviating hepatic oxidative stress. These proteins have been demonstrated to be regulated by Nrf2, the main regulator of the antioxidant defense system. In addition, Nrf2 activation has been reported to have a protective role against hepatocyte oxidative stress in NAFLD models. To test whether the anti-lipogenic effects of KMF is mediated through regulating the oxidative stress process, we evaluated several markers of oxidative stress in HepG2 cells. Kaempferol pretreatment to PA exposed cells, reduced ROS production and MDA level. Pretreatment with KMF led to nuclear translocation of Nrf2 and subsequently increased both the expression and activities of its target genes HO-1, SOD and GPx. In support of these findings, the protective role of KMF against oxidative stress in other cells has been previously reported., To obtain more insight into mechanisms by which KMF activates Nrf2, we performed luciferase reporter assay and found that Nrf2 is activated following KMF treatment. Furthermore, in PA-exposed cells, silencing of Nrf2 using siRNA significantly suppresses the upregulation of antioxidant genes and reduction of ROS and MDA levels by KMF. These findings confirm the crucial role of the Nrf2 signaling in control of oxidative stress in NAFLD.
Considering the inhibitory function of KMF on oxidative stress through upregulating the expression and activity of antioxidative proteins and because KMF suppresses PA-lipid accumulation, we speculated that KMF might inhibit lipid accumulation by activating the Nrf2 signaling pathway. To assess this hypothesis, we evaluated PA-induced lipid accumulation in hepatocytes with reduced Nrf2 level by siRNA. We observed that reduction in intracellular TG levels by KMF were abolished by transfection with Nrf2 siRNA. Next, it was verified that KMF did not down-regulate SREBP1, FAS, and ACC mRNA and protein levels in Nrf2 siRNA transfected HepG2 cells. In addition, KMF could not upregulated the mRNA and protein levels of genes involved in β-oxidation when Nrf2 activation was blocked. These data suggest that the enhancement of the antioxidant defenses via Nrf2 activation is a mechanism by which KMF prevents lipid accumulation in hepatocytes exposed to PA. In support of this idea, the beneficial anti-lipogenic function of several polyphenols through the activation of the Nrf2 signaling has been previously reported. A previous study reported that the prolonged activation of HO-1 with the use of a Nrf2-specific activator compensated PA-induced oxidative stress. Study by Valdecantos et al. revealed that Nrf2 is a key player in suppression of oxidative stress by α-lipoic acid leading to protection against PA-mediated lipoapoptosis. In another study, It was also suggested that punicalagin ameliorated PA-mediated lipotoxicity in HepG2 cells by enhancing the activity of the Keap1-Nrf2 antioxidant defense system.
In summary, the findings of the present study provided the evidence that KMF can activate the Nrf2-mediated antioxidative system and suppresses PA-induced ROS overproduction and subsequently relieves hepatic lipid accumulation by down regulating lipogenic genes and upregulating the expression of genes related to β-oxidation. These data suggest that KMF has a therapeutic potential to be used for the improvement of NAFLD.
Author contributions L.Z: Performed the experiments, wrote the manuscript with support from L.Y and A.M. L.Y: Conceived and planned the experiments, Supervised the project. K.M: Performed the experiments, contributed to the interpretation of the results, wrote the paper
Declaration of conflicting interests The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding The author(s) received no financial support for the research, authorship, and/or publication of this article.
Data availability statements All data generated or analyzed during this study are included in this published article
Supplemental Material Supplemental Material for this article is available online
References
- 1. Paschos P, Paletas K. Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia 2009; 13: 9.
- 2. Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell and Molecular Life Sciences 2018; 75: 3313–3327.
- 3. Shen B, Zhao C, Wang Y, et al. Aucubin inhibited lipid accumulation and oxidative stress via Nrf2/HO-1 and AMPK signalling pathways. J Cellular and Molecular Medicine 2019; 23: 4063–4075.
- 4. Geng Y, Villanueva AH, Oun A, et al. Protective effect of metformin against palmitate-induced hepatic cell death. Biochim Biophys Acta (BBA)-Molecular Basis Dis 2020; 1866: 165621.
- 5. Shimano H. Sterol regulatory element-binding proteins (SREBPs): transcriptional regulators of lipid synthetic genes. Prog Lipid Research 2001; 40: 439–452.
- 6. Bougarne N, Weyers B, Desmet SJ, et al. Molecular actions of PPAR α in lipid metabolism and inflammation. Endocr Reviews 2018; 39: 760–802.
