INTRODUCTION
Adrenal corticosteroids and androgens are involved in several metabolic and direct cellular functions under the strenuous regulation of hypothalamic–pituitary adrenal (HPA) axis. Faulty steroidogenesis may result in several disorders, out of which congenital adrenal hyperplasia (CAH) is the major cause of primary adrenal insufficiency.[] These enzyme deficiencies result in a clinical spectrum of abnormalities that ranges from atypical genitalia and infertility to severe life-threatening conditions.[] The enzyme 21-hydroxylase (21-OH), critical for cortisol synthesis when impaired, accounts for 90% of patients with CAH.[]
Although CAH is a monogenic disorder, the phenotypic manifestations are highly heterogeneous. Based on clinical severity, CAH is classified into the classical form: salt wasting (SW), simple virilizing (SV) and the non-classical forms. The classical SW is the most severe form, which expresses itself early in life with severe electrolyte imbalance and shock.[] Followed by this is the SV type with atypical genitalia and a markedly increased secretion of adrenal androgens.[] The non-classical type (NCCAH), which has been referred to as late-onset CAH (LOCAH), bears similarity to the SV type manifesting peri-pubertally with hirsutism, oligomenorrhoea or virilization in girls and, at times, diagnosed during evaluation for infertility later in life.
CLINICAL DIAGNOSIS
In newborns presenting with SW crisis or atypical genitalia and adolescents or adults presenting with oligomenorrhoea, amenorrhoea, hirsutism, or infertility, hormonal testing is usually carried out for diagnosis. Elevated 17-hydroxy progesterone (17-OHP), the major substrate for 21-hydroxylase (21-OH), is the hallmark for diagnosis of CAH due to 21-hydroxylase deficiency (21-OHD). Measurement of basal 17-OHP early in the morning is usually done for CAH screening, and a value of >10 ng/ml is diagnostic of 21-OHD, while a value less than 2ng/ml excludes 21-OHD.[] An intermediate value of 2–10 ng/ml requires cosyntropin stimulation. First-tier newborn screening (NBS) is done by measuring 17-OHP in dried blood spots by heel prick after 24–74 hours of life. Preterm newborns should preferably be screened 2–4 weeks after birth before discharge. The diagnostic cut-offs in neonates born preterm weighing <2.5 kg are 53.5 ng/ml (≤32 weeks) and 27.7 ng/ml (32–36 weeks) and in those weighing ≥2.5 kg, the cut-offs are 33.7 ng/ml (≤32 weeks) and 24.7 ng/ml (32–36 weeks). The cut-off is ≥24.7 ng/ml for those born at term.[] Those whose mothers received glucocorticoids during the antenatal period have to be tested again after 2 weeks. However, 17-OHP measured by ELISA yields high false-positive results due to assay interference by other steroid intermediates.[] Therefore, in most cases, second-tier confirmation with liquid chromatography-mass spectrometry (LC-MS/MS) is carried out.[,] Adrenocortical steroid profiling post cosyntropin stimulation is recommended when 17-OHP values are borderline to biochemically confirm 21-OHD and to differentiate from other enzyme defects.[,] Commercially available adrenal steroid profiling in India includes cortisol (mg/dl), 17-OHP (ng/dl), testosterone (ng/dl), androstenedione (ng/dl), Dehydroepiandrosterone sulfate (DHEAS) (mg/dl), DHEA (ng/ml), progesterone (ng/ml), 11-deoxycortisol (ng/dl), corticosterone (ng/dl), estradiol (pg/ml) and aldosterone (ng/dl).[] Also, several studies have evaluated the utility of 21-deoxycortisol in the diagnosis of CAH, suggesting it as the key biochemical marker in replacement of 17-OHP measurements.[]Table 1 summarizes the biochemical profiling in 21-OHD and other forms of CAH.[]
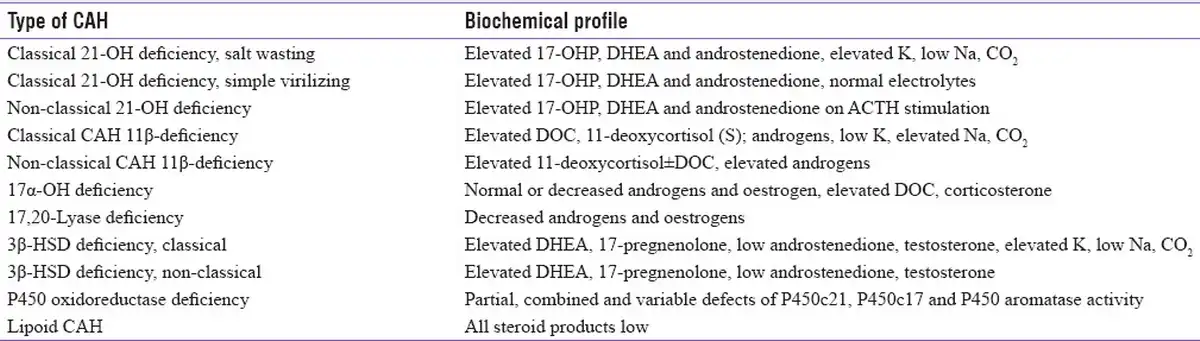
Table 1
Summary of biochemical profiling in different forms of CAH, adapted from Yau M, 2000
Genetics of 21-Hydroxylase Deficiency
Steroid 21-OH, a member of the cytochrome p450 family, is encoded by the 3.2 kb long CYP21A2 (cytochrome P450 family 21 subfamily A member 2) gene with 10 exons on chromosome 6 with a complex tandem arrangement in the MHC class III region. Located 30 kb away from its non-processed pseudogene CYP21A1P (cytochrome P450 family 21 subfamily A member 1 pseudogene), it alternates with C4B and C4A, RP1 (STK19) and RP2 (STK19B), and TNXA and TNXB genes.[] This region comprising the RCCX module can be monomodular, bimodular, or trimodular in genomic arrangement.[] A non-functional pseudogene (98% homologous to the functional gene) in the bimodular variation of the RCCX module forms the ground for unequal crossing over and gene conversion events resulting in disease-causing mutations.[] The pseudogene itself is functionally inactivated by several pathogenic mutations within its sequence.
Upto 95% of mutations in 21 OHD are transferred from pseudogene to functional gene through non-allelic homologous recombination.[,] These are broadly grouped into two: (1) structural rearrangement from unequal meiotic crossing over; this facilitates large deletions, duplications and chimeras in up to 30% of the cases.[,] The 30 kb deletion is the most frequent and leads to chimeric genes. Nine different chimeras (CH1–CH9) have been identified so far, seven of which are classical chimeras associated with classical forms and the other two are attenuated resulting in non-classical forms. Followed by large deletions are gene conversions mediating comparatively shorter sequence transfers while other rearrangements are less prevalent.[] (2) Point mutations by micro conversion; several point mutations are passed on from pseudogene to functional gene during micro conversion in meiosis and is reported in 70–75% of the cases.[] Such recurrent micro conversion mutations include I2G splice mutation, P30L, 8BPDEL, I172N, E6CLUS (I235N, V236E, and M238K), V281L, Q318X, and R356W. Figure 1 depicts the molecular mechanisms and the most common pseudogene-derived mutations in 21-OHD.
