Introduction
Amyloidosis is a group of systemic disorders characterized by the deposition of abnormal misfolded proteins in the extracellular space of various organs. It can involve multiple organs like the liver, kidney, heart, and peripheral nerves, causing organ dysfunction leading to varied clinical manifestations. Cardiac amyloidosis refers to insoluble deposits of misfolded proteins in the extracellular matrix of the heart. About 20% of patients with systemic amyloidosis have cardiac involvement. Cardiac amyloidosis can present with symptoms related to diastolic dysfunction, heart failure, and arrhythmias due to amyloid deposits in the atrium and conducting tissues. It has been an underdiagnosed condition due to a lack of specific clinical manifestations and the necessity of biopsy to demonstrate amyloid deposition. However, the disease is being recognized early with advances in cardiovascular imaging techniques. In this case report, we discuss a patient presenting with heart failure with subsequent workup leading to a diagnosis of AL-type cardiac amyloidosis.
Case Report
A 55-year-old male patient, with a history of type 2 diabetes mellitus for 20 years, hypertension for 20 years, and chronic kidney disease (baseline creatinine—1.3 mg dl−1) for 5 years, was presented to us with a complaint of shortness of breath and bilateral lower limb swelling for last 4 months. He also had abdominal distension for 15 days. Shortness of breath was insidious in onset and gradually progressed over 4 months from breathlessness on exertion to breathlessness at rest. There was no diurnal, postural, or seasonal variation in breathlessness. There was no history of chest pain, orthopnea, paroxysmal nocturnal dyspnea, fever, cough, yellowish discoloration of eyes or skin, or reduced urine output. The swelling started in bilateral feet and gradually progressed to involve both lower limbs up to the knees. On examination, he was afebrile, pulse rate was 80 beats per minute, blood pressure was 110/70 mmHg in the right arm supine position, and saturation was 98% on room air. Jugular venous pressure was raised, and bilateral pitting edema was present up to the knees. On systemic examination, bilateral inspiratory crepitations were present in the infrascapular areas on chest auscultation. The abdomen was distended with shifting dullness, and the liver was palpable 5 cm below the right costal margin.
A clinical diagnosis of chronic congestive heart failure was made, and the patient was started on diuretics and investigated. Creatinine was 1.57 mg dl−1 with normal complete blood count and liver function tests (Table 1). Cardiac biomarkers revealed hs-tropI—383.7 ng l−1, Ck-MB—20 IU l−1, and NT-proBNP—8665 pg ml−1. Electrocardiogram (ECG) showed low voltage complexes in arm leads (Figure 1). Two-dimensional echocardiogram (2D ECHO) findings showed marked concentric left ventricular hypertrophy (LVH), LVEF—60%, abnormal global longitudinal strain pattern (apical sparing of longitudinal strain pattern), and cherry on top appearance. A low voltage ECG with concentric LVH and apical sparing of longitudinal strain pattern on 2D ECHO were suggestive of cardiac amyloidosis. A cardiac magnetic resonance (CMR) was done, which showed diffuse thickening of both ventricles, interatrial septum and interventricular septum, with reversal of the nulling pattern on T1 scout, with the diffuse enhancement of the walls of the thickened ventricles (Figures 2a,b and 3). A workup for primary amyloidosis was done. Serum immunofixation electrophoresis showed a monoclonal band in the beta region in the reference lane corresponding to lambda-only light chain, suggestive of light chain-only gammopathy (Figure 4). Free light-chain assay done showed involved FLC (lambda): uninvolved FLC (kappa) ratio was 14.01. iFLC was 835 mg l−1. dFLC was 775.39 mg dl−1. Serum B2 microglobulin was 5.39 mg l−1 (raised, cutoff—2.34 mg l−1). Total IgG was 14.06 mg l−1 (normal), IgA was 3.86 mg l−1 (normal), and IgM was 0.52 mg l−1 (normal). Flow cytometry immunophenotyping showed about 0.7% lambda-restricted abnormal plasma cells with reduced CD81 and aberrant CD28 expression. Bone marrow aspirate and biopsy were done, which showed trilineage hematopoiesis with myeloid preponderance and increased eosinophils and their precursors; plasma cells constitute about 5.5% of total nucleated cells with no evidence of amyloid deposit in the biopsy. A confirmatory diagnosis required a pericardial biopsy, but the patient refused for the same in view of the risk of adverse events. With cardiac ECHO/CMR findings and free light chain assay, a final diagnosis of AL-type cardiac amyloidosis, restrictive cardiomyopathy and heart failure with preserved ejection fraction (LVEF - 60%), on a background of chronic kidney disease, type 2 diabetes mellitus and hypertension was made. The patient was treated with intravenous Bortezomib (2 mg), Cyclophosphamide (300 mg) and Dexamethansone (40 mg). The patient showed clinical improvement and was discharged but was lost to follow-up.

