Introduction
The Venus von Willendorf, a 13-inch bust of an obese woman’s torso, sitting in the Natural History Museum of Vienna, carbon dates back to 24,000 BCE (Fig. 1). Obesity has been around for a long time, although in ancient times it was a rare occurrence. Throughout the Middle Ages and the Renaissance, obesity was considered a status symbol of affluence (e.g., Rubenesesque body types). The focus on childhood obesity is much more recent and only gained public attention approximately 40 years ago, when it was noted that a stable childhood obesity prevalence curve morphed into a parabolic one. Currently, 19.7% of American children [] and 5.7% of European children [] are obese. Similarly, the number of academic articles on obesity has exhibited a similar parabolic climb over the last 20 years [].

Fig. 1
The Venus von Willendorf, a sculpture housed in the Vienna Natural History Museum, carbon dates to 24,000 BCE. Venus von Willendorf by Matthias Kabel: CC-BY-2.5.
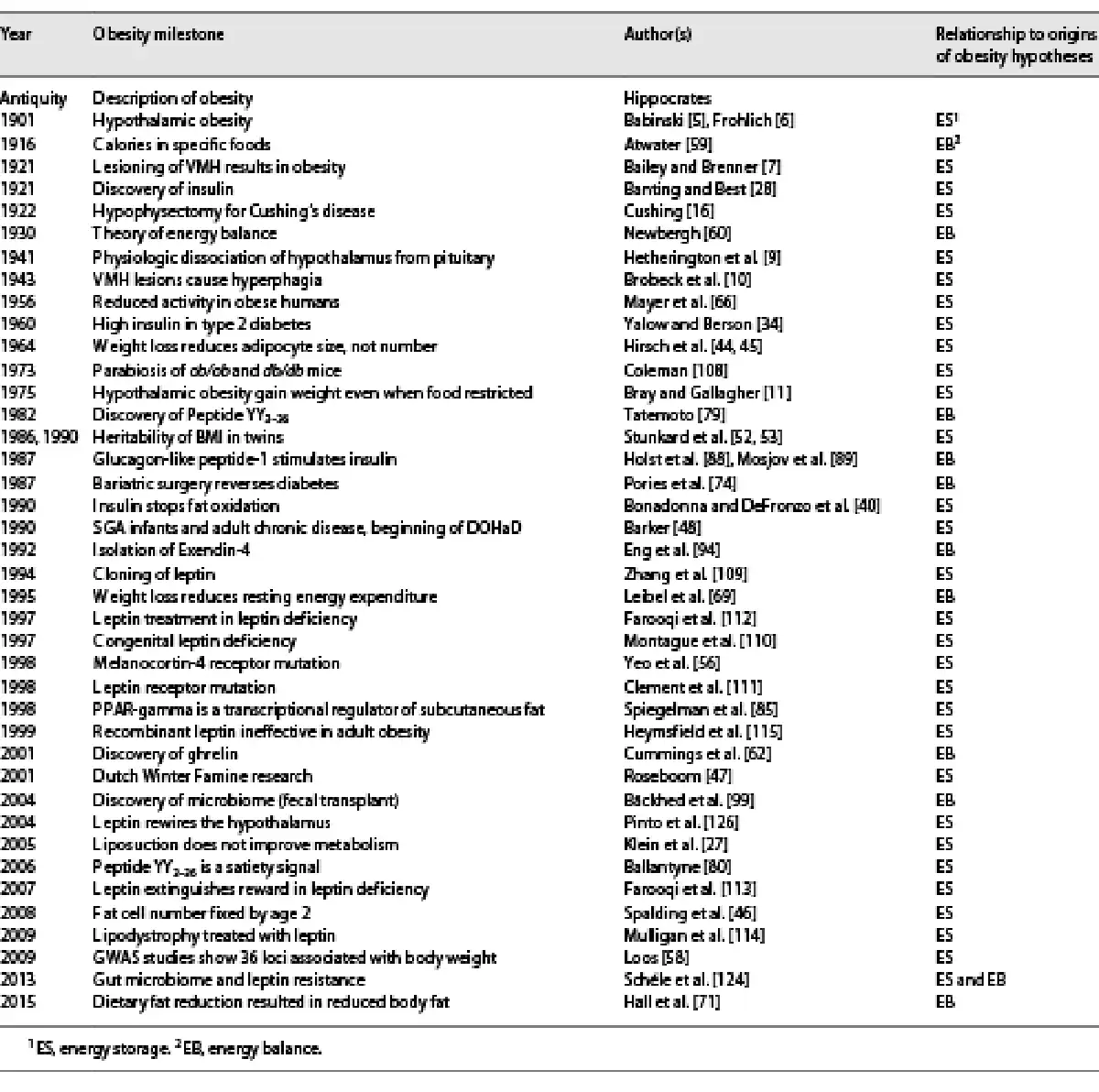
Despite its increasing incidence, prevalence, and severity, we have limited understanding of what causes obesity and an even poorer understanding of best approaches to management. Perhaps the most blatant example of our confusion is that to this day, we still don’t know to what extent, for the vast majority of individuals, obesity is due to a defect in energy storage, which is primarily endocrine in etiology, or due to a defect in energy balance, which is primarily nutritional in etiology. Indeed, in the 20th century, the history of obesity research had taken two divergent paths, with various adherents subscribing to one or the other hypothesis. Table 1 provides an integrated chronological timeline of research progress, underscoring proponents of each hypothesis.
The Energy Storage Hypothesis
In exploring the energy storage hypothesis of obesity, genetic and endocrinologic constructs have provided the primary key insights into the determinants of obesity. Organs involved include the brain, the adrenal gland, the pancreas, and the adipocyte itself.
The Ventromedial Hypothalamus and Obesity
The seminal illustration of the energy storage hypothesis is the clinical syndrome of hypothalamic obesity. The first description dates back to 1840, when a 57-year old woman developed massive obesity within a year of her death, and an autopsy demonstrated a tumor at the base of the brain []. The notion that obesity could have a neuroendocrine (i.e., either brain or hormone) origin dates back to 1900, when Babinski [] and Frohlich [] independently described children who were normal weight until the development of a tumor at the base of the brain, which led to massive obesity that was unresponsive to any intervention. However, both ascribed the obesity to dysfunction of the pituitary. By 1921, Bailey and Brenner [] showed that stereotactic lesioning of the same area in rats led to massive obesity. In 1937, Harris [] localized the hypothalamus as the brain area regulating energy metabolism and other hormonal secretions. In 1940, Hetherington and Ranson [] lesioned the ventromedial hypothalamus (VMH) without damaging the pituitary and recapitulated the same increase in adiposity, and in 1943, Brobeck et al. [] demonstrated that these animals developed severe hyperphagia, thus postulating (incorrectly) that the VMH was the brain’s “satiety center”. However, this was disproven in 1975 when Bray and Gallagher [] demonstrated that even on a calorically restricted diet in-hospital, these patients still gained weight. Rather, in the 1970s, studies in rats by the Bray lab in the USA [] and Rohner-Jeanrenaud and Jeanrenaud [, ] in France showed that either VMH lesions or vagal stimulation increased insulin secretion from the pancreas and resultant hyperphagia, both of which could be blocked by vagotomy. These findings have argued for a VMH-vagus-pancreas-adipose axis that regulates energy storage [].