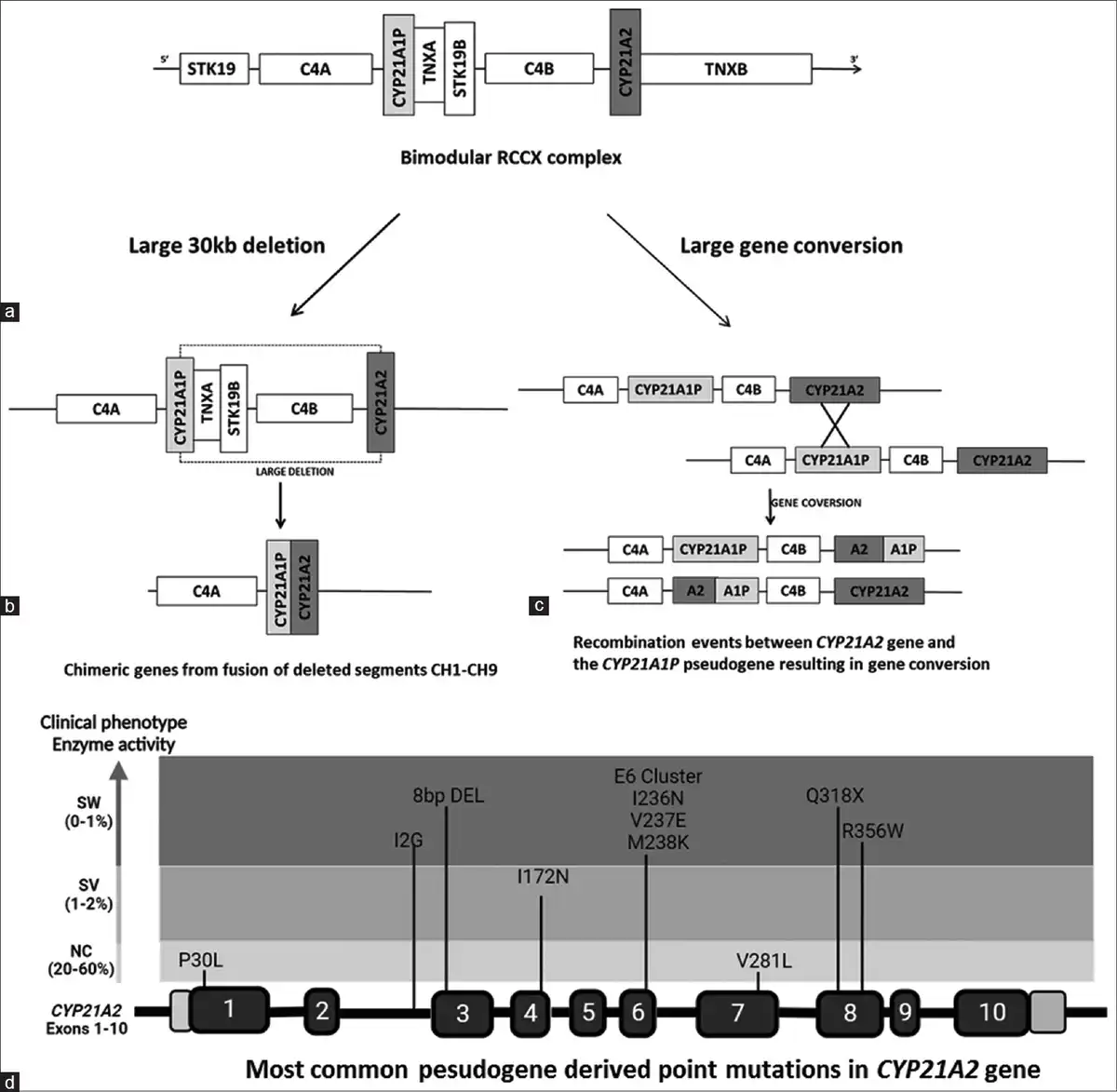
Figure 1
Schematic representation of molecular mechanisms in 21-OH deficiency. a) Bimodular RCCX complex with CYP21A2, C4B, TNXB and STK19B genes. b) Large 30 kb deletion encompassing 5' of CYP21A1P and 3 ' of CYP21A2 genes. The remaining segment of these two genes fuse together to form different chimeric genes. c) Large gene conversion replacing functional gene sequence partly or completely with that of pseudogene. d) The most common pseudogene derived hotspot mutations in CYP21A2 gene
Besides this, around 240 variants have been identified housing several Single-nucleotide variants (SNVs), small deletions, insertions and complex rearrangements.[]De novo mutations are usually rare and reported in less than 5% of the cases.[] Founder effect in non-pseudogene-derived mutations has also been observed.[,]
Genotype–Phenotype Correlation
CAH mutations inherited in an autosomal recessive manner are mostly compound heterozygous.[] These mutations are grouped into four based on clinical severity and functional activity. The ‘null’ mutations result in absent in vitro activity, group A with 1% activity, group B with 2% activity and group C retaining 10–20% activity.[] The mutation spectrum includes Q318X, R356W, E6CLUS and 8BPDEL of the Null group (SW), I2G of group A, I172N and I177T of group B (SV) and V281L, P30L and P453S of group C (NC).[] Null and group A mutations principally promote a SW phenotype with high clinical severity. Following this, group B mutations comprise the SV phenotype and group C produces the mildly affected non-classical type.[] SW mutations induce adverse effects on membrane anchoring and enzyme stability with the loss of 21-OH function. On the other hand, SV mutations negatively impact the transmembrane region and hydrophobic residues with 2% enzyme activity, whereas those that cause a mild NC type interfere with the oxidoreductase function.[] Direct genotype–phenotype concordance has been observed in 90% of the cases.[] Phenotypic variability has been observed in I2G, I172N and P30L mutations.[,]
Molecular Analysis of CYP21A2 Gene
Molecular analysis of CYP21A2 gene is highly advantageous in overcoming the pitfalls with biochemical testing of 17-OHP to confirm the diagnosis; enable carrier screening, prenatal diagnosis (PND), and NBS; and offer genetic counselling. However, genotyping CYP21A2 gene is complicated by pseudogene-derived complex rearrangements and deletions.[] Also, parental screening is often required to differentiate compound heterozygote variants in these rearrangements.[] There are very few reports on molecular analysis of CYP21A2 gene from India. Most of the studies have focused on genotyping the common pseudogene-derived mutations[,] whereas few others have focused on complete gene analysis.[]Table 2a explains the various molecular methods adapted and the most common mutations reported from Indian subjects. There is a wide range of mutations noticed across the studies that could be due to differences in sample size and the various techniques adapted. Conducting CAH genetic studies in large cohorts in the country will thus validate the results and identify the actual spread of these common mutations. Table 2b is a comparison of different techniques utilized in India and their diagnostic yield in CAH to other reported studies across the world.
