Bone sarcomas are the third leading cause of pediatric cancer-related deaths in the United States. Although there have been improvements in survival rates among pediatric cancer patients over the past 30 years, bone sarcomas, unfortunately, remain the exception. Despite considerable advances in characterizing molecular and genetic predispositions for these cancers, survival rates have plateaued overall. Ewing sarcoma (ES) remains the second most common primary bone sarcoma in adolescents, second to osteosarcoma, and serves as a predominant causality for the stagnant survival rates.
ES was originally characterized as a round cell neoplasm of bone and later as a subset of primitive neuroectodermal tumors. The World Health Organization's most recent update defines these tumors as “Ewing sarcoma” with the pathognomonic gene fusions involving the fused in sarcoma and E26 transformation-specific gene families, most classically EWSR1-FLI1, with rare variants containing STAG2, CDKN2A, or TP53 mutations. ES displays a single peak incidence of around 15 years. Treatment typically includes neoadjuvant chemotherapy, wide resection with limb salvage, and adjuvant chemotherapy. However, additional radiation treatment may be employed in select cases or as a means of definitive control in lieu of surgical resection with irresectable tumors. Based on previous epidemiologic reports, patients with localized disease demonstrate 5-year survival rates of approximately 72%, whereas those initially presenting with metastases demonstrate 5-year survival rates of approximately 28%.
Since the initial prognostic improvement with the introduction of systemic chemotherapy in the 1970s, ES survival rates have remained bleak, particularly for those with metastatic and recurrent disease. Despite an improved understanding of the pathogenesis and altered treatment protocols of ES, survival rates overall among this cancer have remained relatively stagnant. Considering clinical trials involving novel targeted therapies, the establishment of updated survival rates and predictors of mortality for pediatric ES patients would be prudent. This investigation sought to determine the most updated 5-year survival rates among these pediatric patients diagnosed with ES and treated within the United States. Secondary outcomes included multivariable determination of patient, tumor-, and treatment-specific effects on survival rates.
Methods
The National Cancer Database (NCDB) was inquired for all pediatric ES cases within the most updated bone and joint public use file available in September 2022 (patients reported from 2008 to 2017). The NCDB contains deidentified data, and therefore was exempted from formal institutional board review as nonhuman subject research. This database includes 34 million patient records with a 90% rate of 5-year follow-up, capturing approximately 70% of all patients newly diagnosed with cancer. Reported pediatric (<18 years of age) sarcomas were extracted using appropriate International Classification Diseases for Oncology coding. The International Classification Diseases for Oncology codes 9260/3 and 9264/3 were used for the identification and separation of ES for subsequent analysis based on reported histologic diagnostic confirmation. Primary tumor locations were narrowed and organized to specific areas of interest, including the face and skull, axial skeleton (spine, ribs, manubrium), pelvis, and extremities for clinical in concordance with the American Joint Committee on Cancer (AJCC) classification (Table 1). Patients were then separated into alive versus deceased cohorts based on reported 5-year vital (i.e. survival) status. Chi-square test or Fisher exact test was used to determine whether an association exists among the categorical variables of interest in these patient cohorts (Table 2). Continuous data were compared between alive versus deceased by t-test or Wilcoxon rank sum test. To avoid violating the proportional hazards assumption, the data thereafter required truncation to the first 10 years reported (which allowed for a minimum 5-year follow-up) for relevant survival analysis. A Cox proportional hazard regression was conducted on both the truncated data and the entire cohort to validate the findings. Hazard ratios (HRs), 95% confidence intervals (CIs), and P values were used to describe the Cox model. Test for proportional hazard assumption after fitting the Cox model was explored based on Schoenfeld residuals. After a univariate analysis was completed (Table 3), a backward stepwise procedure was used to select the optimal set of variables for multivariable (Figure 2) regression models. Significance was considered at P < 0.05. All statistical tests were conducted using STATA (V17).