Cortisol and Obesity
In 1912, Cushing described his experience with the now-famous “Minnie G.” and seven other patients with pituitary basophilic adenoma, “painful” obesity, headache, and hypertension []. Subtemporal decompression reversed many of the symptoms (as well as the obesity), for a time, but most of the patients died years later. However, within the next 2 decades, it became clear that many patients with “Cushing’s Disease” did not have a pituitary tumor but rather an adrenal adenoma [], and that unilateral adrenalectomy would reverse the obesity, suggesting an adrenal hormonal etiology. By 1936, the Kendall lab had isolated cortisone from bovine adrenal glands [], and by 1946, Sarett had succeeded in synthesizing cortisol for pharmaceutical use []. While cortisol turned out to be a “wonder drug” for various inflammatory diseases, its side-effects were undeniable, especially weight gain [].
Numerous studies of adrenal function in general obesity have demonstrated increased absolute daily urinary cortisol excretion; however, when corrected for body mass index or fat mass, these differences are mitigated []. Further investigation, using transgenic mice bearing a mutation in the gene encoding 11β-hydroxysteroid dehydrogenase-1, which prevents the conversion of the active hormone cortisol to the inactive precursor cortisone, by the Flier lab in 2001, demonstrated an increase in visceral obesity and metabolic syndrome []. In another set of transgenic studies, the Zukowska lab in 2007 showed that acute adrenergic activation of the visceral adipose tissue depot resulted in lipolysis, while chronic activation resulted in lipogenesis, due to the adrenergic-blocking effects of the co-localized Neuropeptide Y [].
In clinical studies, increased stress and HPA axis activity have been noted in patients with visceral adiposity [, ]. These findings support a role for stress, chronic adrenergic activation, and cortisol in the growth of the visceral fat depot, exclusive of the subcutaneous fat depot, and with an increased risk for chronic metabolic disease. Conversely, in 2002, the DeFronzo lab showed that thiazolidinediones (TZDs), by binding to the peroxisome proliferator-activated receptor-gamma (PPARγ) increase subcutaneous fat development but not visceral fat, and cause weight gain yet improve insulin sensitivity and reduce risk for metabolic syndrome []. Apparently, the converse is also true, as Klein et al. [] demonstrated in 2005 that liposuction of subcutaneous fat only did nothing to improve the metabolic dysfunction of obese subjects.
Insulin and Obesity
Insulin is a prerequisite for energy storage in adipose tissue. It has long been known that type 1 diabetics lose significant weight and fat mass before succumbing to diabetic ketoacidosis. In 1921, Banting et al. [] discovered insulin; shortly thereafter, patients with type 1 diabetes worldwide started receiving exogenous insulin injections, and it became apparent that recipients rapidly increased their adiposity by shunting glucose and lipid into adipocytes []. Furthermore, studies of patients who manifested increased endogenous insulin release (e.g., congenital hyperinsulinism, Beckwith-Wiedemann syndrome, insulinoma) also had increased adiposity [, ].
In 1939, Himsworth [] first postulated the concept of insulin resistance; this was borne out in 1951 when Bornstein and Lawrence [] first measured insulin levels in patients with type 2 diabetes. In 1959, Yalow and Berson [], in developing the concept and procedures for the radioimmunoassay, showed consistent hyperinsulinemia in those with type 2 diabetes. In 1983, Reaven et al. [] demonstrated that the majority of, but not all, obese persons without diabetes also had increased fasting insulin levels; those that did had associated insulin resistance [], and that they manifested the cluster of signs (“diseases”) which he termed “Syndrome X,” but “metabolic syndrome” was the term that stuck. However, for decades, this paradox of high insulin levels and low insulin effect was explained by the postulate that obesity induced insulin resistance. Furthermore, Knowler et al. [] showed in 2002 that weight loss improved insulin sensitivity and reduced risk for disease.
More recent research argues that insulin is a primary driver of weight gain. Johnson et al. have demonstrated that transgenic animals deficient in insulin secretion gain less adipose tissue and manifest increased longevity []. In humans, the Ludwig lab showed in 2008 that the Quebecois, who exhibit an increased insulin release in response to glucose, manifest increased weight gain in response to a high-carbohydrate diet []. Bonadonna et al. [] showed that insulin inhibits fatty acid beta-oxidation, thus preventing energy utilization. Robert Lustig et al. [, ] suppressed insulin secretion using octreotide and demonstrated weight loss, first in 1999 in patients with hypothalamic obesity, and then in a sub-cohort of obese adults in 2006. These and other revelations have supported the carbohydrate-insulin hypothesis of obesity []; that is, increased carbohydrate diets stimulate insulin release, resulting in energy storage and increasing adiposity.
The Adipocyte and Obesity
Many other hormones affect adipocyte growth (e.g., estrogens, androgens, thyroid hormone), but first those adipocytes have to develop – and once they do, they don’t go away. In 1964, Hirsch and Goldrick [, ] demonstrated that weight loss reduces fat cell size but not fat cell number, suggesting that obese individuals have aberrant adipose tissue development. Spalding et al. [] in 2008 showed that adipose tissue number was set by age 2 years in humans, putting the focus on the fetal and neonatal period. Indeed, documentation of the relationship of small for gestational age with adult obesity and cardiovascular disease started with studies of the Dutch Hunger Winter during World War II and its aftermath. Data on adults born to women exposed to rations of less than 1,000 kcal/day during different periods of gestation identified small size at birth for third trimester exposure with abnormalities of glucose tolerance as adults, while first trimester exposure resulted in increased size at birth with atherogenic lipogenic profile as adults []. These studies supported Barker’s 1990 postulate of the Developmental Origins of Health and Disease (DOHaD) []. We now have data that the in utero environment modulates the risk of obesity and metabolic disease into childhood and adulthood [].
The DOHaD paradigm applies to environmental chemical exposures, including obesogens, which can also lead to obesity later in life, and even across generations []. For example, the U.S. government in 1972 banned the insecticide dichlorodiphenyltrichloroethane (DDT), composed of o,p′-DDT and p,p′-DDT, of which the latter is metabolized to the long-acting p,p′-DDE, which was shown in 2021 to be associated with obesity and earlier age of menarche in the grandchildren of the exposed grandmothers []. These findings demonstrate that there is a window of neural, chemical, and hormonal vulnerability during the fetal and neonatal period, during which risk for obesity can be programmed.
Genetics and Obesity
In 1975, Stunkard et al. [, ] examined the variance in BMI in mono- and dizygotic twins and estimated the heritability of obesity at 40–70%. Further strong evidence of the heritability of BMI came from a study of identical twins separated at or near birth and brought up in different environments. The study demonstrated that as adults, BMI was highly correlated between identical twins but showed little correlation with that of their adoptive parents or siblings. In the mid-1990s, the melanocortin system, consisting of the pro-hormone proopiomelanocortin (POMC) and its metabolites (α-melanocyte stimulating hormone [α-MSH], β-MSH, γ-MSH, and ACTH), as well as the five G-protein melanocortin receptors and their antagonists (e.g., Agouti-related peptide [AgRP]), was associated with feeding behavior and energy homeostasis []. The system acts as a key mediator of a variety of physiologic functions, including adrenal growth and hormone development, erectile responses, natriuresis, melanogenesis, as well as energy homeostasis []. However, despite the physiologic significance of this system and the recognition of its derivation from the POMC gene (as well as the identification of numerous other genes associated with obesity), these genetic loci have had only a limited impact on the epidemic of obesity due to the small numbers of individuals affected by single gene defects. The melanocortin-4 receptor (MC4R) is of greatest epidemiologic significance, being involved in approximately 2.5% of those morbidly obese []; however, all the other monogenetic disorders in the energy balance pathway combined account for less than another 1%. Furthermore, GWAS studies suggest that a minority of weight gain is attributable to other genetic loci. The FTO gene, which encodes alpha-ketoglutarate dependent dioxygenase, is the most prevalent genetic association found thus far (in 14% of children) only accounts for 3.3 kg in extra weight []. While a total of 39 different genes demonstrate linkage with obesity, their collective contribution to the prevalence of obesity is quite small []. Nevertheless, their recognition has led to an enhanced understanding of the physiology of obesity with the interaction of peripheral factors and central factors controlling appetite and energy metabolism.