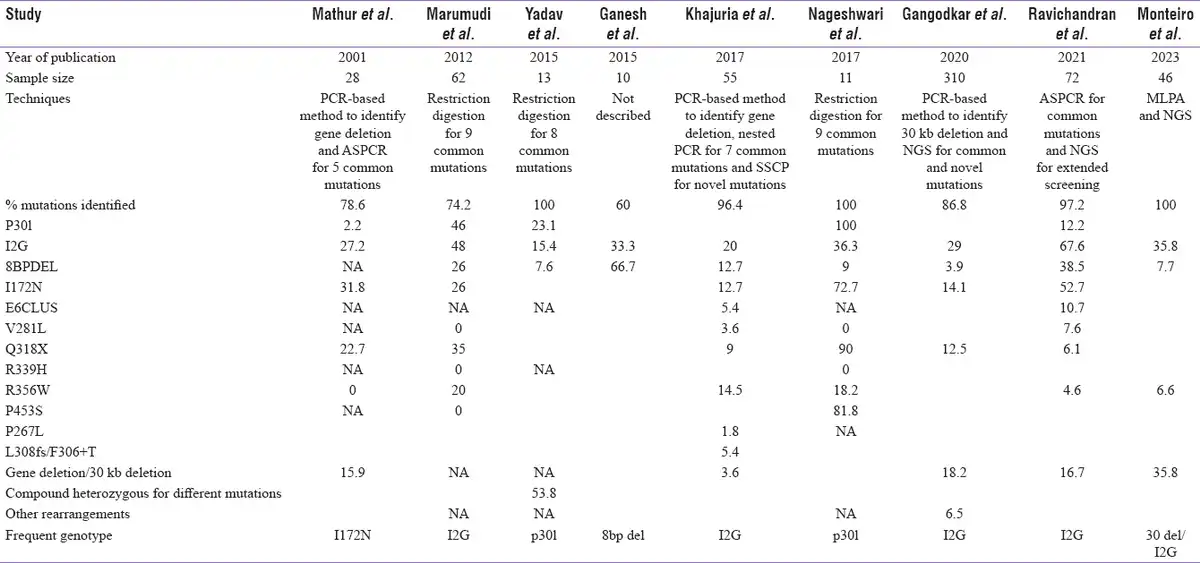
Table 2a
Molecular methods adapted by Indian authors and the results on the most common CYP21A2 mutations from India
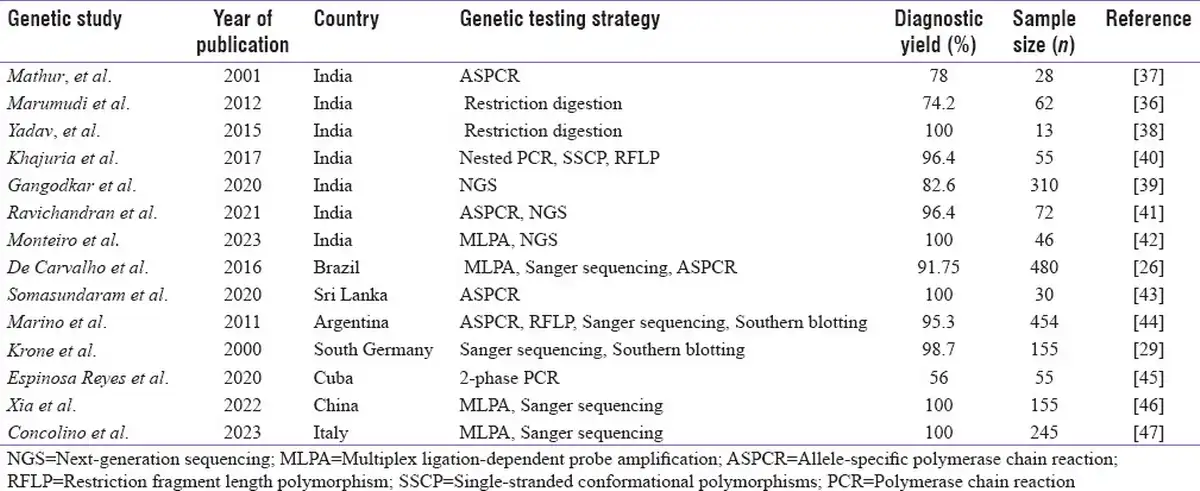
Table 2b
Comparison of molecular techniques and the diagnostic yield reported in Indian studies to other reports across the world
The techniques adapted by the Indian authors and their pros and cons are discussed next.
CYP21A2 Gene Amplification
The key challenge in the molecular diagnosis of 21-OHD is specific amplification of the functional gene CYP21A2. The functional and the pseudogene differ by only 65 nucleotides in the genomic sequence.[] This complicates the primer designing to achieve specific amplification of the functional gene. The most routinely used primers for CYP21A2 amplification include a specific sequence in exon 3 and exon 6.[] However, a large deletion or a mutation in the primer binding site often results in failure of polymerase chain reaction (PCR) amplification. Alternatively, long-range PCRs are utilized to specifically amplify the functional and pseudogene separately.[] Following the PCR amplification, restriction digestion can also give clues for large gene conversion and gene deletion.[] Another strategy is to use four primer sets for locus-specific amplification of CYP21A2, CYP21A1P, 30 kb deletion and gene conversion.[]
Methods for Identification of Pseudogene-Derived Point Mutations
Allele-specific PCR (ASPCR)
By designing primers with wild-type and mutant-specific 3'-terminal mismatches, this technique enables the identification of known mutations in two separate PCR reactions. For the I2G splice mutation, possessing two wild-type and one mutant allele, three PCR reactions are required. It is simple, cost effective and less time-consuming and does not require digestive enzymes or radioactive probes. On the other hand, it involves challenges in primer designing and requires careful standardization to avoid false-positive and negative results. Two studies from India have adapted ASPCR to identify the most common pseudogene-derived pathogenic mutations.[,]
Restriction digestion
For genotyping the common mutations, the amplified CYP21A2 gene product is treated with specific restriction enzymes for each mutation. Many pseudogene-derived hotspot mutations may create or remove restriction sites. However, incomplete digestion can limit the sensitivity.[] The restriction enzymes for CYP21A2 hotspot mutations reported in Indian studies include Hha 1 for P30L and IG, Taq 1 for I172N, ApaL 1 for V281L and R339H, Pst 1 for Q318X, Fnu4 H1 for R356W and Hha1 for P450Ser.[]
Single-stranded conformational polymorphisms (SSCP)
This technique enables the identification of unknown mutations by assessing changes in DNA conformation. The target gene is amplified with conventional PCR. The amplified fragments are denatured and allowed to re-anneal. When run on agarose gels, even a single base change can cause changes in the conformational pattern and a subsequent shift in the electrophoretic mobility. SSCP has been used to identify non-pseudogene-derived novel mutations in the CYP21A2 gene.[] Nevertheless, it is less sensitive for larger fragments and is largely replaced by newer techniques.
Large deletion and duplication analysis
Primary amplification of CYP21A2 and CYP21A1P with long-range PCR, followed by restriction digestion, could yield differential PCR product sizes or digested patterns that provide initial clues for large rearrangements and gene conversion. Multiplex ligation-dependent probe amplification (MLPA), being the gold standard technique for analysing copy number variations, can be utilized for validation of these large deletions and rearrangements in 21-OHD. These assays collectively have replaced Southern blotting for genetic diagnosis of CAH.[] However, results acquired with the combination of techniques require careful interpretation to identify the actual extent of rearrangement and the resulting chimeric genes. Also, differentiating these chimeras into classic and attenuated is essential for genotype–phenotype correlation.[]
Direct sequencing
Following preliminary amplification of CYP21A2 gene, direct sequencing has been mostly employed to detect known hotspot mutations and novel variants and to identify the extent of rearrangements. However, with the advent of next-generation sequencing (NGS) technology, targeted gene/panel sequencing in CAH with these strategies is being evaluated. In terms of clinical utility, there is a need for establishing robust computational pipelines to overcome challenges associated with the data analysis of CYP21A2 gene.[,] The application, pros and cons of these different techniques are given in Table 3.