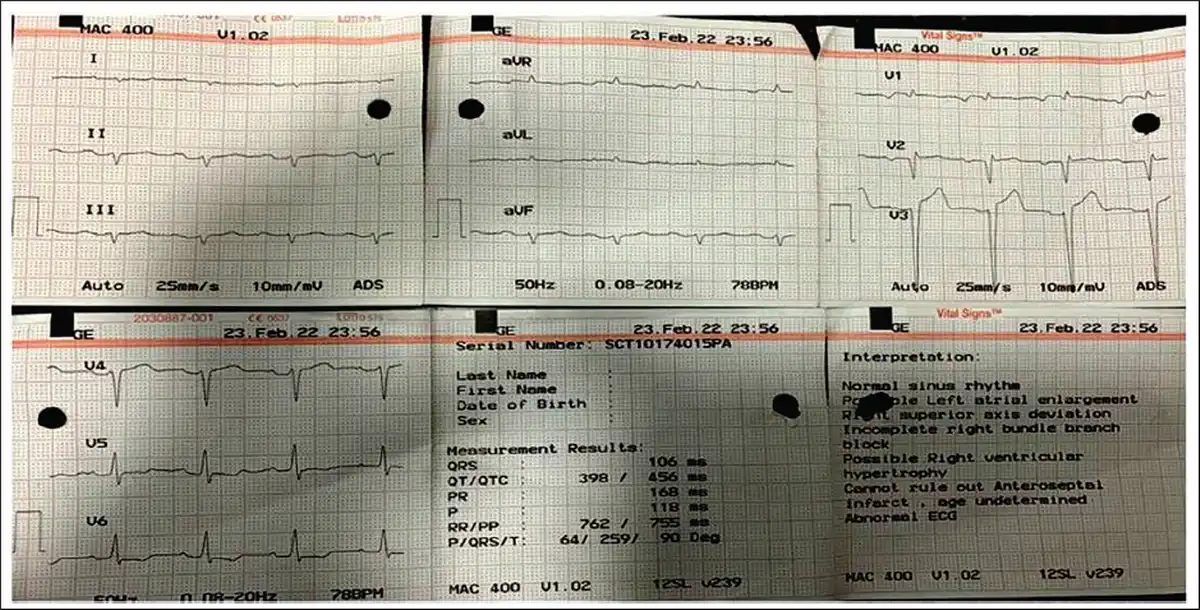
Low Voltage Complexes in Arm Leads and Pseudo-infarct Pattern.