The Energy Balance Hypothesis
The energy balance hypothesis has been around for just as long, and encompasses both control of energy intake and rates of energy expenditure, including thermogenesis. Relevant organs include the liver, muscles, and GI tract.
Energy Intake and Obesity
The conservation of energy by the human body was first posited by Atwater []. He related the unit of combustion, the “calorie”, to the nutrient value of foods, and demonstrated that each calorie consumed was either stored, burned, or excreted. Newburgh and Johnston [] in 1930 proposed that obesity was a simple matter of excessive intake relative to energy expended. He proposed that “All obese persons are, alike in at least one fundamental respect – they literally overeat.” This hypothesis has become a central tenet of obesity management, leading to the focus on dietary restriction as the primary intervention [].
Cummings et al. [] pursued the idea of obesity as an endocrine problem identifying ghrelin, its pre-prandial rise, and role in meal initiation. Ghrelin levels are elevated in patients with Prader-Willi syndrome [] and low in patients with simple obesity [], confounding the idea that abnormalities in ghrelin explained general obesity. To date, ghrelin remains one of a limited number of identified orexigenic hormones, but one without a disease attached to its excess or deficit.
Energy Expenditure and Obesity
The notion that energy expenditure might be dysfunctional in obese individuals was codified by Mayer [], who in 1953 noted that obese mice were more sedentary than controls; a finding he expanded to humans by 1956 [], suggesting that “calories out” played a major role in obesity. This started the “exercise” craze for weight loss in an attempt to balance the “calories in” part of the equation.
However, implementation of this approach to obesity management has demonstrated that weight loss and weight maintenance are difficult to achieve [, ]. Metabolic adaptation to change from baseline weight through changes in energy expenditure during the process of weight loss or gain was demonstrated by Rudolph Leibel et al. [] and shown to persist during weight maintenance by Camps et al. [].
Several approaches to understanding metabolic adaptation have been undertaken. One effort has focused on the value of carbohydrates versus fat or protein for the adaptive response. The questions remain, is a calorie a calorie? And which is better – low fat or low carbohydrate diets? Numerous studies have explored these questions. In a 2015 in-patient metabolic balance study, Hall et al. [] demonstrated that a 30% caloric restriction with stable protein but decreased carbohydrate content of the diet resulted in increased oxidation of fat but decreased carbohydrate oxidation and no appreciable change in body fat; whereas decreasing the fat content of the diet resulted in decreased body fat, suggesting that dietary fat was a greater contributor to body fat because of its calories. These findings were supported by a systematic review and meta-analysis of randomized control trials [].
Nevertheless, the proponents of the energy storage hypothesis have argued that obesity is the result of high-carbohydrate diets, which stimulate insulin release, driving fat into cells for storage, decreasing lipolysis, and producing obesity []. As a result, they argue that the emphasis should be on diet composition rather than calorie counting and energy balance, with the hypothesis that the etiology of obesity is fundamentally an endocrine problem affecting metabolism [].
GI Hormones and Obesity
Bariatric surgery as a modality for severe obesity was introduced as early as 1950. Pories et al. [] were able to demonstrate in 1987 not only weight loss but also control of type 2 diabetes by this procedure, and, in 1992 documented, long-term diabetes remission with up to 10-year follow-up []. Furthermore, improved control of diabetes occurred within days after bariatric surgery and therefore was not likely due to weight loss [], making the initial hypotheses of the metabolic benefit of this procedure due to restriction [] and malabsorption [] less likely as the sole mechanism.
Exploration of mechanisms that might explain this rapid change in diabetes control after bariatric surgery involved analysis of the effects of the procedure on gut hormone secretion including peptide YY (PPY) and glucagon-like peptide 1 (GLP-1). PYY had been initially identified in porcine intestine in 1980 and shown to inhibit exocrine pancreatic secretions []. Secreted by the L-cells of the intestine, PYY is thought to exert its primary effect by acting on the central nervous system to induce satiety by binding to the Y2 receptors on POMC neurons in the VMH []. Obese subjects appear to have reduced PYY secretion and decreased sense of satiety []. After bypass surgery, baseline PYY levels decrease, yet respond better to stimulation by food []. Though PYY is clearly a satiety hormone, its role in obesity management remains an area of ongoing investigation [, ].
Glucagon-like peptide-1 (GLP-1), a 31-amino acid polypeptide secreted primarily by the L-cells of the intestine [], is a splice product of proglucagon [, ], which was shown in 1987 to be a potent stimulator of insulin release [, ]. Nauck et al. [] showed the GLP-1-induced insulin response to be glucose-dependent and absent below a glucose threshold of 4.3 mM. As with PYY, GLP-1 acts both centrally [] and peripherally [] to inhibit food intake. GLP-1 is secreted in response to nutrients [] with a short half-life of activity due to cleavage by dipeptidyl peptidase-4 (DPP-4) and renal clearance. However, with the isolation of Exendin-4 (a long-acting analog of GLP-1) from the Gila monster [], a new world of therapeutics (first for diabetes, then for obesity) began with GLP-1 receptor agonist therapy [].
The Gut Microbiome and Obesity
The gastrointestinal tract is home to trillions of microorganisms collectively known as the microbiome []. Differences in the microbial populations are noted between normal weight and obese individuals [], with less microbial diversity described in those with obesity []; however, it is still not clear whether this is a cause or effect. The Gordon lab was able to show that germ-free mice receiving a fecal microbial transplant (FMT) from conventional mice gained weight despite increased energy expenditure and decreased energy consumption []. By transplanting microbiota from genetically obese ob/obmice into germ-free mice, they were able to demonstrate a 2% increase in energy deposition over calories actually consumed. As a result, they hypothesized that the microbiota of obese animals might more efficiently extract energy []. Several investigators have demonstrated changes in the microbiome paralleling changes in the diet [, ], suggesting that one way diet affects obesity is by altering the microbiome. Thus, the animal model has provided direct evidence that a “predisposed” microbiome might increase the energy intake of individuals with obesity. However, data from humans have been inconsistent [].
Dietary efforts to manipulate this system have focused on prebiotics, “a nondigestible food ingredient that beneficially affects the host by selectively stimulating the growth and/or activity of one or a limited number of bacteria in the colon, and thus improves host health” []. For instance, in 2006 Cani et al. [] administered the indigestible carbohydrate oligofructose to animals, resulting in improved insulin sensitivity, and to humans, resulting in increased satiety and decreased energy intake [], possibly by altering GI hormones. Prebiotics, probiotics, and postbiotics remain an active area of investigation in the treatment of obesity [].