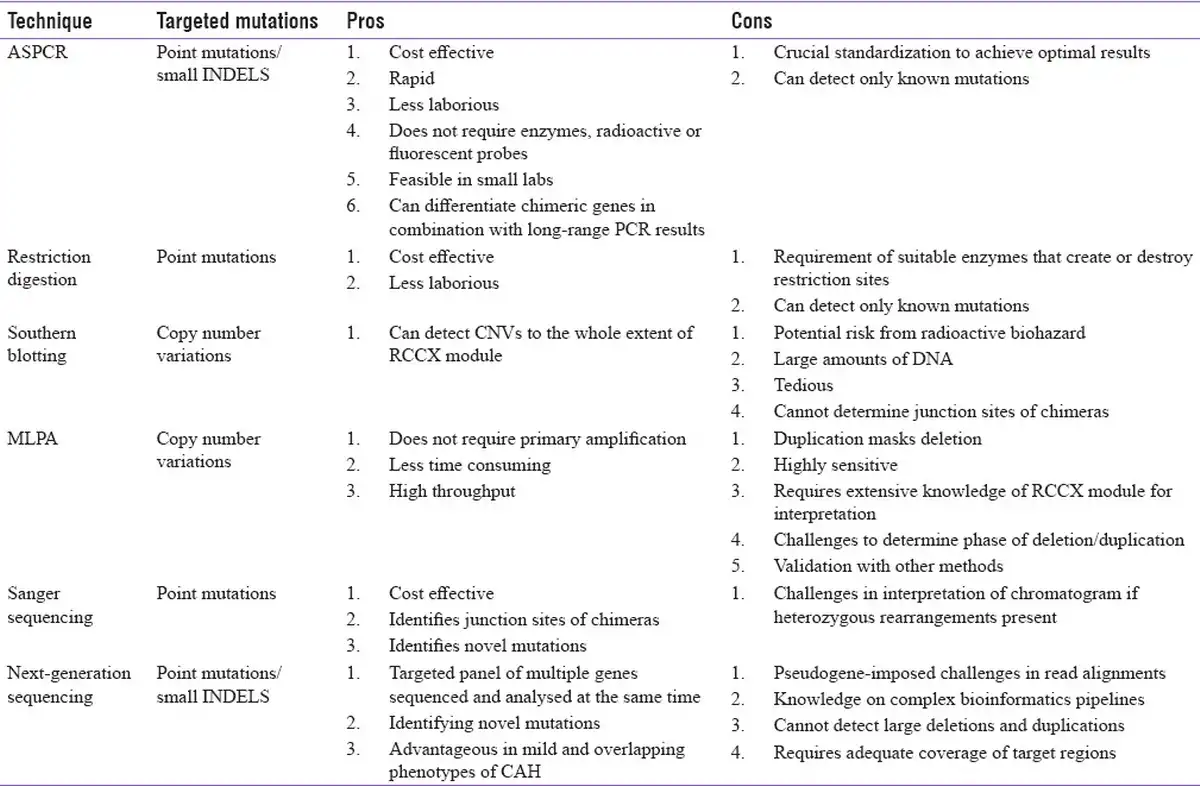
Table 3
The utility, pros and cons of different molecular techniques in the analysis of CYP21A2 gene defects
Genotype–Phenotype Correlation in Indian studies
The I2G mutation is the most prevalent genotype reported by Marumudi et al.[], Khajuria et al.[], Ravichandran et al.[] and Gangodkar et al.[] in their cohorts. Yadav et al.[] have reported p30L to be predominant in their study population whereas Mathur et al.[] have reported I172N. Marumdi et al.[] have also reported p30L in a relatively high frequency in comparison to other studies (46%).
Although genotype–phenotype correlation is high in SW and NCCAH,[,,] there exists heterogeneity in the genotype and phenotype concordance of 21-OHD. Phenotypic variability is commonly observed in P30L, I2G and I172N mutations. Though P30L is predominantly associated with NCCAH, 30% of classical CAH harbour P30L mutation. I2G, a SW mutation, is seen in 20% of SV patients and I172N of SV group is reported in 25% of SW phenotype.[] Phenotypic variability has been observed by Mathur et al.,[]Ravichandran et al.,[]Gangotkar et al. and Khajuria et al.[] in I2G and I172N mutations. Khajuria et al.[] have reported an NCCAH subject with 8 bp deletion.
Novel CYP21A2 Variants Reported from India
Though most of the reports from India focus on the common pathogenic variants, there are few reports identifying novel variants. Khajuria et al.[] have reported two novel missense variants in heterozygous state with other mutations in a cohort of 55 subjects screened using SSCP. Gangodkar et al.[] have identified 9 novel variants in 310 subjects using NGS: 6 missense, 2 frameshift and 1 nonsense variant (4 homozygous and 5 heterozygous variants with known variant on the other allele). Ravichandran et al.[] have reported two novel missense variants using NGS: one homozygous and one compound heterozygous variant. It is interesting to note that 5 out of these 13 novel variants were located in exon 8. The details of these variants are listed in Table 4. Identification of these novel variants is essential to expand the knowledge on the spectrum of CYP21A2 mutation in the Indian population.
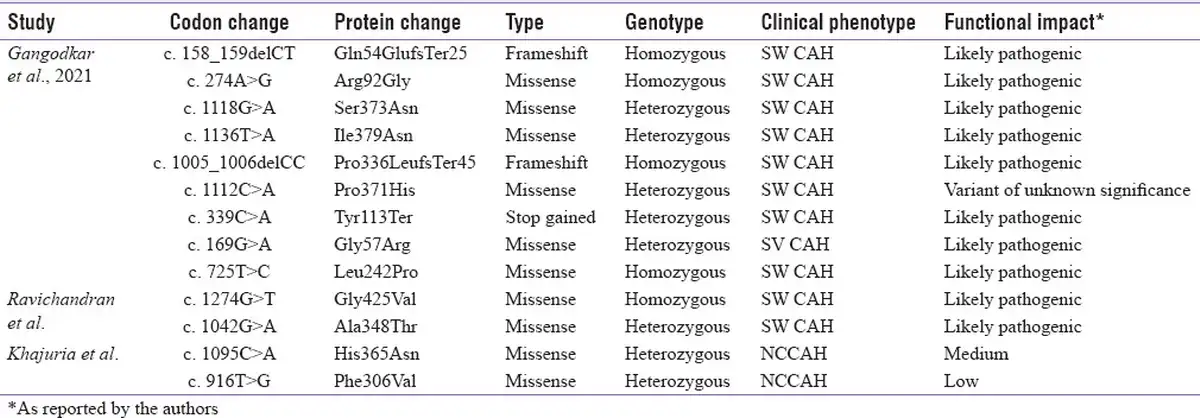
Table 4
Novel CYP21A2 variants reported from India (Transcript ID: NM_000500.9)
Genotyping in Pregnancy and Prenatal Diagnosis
Genotyping pregnant women at risk of delivering offspring with CAH is of high importance to enable early diagnosis and may allow medical intervention to avoid virilization of female foetuses. This will also aid in appropriate and timely genetic counselling. The risk of a classical CAH mother having a CAH affected child when the partner’s genotype is unknown is 120. Conversely, If the mother is affected with non classical CAH, the risk of the child being affected is 1 in 360 [Table 5].[]

Table 5
The risk probability of a CAH mother having a CAH child
These probabilities are based on the carrier frequency data available from the west. There is no report from India on the carrier frequency of classical and non-classical mutations in healthy individuals. The risk can be much higher due to the high rates of consanguinity in the country.[] Thus, genetic diagnosis in affected probands and carrier testing in their partners before planning pregnancy will be extremely beneficial. Continuous follow-up of both the partners being carriers or CAH-affected individuals marrying a carrier will benefit in better management of CAH-associated pregnancies and drastically reduce the incidence of CAH in India.