Table 1
Distribution of Overall Patient-, Tumor-, and Treatment-specific Characteristics for Reported Pediatric Ewing Sarcoma Patients
| Factor | Value |
| N | 1996 |
| Age at diagnosis (yrs), mean (SD) | 11.6 (4.2) |
| Age groups (yrs) | |
| 0-5 | 206 (10.32%) |
| 6-11 | 633 (31.71%) |
| 12-17 | 1157 (57.97%) |
| Sex | |
| Male | 1160 (58.12%) |
| Female | 836 (41.88%) |
| Race | |
| White | 1767 (88.53%) |
| African American/Black | 59 (2.96%) |
| Other | 170 (8.52%) |
| Ethnicity | |
| Non-Hispanic | 1611 (84.79%) |
| Hispanic | 289 (15.21%) |
| Insurance status | |
| Not insured | 50 (2.58%) |
| Private insurance/managed care | 1341 (69.23%) |
| Government | 546 (28.19%) |
| Primary tumor location | |
| Lower extremity | 629 (32.62%) |
| Upper extremity | 243 (12.60%) |
| Facial & skull + mandible-specific | 128 (6.64%) |
| Spine + axial skeleton | 395 (20.49%) |
| Pelvis | 493 (25.57%) |
| Extremity, unspecified + unspecified bone | 40 (2.07%) |
| Tumor size | |
| ≤8 cm | 329 (16.48%) |
| >8 cm | 193 (9.67%) |
| Primary tumors could not be assessed | 873 (43.74%) |
| Missing | 601 (30.11%) |
| Margin status | |
| Negative | 777 (38.93%) |
| Positive | 156 (7.82%) |
| No primary site surgery | 859 (43.04%) |
| Not reported | 204 (10.22%) |
| Metastasis status | |
| No | 1727 (86.52%) |
| Yes | 241 (12.07%) |
| Not reported | 28 (1.40%) |
| Metastasis—bone | |
| No | 714 (35.77%) |
| Yes | 103 (5.16%) |
| Unknown/missing | 1179 (59.07%) |
| Metastasis—lung | |
| No | 639 (32.01%) |
| Yes | 178 (8.92%) |
| Unknown/missing | 1179 (59.07%) |
| Surgical procedure of the primary site | |
| Wide resection (with limb salvage) | 671 (34.32%) |
| Local excision | 332 (16.98%) |
| No surgery performed | 859 (43.94%) |
| Amputation | 93 (4.76%) |
| Surgery | |
| No | 859 (43.27%) |
| Yes | 1126 (56.73%) |
| Chemotherapy | |
| No | 79 (4.01%) |
| Yes | 1892 (95.99%) |
| Radiation | |
| No | 1571 (79.50%) |
| Yes | 405 (20.50%) |
| Treatment type | |
| Chemotherapy and surgery | 1103 (55.26%) |
| Surgery without chemotherapy | 23 (1.15%) |
| Chemotherapy without surgery | 789 (39.53%) |
| No chemotherapy or surgery | 81 (4.06%) |
| Mortality (5 year) | |
| Alive | 1361 (73.41%) |
| Deceased | 493 (26.59%) |
Table 2
Distribution of Characteristics by Mortality Status at 5-Year Follow-up Among Ewing Sarcoma Patients
| Factor | Alive | Deceased | P |
| N | 1361 | 493 | |
| Age at diagnosis, mean (SD) | 11.3 (4.3) | 12.5 (3.8) | <0.001 |
| Last contact or death, months from Dx, median (IQR) | 68.8 (38.8, 107.3) | 27.8 (15.9, 44.9) | <0.001 |
| Age—categorized | <0.001 | ||
| 0-5 | 163 (11.98%) | 27 (5.48%) | |
| 6-11 | 465 (34.17%) | 131 (26.57%) | |