Putting It All Together: The Leptin Era
Throughout the 20th century, these two investigative paradigms seemed destined to continue to disagree about the etiology and pathogenesis of obesity. But two mice solved the conundrum that is obesity – or so one thought. Coleman was the first to postulate hormonal feedback from the adipose tissue to the brain. In 1973, he joined the circulations of an ob/ob and a db/db mouse in a parabiosis experiment; the ob mouse lost weight, while the db mouse did not []. This suggested a circulating factor present in the db mouse and a functional receptor present in the ob mouse. Positional cloning by Friedman’s lab led in 1994 to the identification of the adipocyte hormone leptin [].
Leptin Clinical Studies
In 1997, the O’Rahilly lab identified the first leptin-deficient child [], and in 1998, the first leptin receptor-deficient patient was identified by Clement et al. []. Farooqi et al. [, ] showed in 1999 that leptin therapy worked well in promoting weight loss and in reducing appetite and reward in children with congenital leptin deficiency, and Mulligan et al. [] in 2009 showed similar results in adult patients with lipodystrophy (who also manifest leptin deficiency). But unfortunately, leptin proved to be ineffective in generalized obesity [] because these patients are leptin resistant.
Leptin Resistance
The first clue to the pathogenesis of leptin resistance was gleaned in 1995 by Leibel et al. [], who showed that obese patients who lost weight also reduced their energy expenditure, as they became leptin deficient in addition to being leptin resistant. Thereafter, Levine et al. [] demonstrated one reason why this occurs; patients who gain weight easily have decreased levels of nonexercise activity thermogenesis (NEAT; otherwise known as fidgeting) due to their leptin resistance. However, Rosenbaum et al. [] performed a seminal experiment which showed that in response to weight loss, obese patients regained sensitivity in response to exogenous leptin injection. These findings suggested that some factor was reversibly antagonizing leptin action in the obese patient.
That factor was identified in 2001, when Niswender and Schwartz [] documented overlap and crosstalk among insulin, the insulin receptor, and the leptin receptor in the hypothalamic POMC neuron. Lustig et al. [] demonstrated increased leptin sensitivity upon insulin suppression. Also, the Bruning lab [] and Nazarians-Armavil et al. [] demonstrated that hyperinsulinemia is at least a cause, if not the only cause, of leptin resistance. The question is why?
Leptin is a necessary signal to the VMH for the initiation of high-energy processes such as puberty and pregnancy []. Indeed, both puberty and pregnancy are insulin-resistant states; leptin levels increase acutely; in adulthood or post-partum, insulin levels fall, weight stabilizes or is lost, and leptin levels return toward the baseline state. Insulin antagonism of leptin signal transduction is likely an integral control mechanism to insure reproductive competence. If leptin signaling could not be modulated, the weight accrual required for reproductive competency during puberty and pregnancy would be compromised [].
The gut microbiome may also play a role in leptin resistance. In an experiment using only mice otherwise resistant to obesity, Schéle et al. [] compared mice raised germ-free to conventionally raised mice. The conventionally raised mice had decreased expression of GLP-1 and brain-derived neurotropic factor (BDNF) in both the hypothalamus and brain stem, as well as decreased expression of anorexigenic peptides POMC and CART (cocaine- and amphetamine-regulated transcript), and increased expression of the orexigenic peptides NPY and AgRP in the hypothalamus. Furthermore, the conventionally raised mice had inherent leptin resistance, as leptin administration caused less suppression of NPY and AgRP and less weight loss than in the germ-free mice, a clear signal that the gut microbiota influences central feeding mechanisms.
Leptin and DOHaD
Leptin also exerts effects in the prenatal period. Leptin-deficient mice (ob/ob) exhibit maldevelopment of hypothalamic architecture, with aberrant projections of neurons from the arcuate nucleus (the site of leptin receptors) to the paraventricular nucleus (the site of the melanocortin-4 receptors) []. However, a single leptin injection at birth can restore normal hypothalamic development and neurotransmission []. This effect of neonatal leptin is also seen in a rat model of maternal undernutrition. Offspring of pregnant rats placed on food restriction during gestation are small for gestational age, insulin resistant, and leptin deficient at birth. However, with adequate nutrition after the neonatal period, these animals become obese as adults, particularly when placed on high-fat chow after weaning [], suggesting that the neonatal leptin overrode a developmental programming signal for future obesity.
Conclusions
Despite the knowledge of the insulin-leptin connection, and despite multiple efforts of diet, pharmacotherapy, and surgery to promote weight loss, obesity, insulin resistance, and chronic metabolic disease continue to predominate in the population. Only 12% of adults are metabolically healthy [], and obesity prevalence and severity continue to increase in children, occurring earlier and earlier []. Genetics can increase predisposition to obesity but do not explain this health care debacle. Whether this is due to processed food, reduced activity, stress, addiction, mitochondrial dysfunction, obesogens, developmental programming, epigenetics, microbial dysbiosis, or a combination thereof, remains unclear. One hypothesis is that obesity is a syndrome with different patients manifesting different etiologies for their obesity, and therefore likely responding to different therapies. But what is clear is that none of these etiologies are the patient’s fault – no one chooses to be obese, especially not children. Obesity and its overlapping disease entities are not a personal health issue, but rather a public health crisis, and will ultimately require a rational public health response.
Acknowledgments
The authors are grateful for a special issue of Hormone Research in Pediatrics in December, 2022, in celebration of the 50th Anniversary of the Pediatric Endocrine Society.
Statement of Ethics
Dr. Lustig and Dr. Fennoy state that this work is original, unbiased, and not a work for hire. They also state that this manuscript is not under review at any other journal.
Conflict of Interest Statement
Dr. Lustig and Dr. Fennoy declare no financial or material conflict of interest with respect to this manuscript.
Funding Sources
This work received no funding or financial support.
Author Contributions
Dr. Lustig and Dr. Fennoy contributed equally to the research, composition, and revisions of this manuscript. They have both approved the final draft.
Data Availability Statement
There were no data generated for this report, only historical references.
References
- 1. Centers for Disease Control. Prevalence of childhood obesity in the United States. 2021. Available from:https://www.cdc.gov/obesity/data/childhood.html.
- 2. Garrido-Miguel M, Cavero-Redondo I, Álvarez-Bueno C, Rodríguez-Artalejo F, Moreno LA, Ruiz JR, et al. Prevalence and trends of overweight and obesity in European children from 1999 to 2016: A systematic review and meta-analysis. JAMA Pediatr. 2019;173(10):e192430. https://doi.org/10.1001/jamapediatrics.2019.2430.
- 3. Zhao N, Tao K, Wang G, Xia Z. Global obesity research trends during 1999 to 2017: A bibliometric analysis. Medicine. 2019;98(4):e14132. https://doi.org/10.1097/MD.0000000000014132.
- 4. Mohr B. Hypertrophie (markschwammige Entartung?) der hypophysis cerebri und dadurch bedingter Druck auf die Hirngrund-fläche, insbesondere auf die Sehnerven, das Chiasma derselben und den linkseitigen Hirnschenkel. Wochenschift für die Gesammte Heilkunde. 1840;6:565–71.
- 5. Babinski J. Tumeur du corps pituitaire san acromegalie et avec arret de developpement des organes genitaux. Rev Neurol. 1900;8:531–33.
- 6. Fröhlich A. Ein fall von tumor der hypophysis cerebri ohne akromegalie. Wiener Klinische Rundschau. 1901;15:883–6.