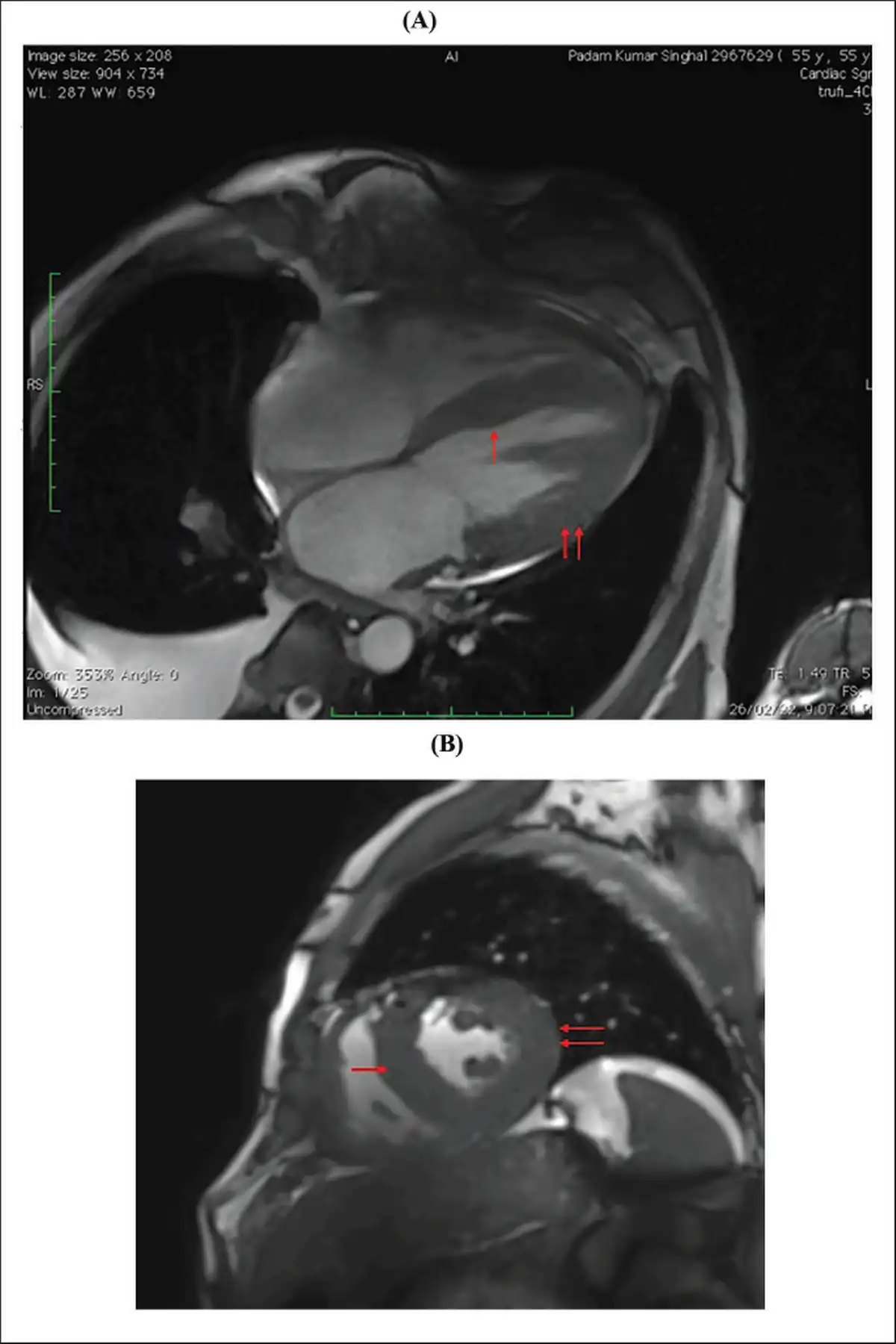
(A and B) Cardiac MR Showing Diffuse Thickening of Both Ventricles and Interventricular Septum (Red Arrows, Respectively).
Note: MR, magnetic resonance.