Following genotyping of the parents, PND is carried out with chorionic villi sampling (CVS) or amniocentesis sampling in the growing foetus. Since 17-OHP measurement is generally unreliable if the mother is already on dexamethasone,[] genotyping is highly advantageous in not only making an accurate diagnosis but also distinguishing between foetus affected with SW, SV, and NCCAH mutations. The PND sample is usually sent for karyotyping and variable number of tandem repeats (VNTR) analysis to check for maternal contamination followed by genotyping of CAH mutations. If the foetus is of 46 XX karyotype and with a positive genotype, prenatal therapy with dexamethasone is possible to prevent virilization.[] Simultaneously, if the foetus is unaffected, unnecessary administration of dexamethasone can be avoided. Dubey et al. in 2017[] have performed genetic screening in 15 foetuses at risk for CAH using MLPA and Sanger sequencing and identified six foetuses with biallelic mutations, six heterozygote carriers and three healthy foetuses with homozygous wild-type alleles that allowed informed decisions on further management of pregnancy. The use of non-invasive prenatal testing with cell-free DNA that can be done in the fourth week of pregnancy may allow for early clinical intervention.[,]
Newborn Screening in CAH
CAH satisfies the NBS criteria (Wilson and Junger) and is recommended with 17-OHP measurements for first-tier testing.[] This NBS recommendation is to provide early diagnosis to reduce mortality in severe cases with adrenal crisis, proper gender assignment and prevent the missing of detection of CAH males.[] First-tier biochemical screening of 17-OHP is usually done by immunoassays like radioimmunoassay (RIA) and enzyme-linked immunosorbent assay (ELISA).[] NBS for CAH has been implemented in all 50 states of America[] and in most other developing countries. In a screening study of 6.5 million newborns worldwide, the overall incidence of CAH was reported to be 1 in 15,000 live births.[] The incidence of classical 21-OHD in the general population varies from 1 in 10,000 to 20,000[] whereas the non-classical incidence is 1 in 1000.[] The reported prevalence is 1:10,000–16,000 in the United States and Europe, 1:21,000 in Japan and 1:23,000 in New Zealand.[]
The infant mortality rate (IMR) in India is 30.6 as per recent report from UNICEF.[] Even with a high IMR, there is no national policy for CAH-NBS in India. There is a pressing need for implementing a uniform nationwide NBS programme for CAH. Several small-scale NBS studies have been carried out using 17-OHP screening in India. The first expanded NBS study was carried out in Hyderabad on 18,300 babies, which showed a prevalence of 1 in 2575.[] Since then, several studies have been conducted across the country by ICMR task force in which 104,066 newborns were screened.[] In comparison to the west, the incidence of CAH in India is high – 1 in 5762 – with remarkable regional differences. This study has also reported an incidence of 1 in 6934 in SW CAH and 1 in 20,801 in SV CAH. The highest incidence has been observed in Chennai (1:2036) and the least in Mumbai (1:9983). Another single-centre study has reported an incidence of 1 in 2800 (n = 11,200 newborns screened) in south India.[] The increased rate of incidence reported in Indian studies in comparison to the west can be attributed to high rate of consanguineous marriages in the country. Following the ICMR pilot study, three states in India – Kerala, Goa and Chandigarh – have initiated government-funded NBS for CAH. These states exhibit an IMR well below the Indian average of 33.[]
NBS with 17-OHP measurements provide the necessary data suggesting a high incidence of CAH in the country; however, several factors limit the sensitivity of 17-OHP assay, so this assay cannot be considered as a confirmed diagnostic test. Reference range varies based on age, birth weight and sex. Though cord blood for measuring 17-OHP in newborns is non-invasive and easily available, it is not the ideal source as 17-OHP values are significantly higher in the first few days after birth.[] The recommended sample collection is usually after 24 hours of birth to 7 days. The other factors that limit the sensitivity of 17-OHP results being used as a diagnostic marker are mode of delivery, gestational age and gender.[] Immature adrenal function in preterm babies, conditions that induce stress and differences in methodologies adapted by the laboratories may lead to false-positive results whereas high exposure of the foetus to maternal cortisol and glucocorticoid treatments before delivery leads to false-negative results.[] High false-positive rates ranging from 9% to 100% in first-tier screening have been reported in NBS for CAH in Indian studies.[] This decreases the positive predictive value of first-tier testing,[] insisting the need for better testing strategies.
Overcoming the Challenges in CAH-NBS
Major concerns associated with undiagnosed early infant deaths and wrong gender assignment in virilized females drive the need for immediate implementation of CAH-NBS in India. However, several logistics like resources, cost effectiveness, and widespread availability of reliable second-tier screening should also be addressed.
To overcome the challenges with 17-OHP testing by LC-MS/MS, genetic testing for the most common mutations (screening for hotspot mutations) can be considered in those with elevated 17-OHP on first-line NBS with a turnaround time of 5–7 days.[] This can be a suitable alternative for the second-tier LC-MS for accurate and cost-effective diagnosis of 21-OHD.
Cost-Effective Genetic Testing in CAH
In a developing country like India, cost effectiveness is one of the key factors determining the availability of genetic testing to a larger extent. High CAH prevalence in the country, high consanguinity rates and the importance for genetic testing in diagnosis, carrier screening, PND and NBS drive the need for affordable genetic testing with highly sensitive and specific molecular strategies. Although the cost of whole genome/exome sequencing (WGS/WES) has drastically come down over the years[] and the current pricing ranges from 20,000 to 25,000 INR,[] it is still expensive for developing countries like India. Also WGS/WES cannot be directly beneficial in many cases with CAH because of the low target coverage and complicated data analysis. Since several rearrangements and chimeras are involved, often a combination of techniques is required to accurately identify mutations on both alleles of CYP21A2 gene. The reported cost of CAH genetic testing in India varies from 5000 to 36,000 INR.[] The cost largely depends on the techniques involved and the extent of mutations covered. Since identifying pseudogene-derived common point mutations can also cover micro conversions and give clues on chimeras, genotyping assays can be an effective first-stage assay in CAH genetic testing. A model algorithm of step-wise and comprehensive genetic screening in 21-OHD is shown in Figure 2.