- 7. Bailey P, Brenner F. Experimental diabetes insipidus. Arch Int Med. 1921;28(6):773–803. https://doi.org/10.1001/archinte.1921.00100180091006.
- 8. Harris GW. The induction of ovulation in the rabbit, by electrical stimulation of the hypothalamo-hypophysial mechanism. Proc R Soc Ser B. 1937;122:374–94.
- 9. Hetherington AW, Ranson SW. Hypothalamic lesions and adiposity in the rat. Endocrinology. 1940;26(2):264–8.
- 10. Brobeck JR, Tepperman J, Long CN. Experimental hypothalamic hyperphagia in the albino rat. Yale J Biol Med. 1943;15(6):831–53.
- 11. Bray GA, Gallagher TF. Manifestations of hypothalamic obesity in man: a comprehensive investigation of eight patients and a reveiw of the literature. Medicine. 1975;54(4):301–30. https://doi.org/10.1097/00005792-197507000-00002.
- 12. Inoue S, Bray GA. The effects of subdiaphragmatic vagotomy in rats with ventromedial hypothalamic obesity. Endocrinology. 1977;100(1):108–14. https://doi.org/10.1210/endo-100-1-108.
- 13. Rohner-Jeanrenaud F, Jeanrenaud B. Consequences of ventromedial hypothalamic lesions upon insulin and glucagon secretion by subsequently isolated perfused pancreases in the rat. J Clin Invest. 1980;65(4):902–10. https://doi.org/10.1172/JCI109744.
- 14. Ionescu E, Rohner-Jeanrenaud F, Berthoud HR, Jeanrenaud B. Increases in plasma insulin levels in response to electrical stimulation of the dorsal motor nucleus of the vagus nerve. Endocrinology. 1983;112(3):904–10. https://doi.org/10.1210/endo-112-3-904.
- 15. Bray GA, York DA. Hypothalamic and genetic obesity in experimental animals: an autonomic and endocrine hypothesis. Physiol Rev. 1979;59(3):719–809. https://doi.org/10.1152/physrev.1979.59.3.719.
- 16. Cushing H. The pituitary body and its disorders. JAMA. 1912;1912(21):1912.
- 17. Walters W, Wilder RM, Kepler EJ. The suprarenal cortical syndrome. Ann Surg. 1934;100(4):670–88. https://doi.org/10.1097/00000658-193410000-00010.
- 18. Mason HL, Myers CS, Kendall EC. Chemical studies of the supra-renal cortex. II. The identification of a substance which possesses the qualitative action of cortin: its conversion into a diketone closely related to androstenedione. J Biol Chem. 1936;116(1):267–76. https://doi.org/10.1016/s0021-9258(18)74681-4.
- 19. Sarett LH. Partial synthesis of pregnene-4-triol-17β, 20β, 21-dione-3, 11 and pregnene-4-diol-17β, 21-trione-3, 11, 20 monoacetate. J Biol Chem. 1946;162(3):601–31. https://doi.org/10.1016/s0021-9258(17)41405-0.
- 20. Bollet AJ, Black R, Bunim JJ. Major undesirable side-effects resulting from prednisolone and prednisone. JAMA. 1955;158(6):459–63. https://doi.org/10.1001/jama.1955.02960060017005.
- 21. Björntorp P, Rosmond R. Obesity and cortisol. Nutrition. 2000;16(10):924–36. https://doi.org/10.1016/s0899-9007(00)00422-6.
- 22. Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, et al. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001;294(5549):2166–70. https://doi.org/10.1126/science.1066285.
- 23. Kuo LE, Kitlinska JB, Tilan JU, Li L, Baker SB, Johnson MD, et al. Neuropeptide Y acts directly in the periphery on fat tissue and mediates stress-induced obesity and metabolic syndrome. Nat Med. 2007;13(7):803–11. https://doi.org/10.1038/nm1611.
- 24. Incollingo Rodriguez AC, Epel ES, White ML, Standen EC, Seckl JR, Tomiyama AJ. Hypothalamic-pituitary-adrenal axis dysregulation and cortisol activity in obesity: a systematic review. Psychoneuroendocrinology. 2015 Dec;62:301–18. https://doi.org/10.1016/j.psyneuen.2015.08.014.
- 25. Kubera B, Leonhard C, Rößler A, Peters A. Stress-related changes in body form: results from the Whitehall II study. Obesity. 2017;25(9):1625–32. https://doi.org/10.1002/oby.21928.
- 26. Miyazaki Y, Mahankali A, Matsuda M, Mahankali S, Hardies J, Cusi K, et al. Effect of pioglitazone on abdominal fat distribution and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab. 2002;87(6):2784–91. https://doi.org/10.1210/jcem.87.6.8567.
- 27. Klein S, Fontana L, Young VL, Coggan AR, Kilo C, Patterson BW, et al. Absence of an effect of liposuction on insulin action and risk factors for coronary heart disease. N Engl J Med. 2004;350(25):2549–57. https://doi.org/10.1056/NEJMoa033179.
- 28. Banting FG, Best CH, Collip JB, Macleod JJR, Noble EC. The effect of pancreatic extract (insulin) on normal rabbits. Am J Physiol. 1922;62(1):162–76.
- 29. Diabetes Control and Complications Trial Research Group. Adverse events and their association with treatment regimens in the Diabetes Control and Complications Trial. Diabetes Care. 1995;18(11):1415–27.
- 30. Schäfer-Graf UM, Dupak J, Vogel M, Dudenhausen JW, Kjos SL, Buchanan TA, et al. Hyperinsulinism, neonatal obesity and placental immaturity in infants born to women with one abnormal glucose tolerance test value. J Perinatal Med. 1998;26(1):27–36. https://doi.org/10.1515/jpme.1998.26.1.27.
- 31. Dizon AM, Kowalyk S, Hoogwerf BJ. Neuroglycopenic and other symptoms in patients with insulinomas. Am J Med. 1999;106(3):307–10. https://doi.org/10.1016/s0002-9343(99)00021-2.
- 32. Himsworth HP. The mechanism of diabetes mellitus. Lancet. 1939;234(6044):1–6. https://doi.org/10.1016/s0140-6736(00)71715-6.
- 33. Bornstein J, Lawrence DD. Plasma insulin in human diabetes mellitus. BMJ. 1951;2(4747):1541–44. https://doi.org/10.1136/bmj.2.4747.1541.
- 34. Yalow R, Berson S. Immunoassay of endogenous plasma insulin in man. J Clin Invest. 1960;39(7):1157–75. https://doi.org/10.1172/JCI104130.
- 35. Reaven GM, Moore J, Greenfield M. Quantification of insulin secretion and in vivo insulin action in nonobese and moderately obese individuals with normal glucose tolerance. Diabetes. 1983;32(7):600–4. https://doi.org/10.2337/diab.32.7.600.
- 36. Hollenbeck CB, Chen N, Chen YD, Reaven GM. Relationship between the plasma insulin response to oral glucose and insulin-stimulated glucose utilization in normal subjects. Diabetes. 1984;33(5):460–3. https://doi.org/10.2337/diab.33.5.460.
- 37. Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393–403. https://doi.org/10.1056/NEJMoa012512.
- 38. Mehran AE, Templeman NM, Brigidi GS, Lim GE, Chu KY, Hu X, et al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 2012;16(6):723–37. https://doi.org/10.1016/j.cmet.2012.10.019.