| 12-17 | 733 (53.86%) | 335 (67.95%) | |
| Sex | 0.96 | ||
| Male | 789 (57.97%) | 285 (57.81%) | |
| Female | 572 (42.03%) | 208 (42.19%) | |
| Race | 0.75 | ||
| White | 1204 (88.46%) | 441 (89.45%) | |
| African American/Black | 40 (2.94%) | 15 (3.04%) | |
| Other | 117 (8.60%) | 37 (7.51%) | |
| Hispanic status | 0.06 | ||
| Non-Hispanic | 1110 (85.52%) | 383 (81.84%) | |
| Hispanic | 188 (14.48%) | 85 (18.16%) | |
| Insurance status | 0.37 | ||
| Not insured | 34 (2.57%) | 15 (3.17%) | |
| Private insurance/managed care | 927 (69.96%) | 315 (66.60%) | |
| Government | 364 (27.47%) | 143 (30.23%) | |
| Primary tumor locations | <0.001 | ||
| Lower extremity | 454 (34.37%) | 138 (29.30%) | |
| Upper extremity | 176 (13.32%) | 46 (9.77%) | |
| Facial and skull + mandible specific | 101 (7.65%) | 14 (2.97%) | |
| Spine + axial skeleton | 279 (21.12%) | 89 (18.90%) | |
| Pelvis | 282 (21.35%) | 176 (37.37%) | |
| Extremity, unspecified + unspecified bone | 29 (2.20%) | 8 (1.70%) | |
| Tumor size | <0.001 | ||
| ≤8 cm | 253 (18.59%) | 40 (8.11%) | |
| >8 cm | 121 (8.89%) | 58 (11.76%) | |
| Primary tumors cannot be assessed | 562 (41.29%) | 290 (58.82%) | |
| Missing | 425 (31.23%) | 105 (21.30%) | |
| Metastasis status | <0.001 | ||
| No | 1203 (88.39%) | 382 (77.48%) | |
| Yes | 139 (10.21%) | 102 (20.69%) | |
| Unknown/NA | 19 (1.40%) | 9 (1.83%) | |
| Metastasis—bone | <0.001 | ||
| No | 589 (43.28%) | 125 (25.35%) | |
| Yes | 41 (3.01%) | 62 (12.58%) | |
| Unknown/missing | 731 (53.71%) | 306 (62.07%) | |
| Metastasis—lung | <0.001 | ||
| No | 518 (38.06%) | 121 (24.54%) | |
| Yes | 112 (8.23%) | 66 (13.39%) | |
| Unknown/missing | 731 (53.71%) | 306 (62.07%) | |
| Margin status | <0.001 | ||
| Negative | 600 (44.09%) | 119 (24.14%) | |
| Positive | 109 (8.01%) | 38 (7.71%) | |
| No primary site surgery | 509 (37.40%) | 291 (59.03%) | |
| Unknown/missing | 143 (10.51%) | 45 (9.13%) | |
| Surgical procedure of the primary site | <0.001 | ||
| Wide resection (with limb salvage) | 510 (38.23%) | 111 (22.98%) | |
| Local excision | 248 (18.59%) | 62 (12.84%) | |
| No surgery performed | 509 (38.16%) | 291 (60.25%) | |
| Amputation | 67 (5.02%) | 19 (3.93%) | |
| Surgery | <0.001 | ||
| No | 509 (37.59%) | 291 (59.39%) | |
| Yes | 845 (62.41%) | 199 (40.61%) | |
| Chemotherapy | 0.50 | ||
| No | 51 (3.80%) | 22 (4.53%) | |
| Yes | 1292 (96.20%) | 464 (95.47%) | |
| Radiation | 0.39 | ||
| No | 1065 (79.06%) | 397 (81.02%) | |
| Yes | 282 (20.94%) | 93 (18.98%) | |
| Treatment type | <0.001 | ||
| Chemotherapy and surgery | 827 (60.76%) | 194 (39.35%) | |
| Surgery without chemotherapy | 18 (1.32%) | 5 (1.01%) | |
| Chemotherapy without surgery | 465 (34.17%) | 270 (54.77%) | |