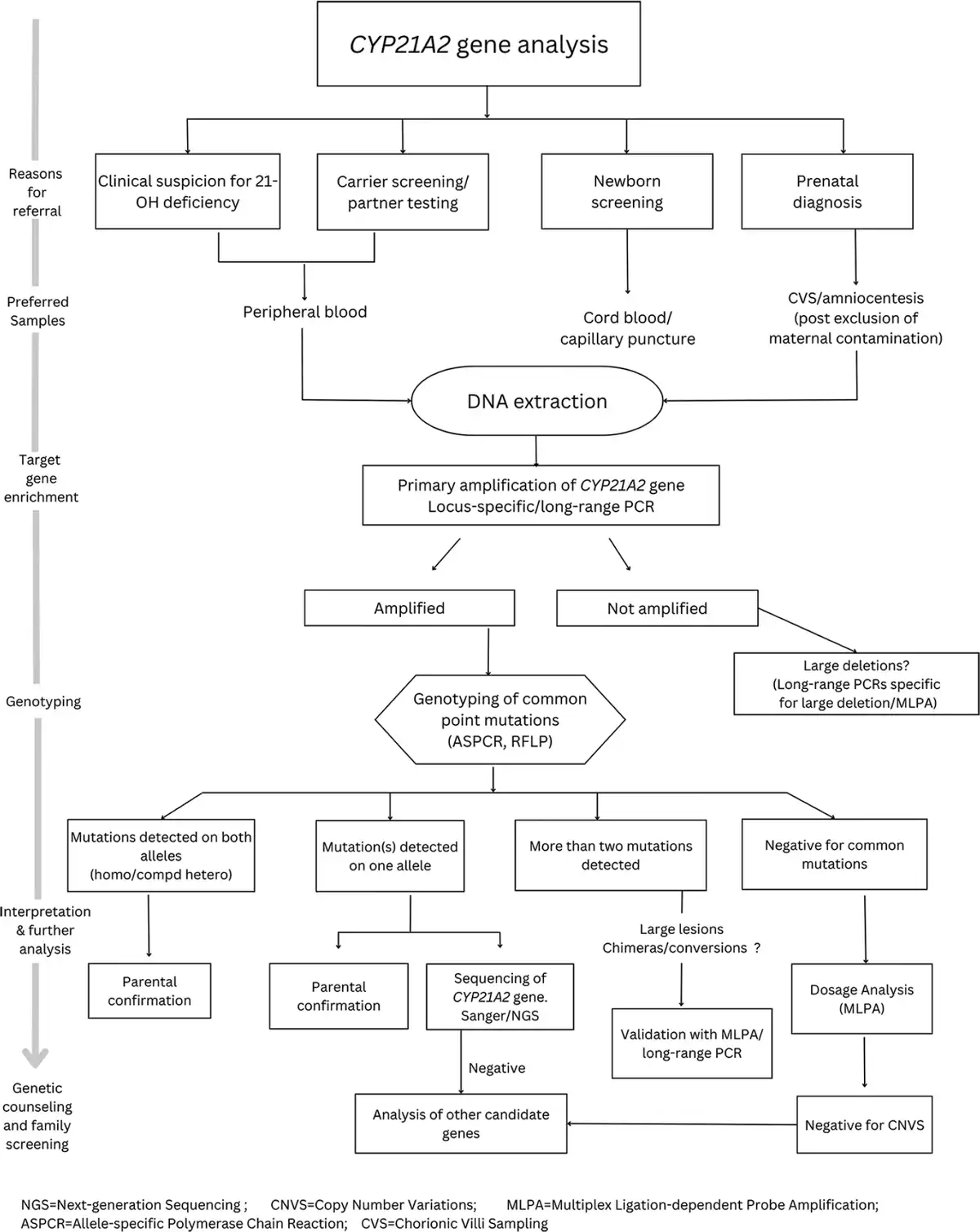
Figure 2
Flow chart on step-wise approach for comprehensive genetic testing in 21-OH deficiency
At the author’s centre, a cost-effective genotyping assay with ASPCR (cost 2600 INR) for the common hotspot mutations and chimeras has been developed that is beneficial in >85% of the subjects with CAH.[] This simple PCR-based analysis does not require radioactive probes or enzymes like other genotyping assays. ASPCR does not also require instrumentation facility for fragment separation as in the case of MLPA. Therefore, this can be easily established even in resource-poor settings, enabling affordable genetic testing and identifying disease-causing CAH mutations in 85–90% of the cases. This can also provide a means for efficient second-tier testing to validate the positive results of 17-OHP-based NBS, carrier testing and community-based screening in CAH.
Treatment and Management
Treatment usually involves lifelong supplementation with mineralocorticoids and glucocorticoids to replace adrenal steroid hormone deficiency, ensure adequate growth in children, suppress excessive androgen secretion in children and adolescents and manage infertility and other long-term consequences. With the recent advances in technology, several studies are in progress to develop alternative therapeutic approaches. They include trials with administration of corticotrophin-releasing hormone (CRH) receptor and melanocortin-2 receptor (MC2R) antagonists, adrenolytic agents and corticotrophin antibodies.[] However, results from on-going research on gene therapy in animal models include adenoviral-CYP21A1 vectors[] and fibroblasts expressing 21-OH in mice,[] which can temporarily enhance glucocorticoid and mineralocorticoid hormone synthesis. Nevertheless, there is no report on clinical trials to witness translational success of these approaches in patient care.
CONCLUSION
Diagnosing and managing CAH presents formidable challenges when not promptly identified and treated. This condition not only carries the risk of life-threatening consequences but also subjects individuals to social stigmatization as they progress through life. Given the high prevalence of CAH and elevated rates of consanguinity in India, the development of widely available, comprehensive and cost effective genetic testing approaches become paramount in achieving a definitive diagnosis for this disorder.
Furthermore, a holistic strategy encompassing genetic screening for carrier status, identification of at-risk communities, genetic counselling for affected families, and advocacy for government policies promoting nationwide NBS programmes is imperative. This multi-faceted approach is essential for early diagnosis, enhanced patient care and implementation of community-based management strategies.
Authors’ contribution
Study concepts and design: Lavanya Ravichandran and Aaron Chapla.
Literature search: Lavanya Ravichandran.
Data acquisition and analysis: Lavanya Ravichandran and Aaron Chapla.
Manuscript preparation: Lavanya Ravichandran and Aaron Chapla.
Manuscript review: Asha H.S, Sarah Mathai, Nihal Thomas, Aaron Chapla.
Manuscript editing: Asha H.S, Sarah Mathai, Nihal Thomas, Aaron Chapla.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
The study on CAH genetics and the assays established were supported by the Indian Council of Medical Research (ICMR) as an ad hoc project (F.No. 54/08/2022-HUM/BUS, PI: Dr. Aaron Chapla). This study was carried out as a part of the Ph.D. thesis of Ms. Lavanya Ravichandran. We also acknowledge Regional Centre for Biotechnology, Faridabad, to which the Ph.D. programme is affiliated. We also thank DST-INSPIRE for the research fellowship.
REFERENCES
1.
Huecker MR, Bhutta BS, Dominique E. Adrenal Insufficiency. StatPearls [Internet] Treasure Island (FL) StatPearls Publishing 2021 Available from:http://www.ncbi.nlm.nih.gov/books/NBK441832/ Last accessed on 2021 Jun 17.2.
New MI, Lekarev O, Mancenido D, Parsa A, Yuen T. Congenital Adrenal Hyperplasia Owing to 21-Hydroxylase Deficiency . Elsevier;2014 29–51 Available from:https://linkinghub.elsevier.com/retrieve/pii/B978012416006400003X Last accessed on 2019 Jul 22.3.
White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev 2000;21:245–91.4.
Globerman H, Rösler A, Theodor R, New MI, White PC. An inherited defect in aldosterone biosynthesis caused by a mutation in or near the gene for steroid 11-hydroxylase. N Engl J Med 1988;319:1193–7.5.
Hindmarsh PC, Geertsma K. Congenital Adrenal Hyperplasia:A Comprehensive Guide London Elsevier/Academic Press 2017 478.6.
Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency:An endocrine society clinical practice guideline. J Clin Endocrinol Metab 2018;103:4043–88.7.
Vats P, Dabas A, Jain V, Seth A, Yadav S, Kabra M, et al. Newborn screening and diagnosis of infants with congenital adrenal hyperplasia. Indian Pediatr 2020;57:49–55.8.
Ambroziak U, Kępczyńska-Nyk A, Kuryłowicz A, Małunowicz EM, Wójcicka A, Miśkiewicz P, et al. The diagnosis of nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency, based on serum basal or post-ACTH stimulation 17-hydroxyprogesterone, can lead to false-positive diagnosis. Clin Endocrinol (Oxf) 2016;84:23–9.9.
Dabas A, Bothra M, Kapoor S. CAH newborn screening in India:Challenges and opportunities. Int J Neonatal Screen 2020;6:70.10.
Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest 1992;90:584–95.11.
Sarathi V, Atluri S, Pradeep TVS, Rallapalli SS, Rakesh CV, Sunanda T, et al. Utility of a commercially available blood steroid profile in endocrine practice. Indian J Endocrinol Metab 2019;23:97–101.12.
Miller WL. Congenital adrenal hyperplasia:Time to replace 17OHP with 21-deoxycortisol. Horm Res Paediatr 2019;91:416–20.13.
Held PK, Bialk ER, Lasarev MR, Allen DB. 21-deoxycortisol is a key screening marker for 21-hydroxylase deficiency. J Pediatr 2022;242:213–9.e1.14.