- 7. Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatology 2015; 62: 720–733.
- 8. Hybertson BM, Gao B, Bose SK, et al. Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Mol Aspects Medicine 2011; 32: 234–246.
- 9. Jin M, Feng H, Wang Y, et al. Gentiopicroside ameliorates oxidative stress and lipid accumulation through nuclear factor erythroid 2-related factor 2 activation. Oxidative Med and Cell Longevity 2020; 2020: 2940746.
- 10. Kitteringham NR, Abdullah A, Walsh J, et al. Proteomic analysis of Nrf2 deficient transgenic mice reveals cellular defence and lipid metabolism as primary Nrf2-dependent pathways in the liver. J Proteomics 2010; 73: 1612–1631.
- 11. Chowdhry S, Nazmy MH, Meakin PJ, et al. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radical Biology Medicine 2010; 48: 357–371. DOI: .
- 12. Zhang YK, Yeager RL, Tanaka Y, et al. Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine- and choline-deficient diet. Toxicol and Applied Pharmacology 2010; 245: 326–334. DOI: .
- 13. Valenzuela R, Videla LA. Crosstalk mechanisms in hepatoprotection: thyroid hormone-docosahexaenoic acid (DHA) and DHA-extra virgin olive oil combined protocols. Pharmacol Research 2018; 132: 168–175.
- 14. Li J, Wang T, Liu P, et al. Hesperetin ameliorates hepatic oxidative stress and inflammation via the PI3K/AKT-Nrf2-ARE pathway in oleic acid-induced HepG2 cells and a rat model of high-fat diet-induced NAFLD. Food Funct 2021; 12: 3898–3918.
- 15. Alam W, Khan H, Shah MA, et al. Kaempferol as a dietary anti-inflammatory agent: current therapeutic standing. Molecules 2020; 25: 4073.
- 16. Ashrafizadeh M, Tavakol S, Ahmadi Z, et al. Therapeutic effects of kaempferol affecting autophagy and endoplasmic reticulum stress. Phytotherapy Res 2020; 34: 911–923.
- 17. Du W, An Y, He X, et al. Protection of kaempferol on oxidative stress-induced retinal pigment epithelial cell damage. Oxidative Med and Cell Longevity 2018; 2018: 1610751.
- 18. Sharma D, Gondaliya P, Tiwari V, et al. Kaempferol attenuates diabetic nephropathy by inhibiting RhoA/Rho-kinase mediated inflammatory signalling. Biomed Pharmacother 2019; 109: 1610–1619.
- 19. Zhang J, Zhang S-d, Wang P, et al. Pinolenic acid ameliorates oleic acid-induced lipogenesis and oxidative stress via AMPK/SIRT1 signaling pathway in HepG2 cells. Eur Journal Pharmacology 2019; 861: 172618.
- 20. Liu P, Wu P, Yang B, et al. Kaempferol prevents the progression from simple steatosis to non-alcoholic steatohepatitis by inhibiting the NF-κB pathway in oleic acid-induced HepG2 cells and high-fat diet-induced rats. J Funct Foods 2021; 85: 104655.
- 21. Xiang H, Shao M, Lu Y, et al. Kaempferol alleviates steatosis and inflammation during early non-alcoholic steatohepatitis associated with liver X receptor α-lysophosphatidylcholine acyltransferase 3 signaling pathway. Front Pharmacol 2021; 12: 1619.
- 22. Rada P, González-Rodríguez Á, García-Monzón C, et al. Understanding lipotoxicity in NAFLD pathogenesis: is CD36 a key driver? Cell Death Disease 2020; 11: 1–15.
- 23. Lee B, Kwon M, Choi JS, et al. Kaempferol isolated from Nelumbo nucifera inhibits lipid accumulation and increases fatty acid oxidation signaling in adipocytes. J Medicinal Food 2015; 18: 1363–1370.
- 24. Zhu G, Liu X, Li H, et al. RETRACTED: Kaempferol inhibits proliferation, migration, and invasion of liver cancer HepG2 cells by down-regulation of microRNA-21. Int Journal Immunopathology and Pharmacology 2018; 32: 2058738418814341.
- 25. Feng X-T, Wang T-Z, Leng J, et al. Palmitate contributes to insulin resistance through downregulation of the Src-mediated phosphorylation of Akt in C2C12 myotubes. Biosci Biotechnology, and Biochemistry 2012; 76: 1356–1361.