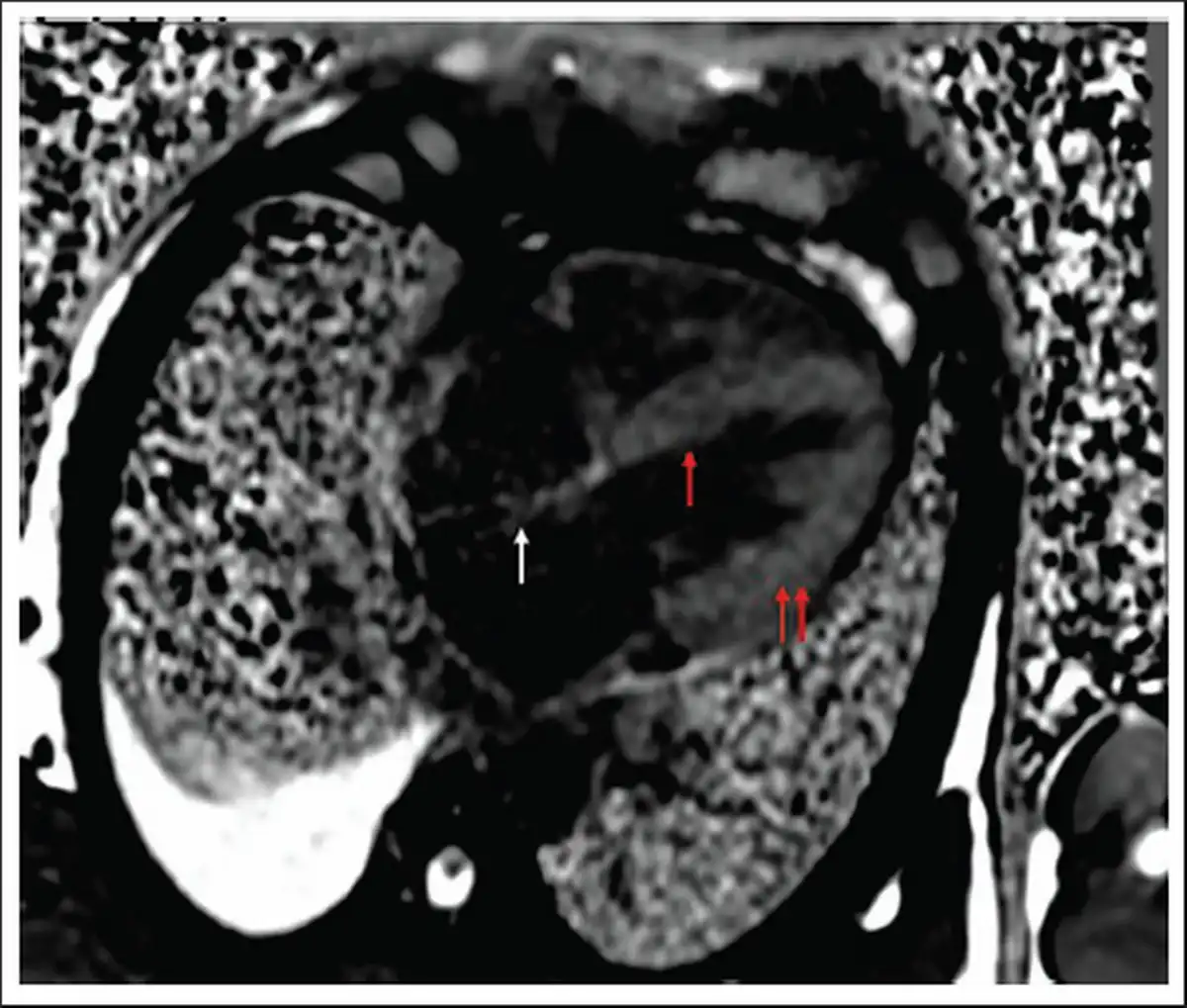
Visible Thickening of Interatrial Septum, Interventricular Septum, and Both Ventricular Walls on Cardiac MR.
Note: MR, magnetic resonance.