- 39. Chaput JP, Tremblay A, Rimm EB, Bouchard C, Ludwig DS. A novel interaction between dietary composition and insulin secretion: effects on weight gain in the Quebec family study. Am J Clin Nutr. 2008 Feb;87(2):303–9. https://doi.org/10.1093/ajcn/87.2.303.
- 40. Bonadonna RC, del Prato S, Bonora E, Gulli G, Solini A, DeFronzo RA. Effects of physiological hyperinsulinemia on the intracellular metabolic partition of plasma glucose. Am J Physiol. 1993;265(6 Pt 1):E943–53. https://doi.org/10.1152/ajpendo.1993.265.6.E943.
- 41. Lustig RH, Hinds PS, Ringwald-Smith K, Christensen RK, Kaste SC, Schreiber RE, et al. Octreotide therapy of pediatric hypothalamic obesity: a double-blind, placebo-controlled trial. J Clin Endocrinol Metab. 2003;88(6):2586–92. https://doi.org/10.1210/jc.2002-030003.
- 42. Lustig RH, Greenway F, Velasquez-Mieyer P, Heimburger D, Schumacher D, Smith D, et al. A multicenter, randomized, double-blind, placebo-controlled, dose-finding trial of a long-acting formulation of octreotide in promoting weight loss in obese adults with insulin hypersecretion. Int J Obesity. 2006;30(2):331–41. https://doi.org/10.1038/sj.ijo.0803074.
- 43. Ludwig DS, Aronne LJ, Astrup A, de Cabo R, Cantley LC, Friedman MI, et al. The carbohydrate-insulin model: a physiological perspective on the obesity pandemic. Am J Clin Nutr. 2021;114(6):1873–85. https://doi.org/10.1093/ajcn/nqab270.
- 44. Goldrick RB, Hirsch J. Serial studies on the metabolism of human adipose tissue. II. Effects of caloric restriction and refeeding on lipogenesis, and the uptake and release of free fatty acids in obese and nonobese individuals. J Clin Invest. 1964;43(9):1793–804. https://doi.org/10.1172/JCI105053.
- 45. Hirsch J, Goldrick RB. Serial studies on the metabolism of human adipose tissue. I. Lipogenesis and free fatty acid uptake and release in small aspirated samples of subcutaneous fat. J Clin Invest. 1964;43(9):1776–92. https://doi.org/10.1172/JCI105052.
- 46. Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453(7196):783–7. https://doi.org/10.1038/nature06902.
- 47. Roseboom TJ, van der Meulen JH, Ravelli AC, Osmond C, Barker DJ, Bleker OP. Effects of prenatal exposure to the Dutch famine on adult disease in later life: an overview. Mol Cell Endocrinol. 2001;185(1–2):93–8. https://doi.org/10.1016/s0303-7207(01)00721-3.
- 48. Barker DJ. The fetal and infant origins of adult disease. BMJ. 1990;301(6761):1111. https://doi.org/10.1136/bmj.301.6761.1111.
- 49. Oken E, Gillman MW. Fetal origins of obesity. Obes Res. 2003;11(4):496–506. https://doi.org/10.1038/oby.2003.69.
- 50. Heindel JJ, Howard S, Agay-Shay K, Arrebola JP, Audouze K, Babin PJ, et al. Obesity II: establishing causal links between chemical exposures and obesity. Biochem Pharmacol. 2022;199:115015. https://doi.org/10.1016/j.bcp.2022.115015.
- 51. Cirillo PM, La Merrill MA, Krigbaum NY, Cohn BA. Grandmaternal perinatal serum DDT in relation to granddaughter early menarche and adult obesity: Three generations in the child health and development studies cohort. Cancer Epidemiol Biomarkers Pre. 2021;30(8):1480–88. https://doi.org/10.1158/1055-9965.EPI-20-1456.
- 52. Stunkard AJ, Foch TT, Hrubec Z. A twin study of human obesity. JAMA. 1986;256(1):51–4. https://doi.org/10.1001/jama.1986.03380010055024.
- 53. Stunkard AJ, Harris JR, Pedersen NL, McClearn GE. The body-mass index of twins who have been reared apart. N Engl J Med. 1990;322(21):1483–7. https://doi.org/10.1056/NEJM199005243222102.
- 54. Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the Agouti obesity syndrome. Nature. 1997;385(6612):165–8. https://doi.org/10.1038/385165a0.
- 55. Cone RD. Studies on the physiological functions of the melanocortin system. Endocr Rev. 2006;27(7):736–49. https://doi.org/10.1210/er.2006-0034.
- 56. Yeo GS, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O’Rahilly S. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. 1998;20(2):111–2. https://doi.org/10.1038/2404.
- 57. Cecil JE, Tavendale R, Watt P, Hetherington MM, Palmer CNA. An obesity-associated FTO gene variant and increased energy intake in children. N Engl J Med. 2008;359(24):2558–66. https://doi.org/10.1056/NEJMoa0803839.
- 58. Loos RJ. Recent progress in the genetics of common obesity. Br J Clin Pharmacol. 2009;68(6):811–29. https://doi.org/10.1111/j.1365-2125.2009.03523.x.
- 59. Atwater WO. Principles of nutrition and nutritive value of foods. Farmer’s Bulletin. Washington, DC: U.S. Dept of Agriculture, USDA; 1918. Vol. 142.
- 60. Newburgh LH, Johnston MW. The nature of obesity. J Clin Invest. 1930;8(2):197–213. https://doi.org/10.1172/JCI100260.
- 61. Howell S, Kones R. “Calories in, calories out” and macronutrient intake: The hope, hype, and science of calories. Am J Physiol Endocrinol Metab. 2017;313(5):E608–12. https://doi.org/10.1152/ajpendo.00156.2017.
- 62. Cummings DE, Purnell JQ, Frayo RS, Schmidova K, Wisse BE, Weigle DS. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes. 2001;50(8):1714–9. https://doi.org/10.2337/diabetes.50.8.1714.
- 63. Cummings DE, Clement K, Purnell JQ, Vaisse C, Foster KE, Frayo RS, et al. Elevated plasma ghrelin levels in Prader-Willi syndrome. Nature Med. 2002;8(7):643–44. https://doi.org/10.1038/nm0702-643.
- 64. Haqq AM, Farooqi IS, O’Rahilly S, Stadler DD, Rosenfeld RG, Pratt KL, et al. Serum ghrelin levels are inversely correlated with body mass index, age, and insulin concentrations in normal children and are markedly increased in Prader-Willi syndrome. J Clin Endocrinol Metab. 2003;88(1):174–8. https://doi.org/10.1210/jc.2002-021052.
- 65. Mayer J. Decreased activity and energy balance in the hereditary obesity-diabetes syndrome of mice. Science. 1953;117(3045):504–5. https://doi.org/10.1126/science.117.3045.504.
- 66. Mayer J, Roy P, Mitra KP. Relation between caloric intake, body weight, and physical work: studies in an industrial male population in West Bengal. Am J Clin Nutr. 1956;4(2):169–75. https://doi.org/10.1093/ajcn/4.2.169.
- 67. Wing RR, Phelan S. Long-term weight loss maintenance. Am J Clin Nutr. 2005;82(Suppl 1):222S–5S. https://doi.org/10.1093/ajcn/82.1.222S.
- 68. Kraschnewski JL, Boan J, Esposito J, Sherwood NE, Lehman EB, Kephart DK, et al. Long-term weight loss maintenance in the United States. Int J Obes. 2010;34(11):1644–54. https://doi.org/10.1038/ijo.2010.94.