Sarafoglou K, Merke DP, Reisch N, Claahsen-van der Grinten H, Falhammar H, Auchus RJ. Interpretation of steroid biomarkers in 21-hydroxylase deficiency and their use in disease management. J Clin Endocrinol Metab 2023;108:2154–75.15.
Yau M, Khattab A, Yuen T, New M. Congenital adrenal hyperplasia Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al. Endotext [Internet] South Dartmouth (MA) MDText.com, Inc. 2000 Available from:http://www.ncbi.nlm.nih.gov/books/NBK278953/ Last accessed on 2023 Jun 19.16.
Carroll MC, Campbell RD, Porter RR. Mapping of steroid 21-hydroxylase genes adjacent to complement component C4 genes in HLA, the major histocompatibility complex in man. Proc Natl Acad Sci U S A 1985;82:521–5.17.
Yu CY. Molecular genetics of the human MHC complement gene cluster. Exp Clin Immunogenet 1998;15:213–30.18.
Higashi Y, Yoshioka H, Yamane M, Gotoh O, Fujii-Kuriyama Y. Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome:A pseudogene and a genuine gene. Proc Natl Acad Sci U S A 1986;83:2841–5.19.
Burdea L, Mendez MD. 21-Hydroxylase Deficiency. In:StatPearls [Internet]. StatPearls Publishing 2023 Available from:https://www.ncbi.nlm.nih.gov/books/NBK493164/ Last accessed on 2023 Sep 15.20.
Carrozza C, Foca L, De Paolis E, Concolino P. Genes and pseudogenes:Complexity of the RCCX locus and disease. Front Endocrinol (Lausanne) 2021;12:709758.21.
Pignatelli D, Carvalho BL, Palmeiro A, Barros A, Guerreiro SG, Macut D. The complexities in genotyping of congenital adrenal hyperplasia:21-hydroxylase deficiency. Front Endocrinol (Lausanne) 2019;10:432.22.
Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med 2003;349:776–88.23.
Massimi A, Malaponti M, Federici L, Vinciguerra D, Manca Bitti ML, Vottero A, et al. Functional and structural analysis of four novel mutations of CYP21A2 gene in Italian patients with 21-hydroxylase deficiency. Horm Metab Res 2014;46:515–20.24.
Kim JH, Kim GH, Yoo HW, Choi JH. Molecular basis and genetic testing strategies for diagnosing 21-hydroxylase deficiency, including CAH-X syndrome. Ann Pediatr Endocrinol Metab 2023;28:77–86.25.
Simonetti L, Bruque CD, Fernández CS, Benavides-Mori B, Delea M, Kolomenski JE, et al. CYP21A2 mutation update:Comprehensive analysis of databases and published genetic variants. Hum Mutat 2018;39:5–22.26.
de Carvalho DF, Miranda MC, Gomes LG, Madureira G, Marcondes JAM, Billerbeck AEC, et al. Molecular CYP21A2 diagnosis in 480 Brazilian patients with congenital adrenal hyperplasia before newborn screening introduction. Eur J Endocrinol 2016;175:107–16.27.
Loidi L, Quinteiro C, Parajes S, Barreiro J, Lestón DG, Cabezas-Agrícola JM, et al. High variability in CYP21A2 mutated alleles in Spanish 21-hydroxylase deficiency patients, six novel mutations and a founder effect. Clin Endocrinol (Oxf) 2006;64:330–6.28.
New MI, Abraham M, Gonzalez B, Dumic M, Razzaghy-Azar M, Chitayat D, et al. Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc Natl Acad Sci U S A 2013;110:2611–6.29.
Krone N, Braun A, Roscher AA, Knorr D, Schwarz HP. Predicting phenotype in steroid 21-hydroxylase deficiency?Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. J Clin Endocrinol Metab 2000;85:1059–65.30.
Nageshwari R, Dhivakar M, Balakrishnan K, Selvan SA, Kumaravel V. Common CYP21A2 gene mutations in South Indian congenital adrenal hyperplasia patients. Int J Hum Genet 2017;17:103–8.31.
Krone N, Rose IT, Willis DS, Hodson J, Wild SH, Doherty EJ, et al. Genotype-phenotype correlation in 153 adult patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency:Analysis of the United Kingdom congenital adrenal hyperplasia adult study executive (CaHASE) cohort. J Clin Endocrinol Metab 2013;98:E346–54.32.
Hannah-Shmouni F, Chen W, Merke DP. Genetics of congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab 2009;23:181–92.33.
Day DJ, Speiser PW, Schulze E, Bettendorf M, Fitness J, Barany F, et al. Identification of non-amplifying CYP21 genes when using PCR-based diagnosis of 21-hydroxylase deficiency in congenital adrenal hyperplasia (CAH) affected pedigrees. Hum Mol Genet 1996;5:2039–48.34.
Speiser PW. Molecular diagnosis of CYP21 mutations in congenital adrenal hyperplasia:Implications for genetic counseling. Am J Pharmacogenomics 2001;1:101–10.35.
Ganesh R, Suresh N, Janakiraman L, Ravikumar K. CYP21A2 gene mutation in South Indian children with congenital adrenal hyperplasia. Indian Pediatr 2015;52:710–1.36.
Marumudi E, Sharma A, Kulshreshtha B, Khadgawat R, Khurana ML, Ammini AC. Molecular genetic analysis of CYP21A2 gene in patients with congenital adrenal hyperplasia. Indian J Endocrinol Metab 2012;16:384–8.37.
Mathur R, Menon PS, Kabra M, Goyal RK, Verma IC. Molecular characterization of mutations in Indian children with congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency. J Pediatr Endocrinol Metab 2001;14:27–35.38.
Yadav S, Birla S, Marumudi E, Sharma A, Khadgawat R, Khurana ML, et al. Clinical profile and inheritance pattern of CYP21A2 gene mutations in patients with classical congenital adrenal hyperplasia from 10 families. Indian J Endocrinol Metab 2015;19:644–8.39.
Gangodkar P, Khadilkar V, Raghupathy P, Kumar R, Dayal AA, Dayal D, et al. Clinical application of a novel next generation sequencing assay for CYP21A2 gene in 310 cases of 21- hydroxylase congenital adrenal hyperplasia from India. Endocrine 2021;71:189–98.40.
Khajuria R, Walia R, Bhansali A, Prasad R. The spectrum of CYP21A2 mutations in congenital adrenal hyperplasia in an Indian cohort. Clin Chim Acta 2017;464:189–94.41.
Ravichandran L, Korula S, Asha HS, Varghese D, Parthiban R, Johnson J, et al. Allele-specific PCR and Next-generation sequencing based genetic screening for congenital adrenal hyperplasia in India. Eur J Med Genet 2021;64:104369.42.
Monteiro A, Pavithran PV, Puthukulangara M, Bhavani N, Nampoothiri S, Yesodharan D, et al. Cost-effective genotyping for classical congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency in resource-poor settings:Multiplex ligation probe amplification (MLPA) with/without sequential next-generation sequencing (NGS). Hormones (Athens) 2023;22:311–20.43.
Somasundaram P, Hewage S, De Silva H. Analysis of common genetic mutations in a cohort of children with salt wasting form of congenital adrenal hyperplasia. Ceylon Med J 2020;65:95–104.44.
Marino R, Ramirez P, Galeano J, Perez Garrido N, Rocco C, Ciaccio M, et al. Steroid 21-hydroxylase gene mutational spectrum in 454 Argentinean patients:Genotype-phenotype correlation in a large cohort of patients with congenital adrenal hyperplasia. Clin Endocrinol (Oxf) 2011;75:427–35.45.