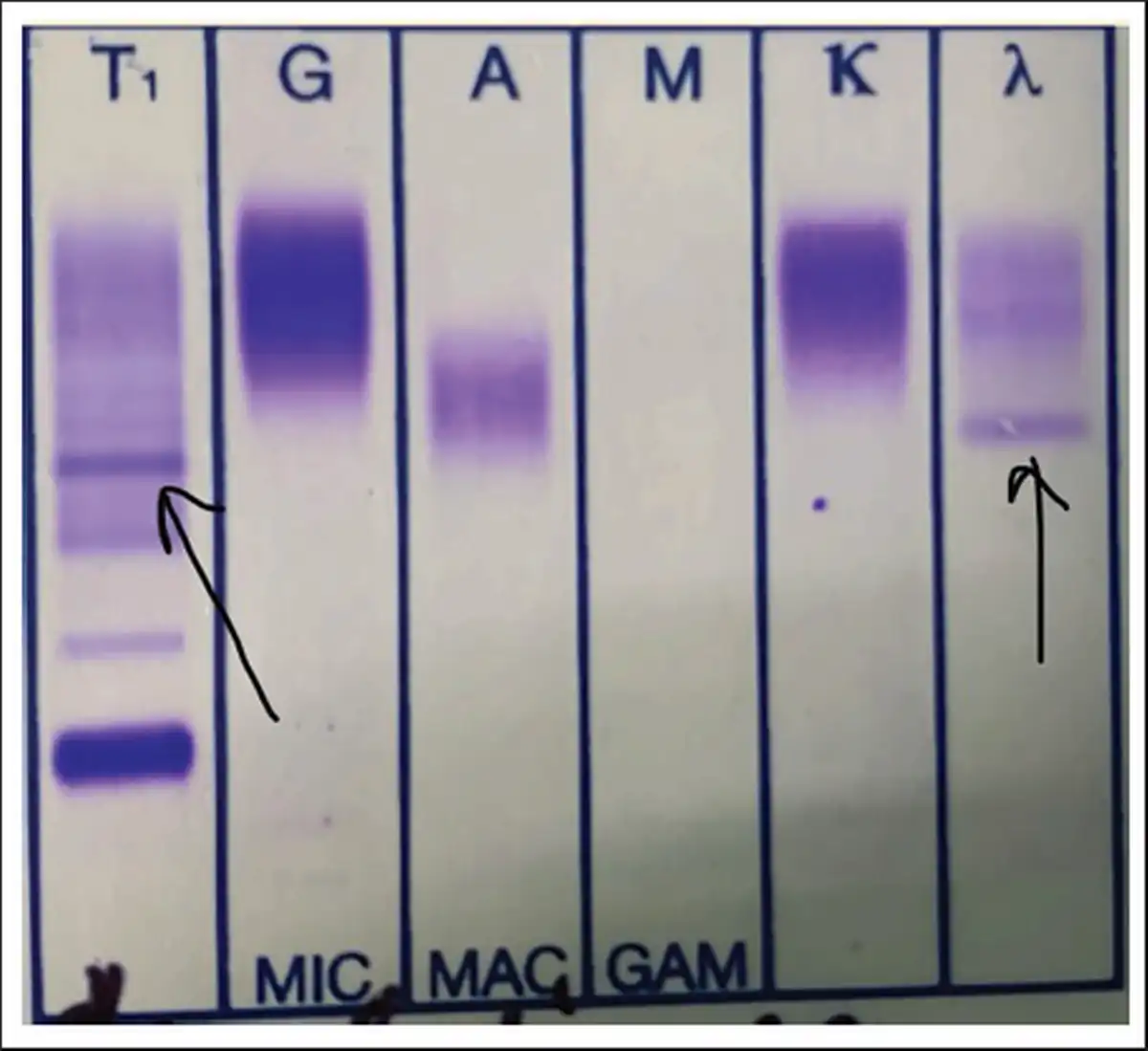
Immunofixation Electrophoresis Showing Monoclonal Band in the Beta Region in the Reference Lane Corresponding to Lambda-only Light Chain.