- 69. Leibel RL, Rosenbaum M, Hirsch J. Changes in energy expenditure resulting from altered body weight. N Engl J Med. 1995;332(10):621–8. https://doi.org/10.1056/NEJM199503093321001.
- 70. Camps SGJA, Verhoef SPM, Westerterp KR. Weight loss, weight maintenance, and adaptive thermogenesis. Am J Clin Nutr. 2013;97(5):990–4. https://doi.org/10.3945/ajcn.112.050310.
- 71. Hall KD, Bemis T, Brychta R, Chen KY, Courville A, Crayner EJ, et al. Calorie for calorie, dietary fat restriction results in more body fat loss than carbohydrate restriction in people with obesity. Cell Metab. 2015;22(3):427–36. https://doi.org/10.1016/j.cmet.2015.07.021.
- 72. Hooper L, Abdelhamid A, Moore HJ, Douthwaite W, Skeaff CM, Summerbell CD. Effect of reducing total fat intake on body weight: Systematic review and meta-analysis of randomised controlled trials and cohort studies. BMJ. 2012;345:e7666. https://doi.org/10.1136/bmj.e7666.
- 73. Ludwig DS, Friedman MI. Increasing adiposity: Consequence or cause of overeating?JAMA. 2014;311(21):2167–8. https://doi.org/10.1001/jama.2014.4133.
- 74. Pories WJ, Caro JF, Flickinger EG, Meelheim HD, Swanson MS. The control of diabetes mellitus (NIDDM) in the morbidly obese with the Greenville gastric bypass. Ann Surg. 1987;206(3):316–23. https://doi.org/10.1097/00000658-198709000-00009.
- 75. Pories WJ, MacDonald KG, Morgan EJ, Sinha MK, Dohm GL, Swanson MS, et al. Surgical treatment of obesity and its effect on diabetes: 10-y follow-up.. Am J Clin Nutr. 1992;55(Suppl 2):582S–5S. https://doi.org/10.1093/ajcn/55.2.582s.
- 76. Pories WJ, Swanson MS, MacDonald KG, Long SB, Morris PG, Brown BM, et al. Who would have thought it? An operation proves to be the most effective therapy for adult-onset diabetes mellitus. Ann Surg. 1995;222(3):339–50; discussion 350–2. https://doi.org/10.1097/00000658-199509000-00011.
- 77. Shin AC, Zheng H, Townsend RL, Patterson LM, Holmes GM, Berthoud HR. Longitudinal assessment of food intake, fecal energy loss, and energy expenditure after Roux-en-Y gastric bypass surgery in high-fat-fed obese rats. Obes Surg. 2013;23(4):531–40. https://doi.org/10.1007/s11695-012-0846-2.
- 78. Odstrcil EA, Martinez JG, Santa Ana CA, Xue B, Schneider RE, Steffer KJ, et al. The contribution of malabsorption to the reduction in net energy absorption after long-limb Roux-en-Y gastric bypass. Am J Clin Nutr. 2010;92(4):704–13. https://doi.org/10.3945/ajcn.2010.29870.
- 79. Tatemoto K. Isolation and characterization of peptide YY (PYY), a candidate gut hormone that inhibits pancreatic exocrine secretion. Proc Natl Acad Sci U S A. 1982;79(8):2514–8. https://doi.org/10.1073/pnas.79.8.2514.
- 80. Ballantyne GH. Peptide YY(1-36) and peptide YY(3-36): Part I. Distribution, release and actions. Obes Surg. 2006;16(5):651–8. https://doi.org/10.1381/096089206776944959.
- 81. Le Roux CW, Batterham RL, Aylwin SJB, Patterson M, Borg CM, Wynne KJ, et al. Attenuated peptide YY release in obese subjects is associated with reduced satiety. Endocrinology. 2006;147(1):3–8. https://doi.org/10.1210/en.2005-0972.
- 82. Jacobsen SH, Olesen SC, Dirksen C, Jørgensen NB, Bojsen-Møller KN, Kielgast U, et al. Changes in gastrointestinal hormone responses, insulin sensitivity, and beta-cell function within 2 weeks after gastric bypass in non-diabetic subjects. Obes Surg. 2012;22(7):1084–96. https://doi.org/10.1007/s11695-012-0621-4.
- 83. Grudell ABM, Camilleri M. The role of peptide YY in integrative gut physiology and potential role in obesity. Curr Opin Endocrinol Diab Obes. 2007;14(1):52–7. https://doi.org/10.1097/MED.0b013e3280123119.
- 84. Lafferty RA, Flatt PR, Irwin N. Established and emerging roles peptide YY (PYY) and exploitation in obesity-diabetes.. Curr Opin Endocrinol Diab Obes. 2021;28(2):253–61. https://doi.org/10.1097/MED.0000000000000612.
- 85. Spiegelman BM. PPAR-gamma: Adipogenic regulator and thiazolidinedione receptor. Diabetes. 1998;47(4):507–14. https://doi.org/10.2337/diabetes.47.4.507.
- 86. Bell GI, Sanchez-Pescador R, Laybourn PJ, Najarian RC. Exon duplication and divergence in the human preproglucagon gene. Nature. 1983;304(5924):368–71. https://doi.org/10.1038/304368a0.
- 87. Orskov C, Holst JJ, Knuhtsen S, Baldissera FG, Poulsen SS, Nielsen OV. Glucagon-like peptides GLP-1 and GLP-2, predicted products of the glucagon gene, are secreted separately from pig small intestine but not pancreas. Endocrinology. 1986;119(4):1467–75. https://doi.org/10.1210/endo-119-4-1467.
- 88. Holst JJ, Orskov C, Nielsen OV, Schwartz TW. Truncated glucagon-like peptide I, an insulin-releasing hormone from the distal gut. FEBS Lett. 1987;211(2):169–74. https://doi.org/10.1016/0014-5793(87)81430-8.
- 89. Mojsov S, Weir GC, Habener JF. Insulinotropin: glucagon-like peptide I (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Invest. 1987;79(2):616–9. https://doi.org/10.1172/JCI112855.
- 90. Nauck MA, Heimesaat MM, Behle K, Holst JJ, Nauck MS, Ritzel R, et al. Effects of glucagon-like peptide-1 on counterregulatory hormone responses, cognitive functions, and insulin secretion during hyperinsulinemic, stepped hypoglycemic clamp experiments in healthy volunteers. J Clin Endocrinol Metab. 2002;87(3):1239–46. https://doi.org/10.1210/jcem.87.3.8355.
- 91. Turton MD, O’Shea D, Gunn I, Beak SA, Edwards CM, Meeran K, et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature. 1996;379(6560):69–72. https://doi.org/10.1038/379069a0.
- 92. Verdich C, Flint A, Gutzwiller JP, Näslund E, Beglinger C, Hellström PM, et al. A meta-analysis of the effect of glucagon-like peptide-1(7-36)amide on ad libitum energy intake in humans. J Clin Endocrinol Metab. 2001;86(9):4382–9. https://doi.org/10.1210/jcem.86.9.7877.
- 93. Elliott RM, Morgan LM, Tredger JA, Deacon S, Wright J, Marks V. Glucagon-like peptide-1(7-36)amide and glucose-dependent insulinotropic polypeptide secretion in response to nutrient ingestion in man: Acute post-prandial and 24-h secretion patterns. J Endocrinol. 1993;138(1):159–66. https://doi.org/10.1677/joe.0.1380159.