| No chemotherapy or surgery | 51 (3.75%) | 24 (4.87%) |
Table 3
Unadjusted Associations of Factors With Survival Status Among Ewing sarcoma Patients
| Factors | HR | 95% CI | P |
| Age groups, yr | |||
| 0-5 | 1 | — | — |
| 6-11 | 1.43 | 0.94-2.16 | 0.09 |
| 12-17 | 2.27 | 1.54-3.37 | <0.001 |
| Sex | |||
| Male | 1 | — | — |
| Female | 1.02 | 0.85-1.22 | 0.82 |
| Race | |||
| White | 1 | — | — |
| African American/Black | 1.14 | 0.68-1.91 | 0.61 |
| Other | 0.95 | 0.68-1.33 | 0.77 |
| Ethnicity | |||
| Non-Hispanic | 1 | — | — |
| Hispanic | 1.33 | 1.05-1.68 | 0.02 |
| Insurance | |||
| Not insured | 1 | — | — |
| Private insurance/managed care | 0.79 | 0.47-1.36 | 0.40 |
| Government | 0.91 | 0.53-1.58 | 0.75 |
| Primary tumor locations | |||
| Lower extremity | 1 | — | — |
| Upper extremity | 0.88 | 0.63-1.22 | 0.44 |
| Facial and skull + mandible specific | 0.53 | 0.30-0.92 | 0.02 |
| Spine + axial skeleton | 1.08 | 0.83-1.41 | 0.58 |
| Pelvis | 1.96 | 1.57-2.46 | <0.001 |
| Extremity, unspecified + unspecified bone | 1.03 | 0.51-2.10 | 0.93 |
| Tumor size | |||
| ≤8 cm | 1 | — | — |
| >8 cm | 2.51 | 1.68-3.75 | <0.001 |
| Primary tumors cannot be assessed | 2.63 | 1.89-3.66 | <0.001 |
| Missing | 1.63 | 1.13-2.34 | 0.01 |
| Metastasis status | |||
| No | 1 | — | — |
| Yes | 2.33 | 1.87-2.91 | <0.001 |
| Unknown/NA | 2.03 | 1.05-3.94 | 0.04 |
| Bone metastasis | |||
| No | 1 | — | — |
| Yes | 5.19 | 3.82-7.04 | <0.001 |
| Unknown/missing | 1.58 | 1.28-1.96 | <0.001 |
| Lung metastasis | |||
| No | — | — | — |
| Yes | 2.28 | 1.69-3.07 | <0.001 |
| Unknown/missing | 1.45 | 1.17-1.80 | 0.001 |
| Margin status | |||
| Negative | 1 | — | — |
| Positive | 1.71 | 1.19-2.47 | 0.004 |
| No primary site surgery | 2.89 | 2.34-3.58 | <0.001 |
| Unknown/missing | 1.66 | 1.18-2.34 | 0.004 |
| Surgical procedure of the primary site | |||
| Wide resection (with limb salvage) | 1 | — | — |
| Local excision | 1.19 | 0.87-1.62 | 0.27 |
| No surgery performed | 2.66 | 2.13-3.30 | <0.001 |
| Amputation | 1.28 | 0.79-2.09 | 0.32 |
| Surgery | |||
| Yes | 1 | — | — |
| No | 2.43 | 2.03-2.91 | <0.001 |
| Chemotherapy | |||
| Yes | 1 | — | — |
| No | 1.86 | 1.21-2.85 | 0.01 |
| Radiation | |||
| No | 1 | — | — |
| Yes | 0.88 | 0.70-1.10 | 0.28 |
| Treatment type | |||
| Chemotherapy and surgery | 1 | — | — |
| Surgery without chemotherapy | 1.56 | 0.64-3.79 | 0.33 |
| Chemotherapy without surgery | 2.39 | 1.99-2.88 | <0.001 |
| No chemotherapy or surgery | 3.28 | 2.13-5.05 | <0.001 |
Results
Overall, 1996 pediatric ES patients were identified within the selected bone and joint public use file, which was truncated to 1854 patients to ensure a reported minimum 5-year follow-up.