Discussion
Amyloidosis is a systemic disorder characterized pathologically by the extracellular deposition of insoluble fibrils composed of misfolded proteins in various organs with resulting alteration in structure and function. The amyloid fibrils are less than 10 nm in diameter with a characteristic apple-green birefringence with Congo-red staining under polarized light microscopy. Light-chain amyloidosis (AL) and transthyretin amyloidosis (ATTR) are responsible for 95% of cases of cardiac amyloidosis. AL occurs due to excessive production of immunoglobulin light chain secondary to plasma cell dyscrasia with associated misfolding and deposition in extracellular tissue. Liver, kidney, and peripheral nerve involvement are common in AL-type amyloidosis in addition to cardiac manifestations.
ATTR is a result of misfolding and deposition of transthyretin, and has the following two subtypes: wild-type amyloidosis and hereditary amyloidosis. Wild-type amyloidosis usually begins after 60 years of age and primarily involves the heart. Heart failure in 10%-15% of older people can be due to undiagnosed wild-type amyloidosis. Hereditary ATTR occurs due to an inherited mutation in the transthyretin gene, causing instability and misfolding of transthyretin followed by deposition. It may also involve peripheral as well as autonomic nervous systems.,
Amyloid fibrils get deposited in the myocardium, leading to ventricular thickening and stiffness on both sides. This further causes impairment in diastolic relaxation and restrictive cardiomyopathy., Typical features of presentation in a case of cardiac amyloidosis are exertional dyspnea and orthopnea. Few patients can also have features of right ventricular failure as the predominant symptom., Fibril deposition in and around smaller vessels of the heart results in narrowing, hence causing ischemia/infarction. Deposition of amyloid in atria and conduction tissue can cause atrial arrhythmias, sinus node dysfunction, or advanced atrioventricular block, but less frequently, ventricular arrhythmia., Pericardial and cardiac tamponade can be observed with advanced cardiac dysfunction.
Diagnosis of cardiac amyloidosis is often missed as it requires a high index of suspicion and low incidence overall. Laboratory tests, electrocardiography, cardiac imaging, and biopsy are required for making a diagnosis. Cardiac amyloidosis ECG pattern includes low voltage in limb leads (46%), due to electrically silent amyloid fibrils, and pseudo-infarct pattern (47%). Some ECGs may also depict arrhythmias, such as atrial fibrillation, AV blocks, and bundle branch blocks. Symmetric biventricular thickened wall with a spectrum of diastolic abnormalities (varying from abnormal relaxation to a restrictive filling pattern) is the hallmark of amyloidosis on 2D ECHO., , In cases where LV wall thickness is >12 mm without any history of hypertension, suspicion of cardiac amyloidosis should be kept high. Left ventricular ejection fraction is usually not affected till a later period in the disease course., As the longitudinal strain decreased on basal and mid-wall segments of the heart as compared to the apex, ECHO shows a pattern of apical sparing pattern, which is specific for cardiac amyloidosis. CMR is a better choice of investigation as it identifies the early stage of a diffuse subendocardial pattern of gadolinium enhancement and the late stage of transmural myocardial enhancement. These changes start to occur before the development of LVH., Mandatory investigations, in a patient with suspected amyloidosis, include serum and urinary protein electrophoresis and immunofixation to identify monoclonal protein., ,
If there is no monoclonal protein but cardiac magnetic resonance imaging is consistent with amyloidosis, this increases the possibility of ATTR. In such cases, bone tracer cardiac scintigraphy using 99m technetium (Tc)-labeled pyrophosphate can be performed to reach a diagnosis of ATTR cardiac amyloidosis. It shows grade 2/3 uptake in ATTR amyloidosis, whereas AL amyloidosis shows absent or grade 1 uptake in scintigraphy., Endomyocardial biopsy remains the gold standard investigation for diagnosing cardiac amyloidosis, and it shows direct evidence of amyloid deposits in cardiac tissue. However, since it is associated with a high risk of adverse events, a combination of noninvasive cardiac imaging (ECHO, CMR, and Tc scintigraphy) with monoclonal protein analysis is done to diagnose amyloidosis as was done in our case. Figure 5 summarizes the diagnostic algorithm for cardiac amyloidosis (Figure 5). Natriuretic peptides and troponins are useful for risk stratification and are also used to evaluate response to treatment and disease progression.