- 94. Eng J, Kleinman WA, Singh L, Singh G, Raufman JP. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem. 1992;267(11):7402–5. https://doi.org/10.1016/s0021-9258(18)42531-8.
- 95. Drucker DJ. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab. 2018;27(4):740–56. https://doi.org/10.1016/j.cmet.2018.03.001.
- 96. Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14(8):e1002533. https://doi.org/10.1371/journal.pbio.1002533.
- 97. Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102(31):11070–5. https://doi.org/10.1073/pnas.0504978102.
- 98. Tagliabue A, Elli M. The role of gut microbiota in human obesity: recent findings and future perspectives. Nutr Met Cardiovasc Dis. 2013;23(3):160–8. https://doi.org/10.1016/j.numecd.2012.09.002.
- 99. Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101(44):15718–23. https://doi.org/10.1073/pnas.0407076101.
- 100. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–31. https://doi.org/10.1038/nature05414.
- 101. Muegge BD, Kuczynski J, Knights D, Clemente JC, González A, Fontana L, et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. 2011;332(6032):970–4. https://doi.org/10.1126/science.1198719.
- 102. David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–63. https://doi.org/10.1038/nature12820.
- 103. Walters WA, Xu Z, Knight R. Meta-analyses of human gut microbes associated with obesity and IBD. FEBS Lett. 2014;588(22):4223–33. https://doi.org/10.1016/j.febslet.2014.09.039.
- 104. Gibson GR, Roberfroid MB. Dietary modulation of the human colonic microbiota: introducing the concept of prebiotics. J Nutr. 1995;125(6):1401–12. https://doi.org/10.1093/jn/125.6.1401.
- 105. Cani PD, Knauf C, Iglesias MA, Drucker DJ, Delzenne NM, Burcelin R. Improvement of glucose tolerance and hepatic insulin sensitivity by oligofructose requires a functional glucagon-like peptide-1 receptor. Diabetes. 2006;55(5):1484–90. https://doi.org/10.2337/db05-1360.
- 106. Cani PD, Joly E, Horsmans Y, Delzenne NM. Oligofructose promotes satiety in healthy human: a pilot study. Eur J Clin Nutr. 2006;60(5):567–72. https://doi.org/10.1038/sj.ejcn.1602350.
- 107. Everard A, Lazarevic V, Derrien M, Girard M, Muccioli GG, Muccioli GM, et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes. 2011;60(11):2775–86. https://doi.org/10.2337/db11-0227.
- 108. Coleman DL. Effects of parabiosis of obese with diabetes and normal mice. Diabetologia. 1973;9(4):294–8. https://doi.org/10.1007/BF01221857.
- 109. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–32. https://doi.org/10.1038/372425a0.
- 110. Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387(6636):903–8. https://doi.org/10.1038/43185.
- 111. Clement K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392(6674):398–401. https://doi.org/10.1038/32911.
- 112. Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341(12):879–884. https://doi.org/10.1056/nejm199909163411204.
- 113. Farooqi IS, Bullmore E, Keogh J, Gillard J, O’Rahilly S, Fletcher PC. Leptin regulates striatal regions and human eating behavior. Science. 2007;317(5843):1355. https://doi.org/10.1126/science.1144599.
- 114. Mulligan K, Khatami H, Schwarz JM, Sakkas GK, DePaoli AM, Tai VW, et al. The effects of recombinant human leptin on visceral fat, dyslipidemia, and insulin resistance in patients with human immunodeficiency virus-associated lipoatrophy and hypoleptinemia. J Clin Endocrinol Metab. 2009;94(4):1137–44. https://doi.org/10.1210/jc.2008-1588.
- 115. Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA. 1999;282(16):1568–75. https://doi.org/10.1001/jama.282.16.1568.
- 116. Levine JA, Eberhardt NL, Jensen MD. Role of nonexercise activity thermogenesis in resistance to fat gain in humans. Science. 1999;283(5399):212–4. https://doi.org/10.1126/science.283.5399.212.
- 117. Rosenbaum M, Goldsmith R, Bloomfield D, Magnano A, Weimer L, Heymsfield S, et al. Low-dose leptin reverses skeletal muscle, autonomic, and neuroendocrine adaptations to maintenance of reduced weight. J Clin Invest. 2005;115(12):3579–86. https://doi.org/10.1172/JCI25977.
- 118. Niswender KD, Schwartz MW. Insulin and leptin revisited: Adiposity signals with overlapping physiological and intracellular signaling capabilities. Front Neuroendocrinol. 2003;24:1–10. https://doi.org/10.1016/s0091-3022(02)00105-x.
- 119. Lustig RH, Sen S, Soberman JE, Velasquez-Mieyer PA. Obesity, leptin resistance, and the effects of insulin reduction.. Int J Obes. 2004;28(10):1344–8. https://doi.org/10.1038/sj.ijo.0802753.
- 120. Klöckener T, Hess S, Belgardt BF, Paeger L, Verhagen LAW, Husch A, et al. High-fat feeding promotes obesity via insulin receptor/PI3K-dependent inhibition of SF-1 VMH neurons. Nat Neurosci. 2011;14(7):911–8. https://doi.org/10.1038/nn.2847.
- 121. Nazarians-Armavil A, Menchella JA, Belsham DD. Cellular insulin resistance disrupts leptin-mediated control of neuronal signaling and transcription. Mol Endocrinol. 2013;27(6):990–1003. https://doi.org/10.1210/me.2012-1338.
- 122. Flier JS. What’s in a name? In search of leptin’s physiologic role. J Clin Endocrinol Metab. 1998;83(5):1407–13. https://doi.org/10.1210/jcem.83.5.4779.
- 123. Könner AC, Brüning JC. Selective insulin and leptin resistance in metabolic disorders. Cell Metab. 2012;16(2):144–52. https://doi.org/10.1016/j.cmet.2012.07.004.
- 124. Schéle E, Grahnemo L, Anesten F, Hallén A, Bäckhed F, Jansson JO. The gut microbiota reduces leptin sensitivity and the expression of the obesity-suppressing neuropeptides proglucagon (Gcg) and brain-derived neurotrophic factor (Bdnf) in the central nervous system. Endocrinology. 2013;154(10):3643–51. https://doi.org/10.1210/en.2012-2151.
- 125. Bouret SG, Draper SJ, Simerly RB. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science. 2004;304(5667):108–10. https://doi.org/10.1126/science.1095004.
- 126. Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, et al. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304(5667):110–5. https://doi.org/10.1126/science.1089459.
- 127. Vickers MH, Gluckman PD, Coveny AH, Hofman PL, Cutfield WS, Gertler A, et al. Neonatal leptin treatment reverses developmental programming. Endocrinology. 2005;146(10):4211–6. https://doi.org/10.1210/en.2005-0581.
- 128. Araújo J, Cai J, Stevens J. Prevalence of optimal metabolic health in American adults: National Health and Nutrition Examination Survey 2009–2016. Metab Syndr Relat Disord. 2019;17(1):46–52. https://doi.org/10.1089/met.2018.0105.
- 129. Cunningham SA, Hardy ST, Jones R, Ng C, Kramer MR, Narayan KMV. Changes in the incidence of childhood obesity. Pediatrics. 2022 Jul 5;150(2):e2021053708. https://doi.org/10.1542/peds.2021-053708.