- 26. Penke M, Schuster S, Gorski T, et al. Oleate ameliorates palmitate-induced reduction of NAMPT activity and NAD levels in primary human hepatocytes and hepatocarcinoma cells. Lipids Health and Disease 2017; 16: 191. DOI: .
- 27. Song YM, Song S-O, Jung Y-K, et al. Dimethyl sulfoxide reduces hepatocellular lipid accumulation through autophagy induction. Autophagy 2012; 8: 1085–1097.
- 28. Lu J, Meng Z, Cheng B, et al. Apigenin reduces the excessive accumulation of lipids induced by palmitic acid via the AMPK signaling pathway in HepG2 cells. Exp and Therapeutic Medicine 2019; 18: 2965–2971.
- 29. Liu P, Shi L, Cang X, et al. CtBP2 ameliorates palmitate-induced insulin resistance in HepG2 cells through ROS mediated JNK pathway. Gen and Comparative Endocrinology 2017; 247: 66–73.
- 30. Mandard S, Müller M, Kersten S. Peroxisome proliferator-activated receptor α target genes. Cell and Mol Life Sci CMLS 2004; 61: 393–416.
- 31. Teimouri M, Hosseini H, Shabani M, et al. Inhibiting miR-27a and miR-142-5p attenuate nonalcoholic fatty liver disease by regulating Nrf2 signaling pathway. IUBMB Life 2020; 72: 361–372.
- 32. Zamani-Garmsiri F, Ghasempour G, Aliabadi M, et al. Combination of metformin and chlorogenic acid attenuates hepatic steatosis and inflammation in high-fat diet fed mice. IUBMB Life 2021; 73: 252–263.
- 33. Aleksunes LM, Manautou JE. Emerging role of Nrf2 in protecting against hepatic and gastrointestinal disease. Toxicologic Pathology 2007; 35: 459–473.
- 34. Bataille A, Manautou J. Nrf2: a potential target for new therapeutics in liver disease. Clin Pharmacol Ther 2012; 92: 340–348.
- 35. Puri P, Wiest MM, Cheung O, et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 2009; 50: 1827–1838.
- 36. Ricchi M, Odoardi MR, Carulli L, et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J Gastroenterology and Hepatology 2009; 24: 830–840.
- 37. Chang CJ, Tzeng T-F, Liou S-S, et al. Kaempferol regulates the lipid-profile in high-fat diet-fed rats through an increase in hepatic PPARα levels. Planta Medica 2011; 77: 1876–1882.
- 38. Tie F, Ding J, Hu N, et al. Kaempferol and kaempferide attenuate oleic acid-induced lipid accumulation and oxidative stress in HepG2 cells. Int J Mol Sci 2021; 22: 8847.
- 39. Lee J-y, Cho H-K, Kwon YH. Palmitate induces insulin resistance without significant intracellular triglyceride accumulation in HepG2 cells. Metabolism 2010; 59: 927–934.
- 40. Ahn JH, Kim MH, Kwon HJ, et al. Protective effects of oleic acid against palmitic acid-induced apoptosis in pancreatic AR42J cells and its mechanisms. The Korean J Physiol Pharmacol Official J Korean Physiol Soc and Korean Soc Pharmacol 2013; 17: 43.
- 41. Valdecantos MP, Prieto-Hontoria PL, Pardo V, et al. Essential role of Nrf2 in the protective effect of lipoic acid against lipoapoptosis in hepatocytes. Free Radic Biol and Med 2015; 84: 263–278.
- 42. Lee J, Park J-S, Roh YS. Molecular insights into the role of mitochondria in non-alcoholic fatty liver disease. Arch Pharmacal Research 2019; 42: 935–946.
- 43. Wang J, Fang X, Ge L, et al. Antitumor, antioxidant and anti-inflammatory activities of kaempferol and its corresponding glycosides and the enzymatic preparation of kaempferol. PLoS One 2018; 13: e0197563.
- 44. Sharma N, Biswas S, Al-Dayan N, et al. Antioxidant role of kaempferol in prevention of hepatocellular carcinoma. Antioxidants 2021; 10: 1419.
- 45. Yan C, Sun W, Wang X, et al. Punicalagin attenuates palmitate-induced lipotoxicity in HepG2 cells by activating the Keap1-Nrf2 antioxidant defense system. Mol Nutrition Food Research 2016; 60: 1139–1149.