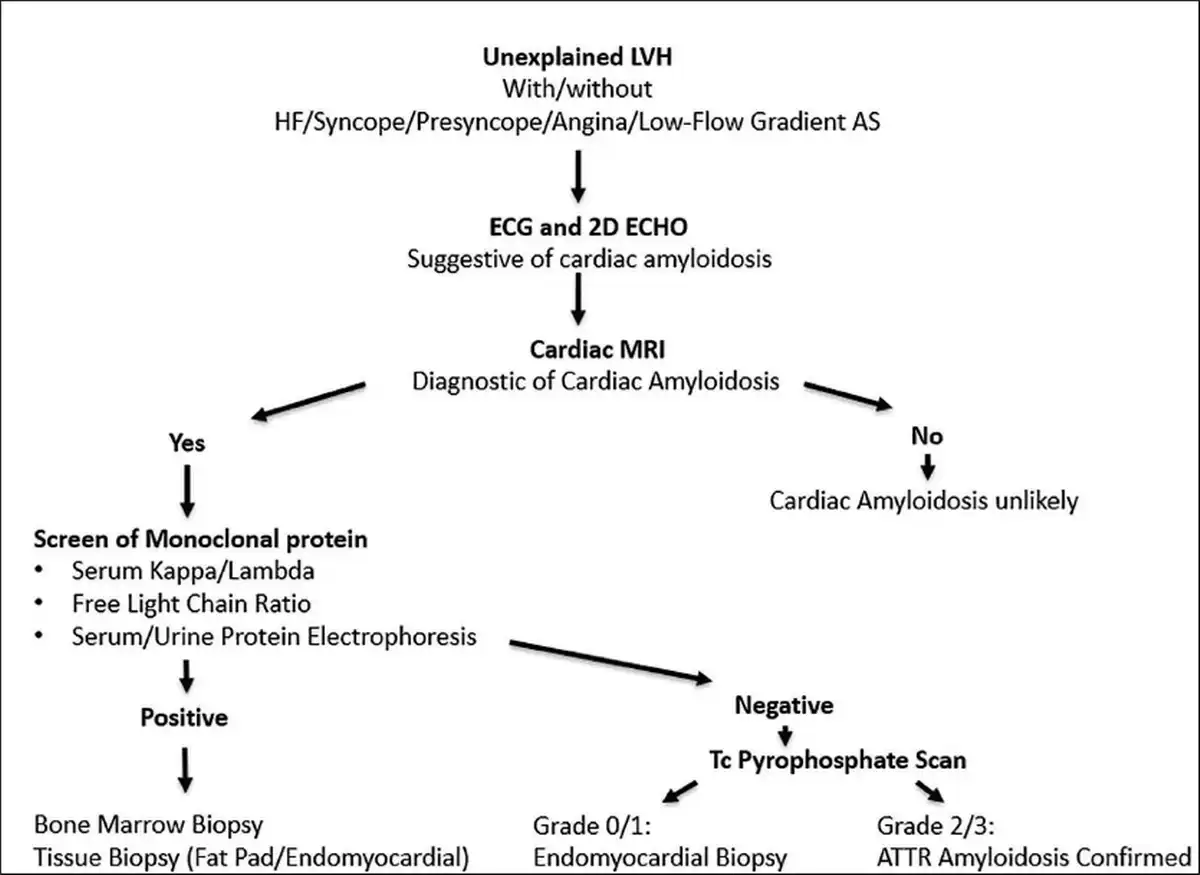
Diagnostic Algorithm for Cardiac Amyloidosis Simplified.a
Cardiac amyloidosis management includes treatment of heart failure with loop diuretics. Drugs like angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and beta blockers are mostly not well tolerated. In patients with severe heart failure, a left ventricular assist device can be used. For atrial fibrillation, rhythm control with antiarrhythmic or catheter ablation, alongside anticoagulation, is the course of action., , , , Patients with conduction system disease may also require permanent pacemakers or implantable cardioverter-defibrillators. Specific disease-modifying treatment for AL comprises chemotherapy succeeded by autologous stem cell transplantation. In cases of ATTR, familial ATTR amyloidosis requires a liver transplant. Tafamidis stabilizes and prevents misfolding of the native TTR tetramer and hence is a potential therapy for senile and familial ATTR amyloidosis. Patisiran and inotersen affect the production of TTR at the level of protein synthesis and can be used in patients with hATTR.–,
Conclusion
A high index of suspicion should be maintained regarding clinical, electrocardiographic, and echocardiographic changes of cardiac amyloidosis, particularly in patients with undiagnosed heart failure. This case also highlights the importance of noninvasive cardiac imaging techniques in diagnosing amyloidosis without invasive procedures like biopsy. Early detection and appropriate treatment can significantly improve outcomes in elderly patients.