Espinosa Reyes TM, Collazo Mesa T, Lantigua Cruz PA, Agramonte Machado A, Domínguez Alonso E, Falhammar H. Molecular diagnosis of patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. BMC Endocr Disord 2020;20:165.46.
Xia Y, Shi P, Gao S, Liu N, Zhang H, Kong X. Genetic analysis and novel variation identification in Chinese patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Steroid Biochem Mol Biol 2022;222:106156.47.
Concolino P, Perrucci A, Carrozza C, Urbani A. Genetic characterization of a cohort of Italian patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Mol Diagn Ther 2023;27:621–30.48.
White PC, New MI, Dupont B. Structure of human steroid 21-hydroxylase genes. Proc Natl Acad Sci U S A 1986;83:5111–5.49.
Xu Z, Chen W, Merke DP, McDonnell NB. Comprehensive mutation analysis of the CYP21A2 gene:An efficient multistep approach to the molecular diagnosis of congenital adrenal hyperplasia. J Mol Diagn 2013;15:745–53.50.
Lee HH. Mutational analysis of CYP21A2 gene and CYP21A1P pseudogene:Long-range PCR on genomic DNA. Methods Mol Biol 2014;1167:275–87.51.
Greene CN, Cordovado SK, Turner DP, Keong LM, Shulman D, Mueller PW. Novel method to characterize CYP21A2 in Florida patients with congenital adrenal hyperplasia and commercially available cell lines. Mol Genet Metab Rep 2014;1:312–23.52.
Finkielstain GP, Chen W, Mehta SP, Fujimura FK, Hanna RM, Van Ryzin C, et al. Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 2011;96:E161–72.53.
New M, Yau M, Lekarev O, Lin-Su K, Parsa A, Pina C, et al. Congenital adrenal hyperplasia Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, et al. Endotext [Internet] South Dartmouth (MA) MDText.com, Inc 2000 Available from:http://www.ncbi.nlm.nih.gov/books/NBK278953/ Last accessed on 2019 Mar 09.54.
Hannah-Shmouni F, Chen W, Merke DP. Genetics of congenital adrenal hyperplasia. Endocrinol Metab Clin North Am 2017;46:435–58.55.
Tippabathani J, Seenappa V, Murugan A, Phani NM, Hampe MH, Appaswamy G, et al. Neonatal screening for congenital adrenal hyperplasia in Indian newborns with reflex genetic analysis of 21-hydroxylase deficiency. Int J Neonatal Screen 2023;9:9.56.
Dubey S, Tardy V, Chowdhury MR, Gupta N, Jain V, Deka D, et al. Prenatal diagnosis of steroid 21-hydroxylase-deficient congenital adrenal hyperplasia:Experience from a tertiary care centre in India. Indian J Med Res 2017;145:194–202.57.
Witchel SF. Congenital adrenal hyperplasia. J Pediatr Adolesc Gynecol 2017;30:520–34.58.
New MI, Tong YK, Yuen T, Jiang P, Pina C, Chan KCA, et al. Noninvasive prenatal diagnosis of congenital adrenal hyperplasia using cell-free fetal DNA in maternal plasma. J Clin Endocrinol Metab 2014;99:E1022–30.59.
Simpson JL, Rechitsky S. Prenatal genetic testing and treatment for congenital adrenal hyperplasia. Fertil Steril 2019;111:21–3.60.
Wilson JMG, Jungner G World Health Organization. Principles and practice of screening for disease 1968 Available from:https://apps.who.int/iris/handle/10665/37650 Last accessed on 2021 Jun 17.61.
Thil'en A, Nordenström A, Hagenfeldt L, von Döbeln U, Guthenberg C, Larsson A. Benefits of neonatal screening for congenital adrenal hyperplasia (21-hydroxylase deficiency) in Sweden. Pediatrics 1998;101:E11.62.
White PC. Neonatal screening for congenital adrenal hyperplasia. Nat Rev Endocrinol 2009;5:490–8.63.
Edelman S, Desai H, Pigg T, Yusuf C, Ojodu J. Landscape of congenital adrenal hyperplasia newborn screening in the United States. Int J Neonatal Screen 2020;6:64.64.
van der Kamp HJ, Wit JM. Neonatal screening for congenital adrenal hyperplasia. Eur J Endocrinol 2004;151 Suppl 3 U71–5.65.
Merke DP, Auchus RJ. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. N Engl J Med 2020;383:1248–61.66.
Nimkarn S, Gangishetti PK, Yau M, New MI. 21-Hydroxylase-Deficient Congenital Adrenal Hyperplasia [Internet. University of Washington, Seattle 2016 Available from:https://www.ncbi.nlm.nih.gov/books/NBK1171/ Last accessed on 2019 Mar 09.67.
UNICEF DATA [Internet]. India (IND)-Demographics, Health and Infant Mortality Available from:https://data.unicef.org/country/ind/ Last accessed on 2023 Jul 13.68.
Rama Devi AR, Naushad SM. Newborn screening in India. Indian J Pediatr 2004;71:157–60.69.
ICMR Task Force on Inherited Metabolic Disorders. Newborn screening for congenital hypothyroidism and congenital adrenal hyperplasia. Indian J Pediatr 2018;85:935–40.70.
Kishore Kumar R, Das H, Kini P. Newborn screening for congenital adrenal hyperplasia in India:What do we need to watch out for?J Obstet Gynaecol India 2016;66:415–9.71.
Mookken T. Universal implementation of newborn screening in India. Int J Neonatal Screen 2020;6:24.72.
Hall K. Suitable specimen types for newborn biochemical screening-A summary. Int J Neonatal Screen 2017;3:17.73.
González EC, Carvajal F, Frómeta A, Arteaga AL, Castells EM, Espinosa T, et al. Newborn screening for congenital adrenal hyperplasia in Cuba:Six years of experience. Clin Chim Acta 2013;421:73–8.74.
Held PK, Bird IM, Heather NL. Newborn screening for congenital adrenal hyperplasia:Review of factors affecting screening accuracy. Int J Neonatal Screen 2020;6:67.75.
Bialk ER, Lasarev MR, Held PK. Wisconsin's screening algorithm for the identification of newborns with congenital adrenal hyperplasia. Int J Neonatal Screen 2019;5:33.76.
. Genome.gov [Internet]. The Cost of Sequencing a Human Genome Available from:https://www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genome-cost Last accessed on 2023 Sep 15.77.
. DNA Labs India [Internet] 2021. What is Whole Exome Sequencing WES Test ? Available from:https://dnalabsindia.com/test/next-generation-whole-exome-sequencing-wes-test-cost Last accessed on 2023 Sep 15.78.
Korula S, Chapla A, Ravichandran L, George A. Comprehensive overview of congenital adrenal hyperplasia and its genetic diagnosis among children and adolescents. J Pediatr Endocrinol Diabetes 2023;2:119–30.79.
Ravichandran L, Varghese D, R P, S AH, Korula S, Thomas N, et al. Allele-specific and multiplex PCR based tools for cost-effective and comprehensive genetic testing in congenital adrenal hyperplasia. Methodsx 2022;9:101748.80.
Tajima T, Okada T, Ma XM, Ramsey W, Bornstein S, Aguilera G. Restoration of adrenal steroidogenesis by adenovirus-mediated transfer of human cytochrome P450 21-hydroxylase into the adrenal gland of 21-hydroxylase-deficient mice. Gene Ther 1999;6:1898–903.81.
Naiki Y, Miyado M, Horikawa R, Katsumata N, Onodera M, Pang S, et al. Extra-adrenal induction of Cyp21a1 ameliorates systemic steroid metabolism in a mouse model of congenital adrenal hyperplasia. Endocr J 2016;63:897–904.