Declaration of Conflicting Interests The authors declared no potential conflicts of interest concerning the research, authorship, and/or publication of this article.
Funding The authors received no financial support for the research, authorship, and/or publication of this article.
Kumar R.
https://orcid.org/0009-0005-5247-0113
- 1. Kyle RA. Amyloidosis: a convoluted story. Br J Haematol. 2001;114:529–538.
- 2. Dubrey SW, Cha K, Anderson J, . The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. QJM. 1998;91:141–157.
- 3. Taiwo AA, Alapati L, Movahed A. Cardiac amyloidosis: a case report and review of literature. World J Clin Cases. 2019;7:742–752.
- 4. Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583–596.
- 5. Donnelly JP, Hanna M. Cardiac amyloidosis: an update on diagnosis and treatment. Cleve Clin J Med. 2017;84:12–26.
- 6. Maleszewski JJ. Cardiac amyloidosis: pathology, nomenclature, and typing. Cardiovasc Pathol. 2015;24:343–350.
- 7. Sperry BW, Ikram A, Hachamovitch R, . Efficacy of chemotherapy for light-chain amyloidosis in patients presenting with symptomatic heart failure. J Am Coll Cardiol. 2016;67:2941–2948.
- 8. Siddiqi OK, Ruberg FL. Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med. 2018;28:10–21.
- 9. Di Giovanni B, Gustafson D, Delgado DH. Amyloid transthyretin cardiac amyloidosis: diagnosis and management. Expert Rev Cardiovasc Ther. 2019;17:673–681.
- 10. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73:2872–2891.
- 11. Bhogal S, Ladia V, Sitwala P, . Cardiac amyloidosis: an updated review with emphasis on diagnosis and future directions. Curr Probl Cardiol. 2018;43:10–34.
- 12. Manolis AS, Manolis AA, Manolis TA, Melita H. Cardiac amyloidosis: an underdiagnosed/underappreciated disease. Eur J Intern Med. 2019;67:1–13.
- 13. Ash S, Shorer E, Ramgobin D, . Cardiac amyloidosis–a review of current literature for the practicing physician. Clin Cardiol. 2021;44:322–331.
- 14. Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12:91–102.
- 15. Tuzovic M, Yang EH, Baas AS, . Cardiac amyloidosis: diagnosis and treatment strategies. Curr Oncol Rep. 2017;19:46.
- 16. Brenner DA, Jain M, Pimentel DR, . Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ Res. 2004;94:1008–1010.
- 17. Takashio S, Yamamuro M, Izumiya Y, . Diagnostic utility of cardiac troponin T level in patients with cardiac amyloidosis. ESC Heart Fail. 2018;5:27–35.
- 18. Garcia-Pavia P, Rapezzi C, Adler Y, . Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2021;42:1554–1568.