Five-Year Survival
Overall, an aggregated 5-year survival rate of 73.92% was found in the entire cohort. Patients with localized cancer had a comparatively improved 5-year survival rate of 84.70% as opposed to those with macrometastatic disease on presentation, with a survival rate of 50.38% (Figure 1).
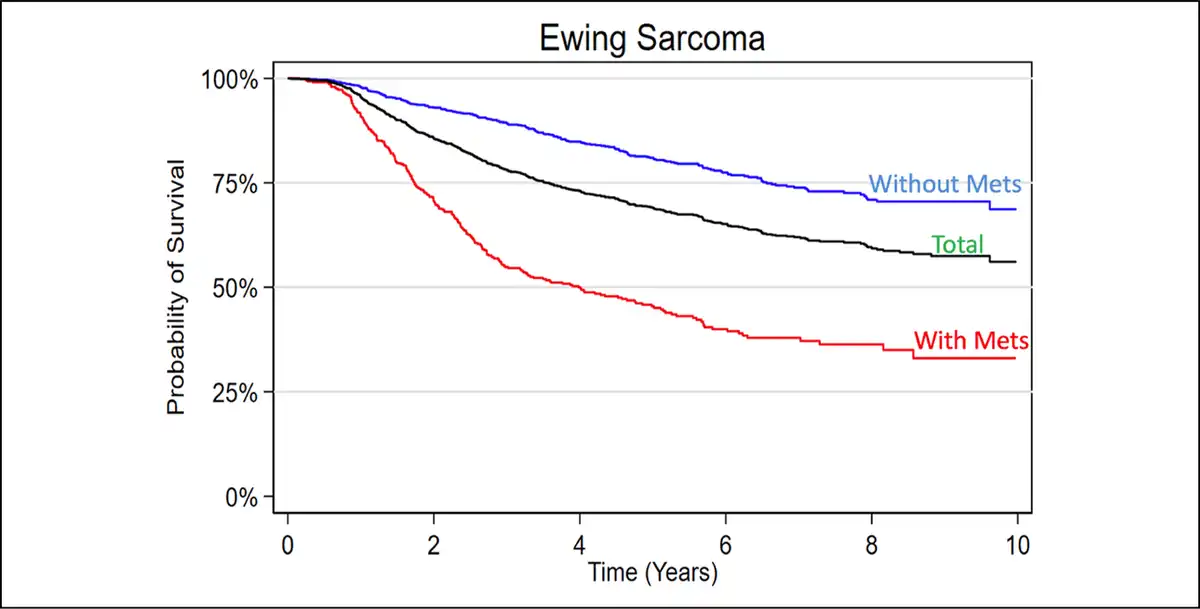
Figure 1
Graph showing Kaplan-Meier 10-year survival analysis of the included Ewing sarcoma patient cohort.
Patient-Specific Prognostic Factors
Most patients were of adolescent age (57.6%) with the majority being non-Hispanic, White males (Table 1). Most patients had private insurance or other form of managed care (69.23%) followed by government-based insurance (28.19%; Table 1). When separated into alive vs deceased groups, adolescent age at diagnosis was proportionally more common in the deceased group (67.95% vs. 53.86), whereas no notable differences were observed between sex, race, Hispanic status, or insurance type observed (Table 2). Unadjusted patient demographic-specific variables associated with worse survival rates included increasing age (12 to 17 years, HR [95% CI] = 2.27 [1.54 to 3.37]) and Hispanic ethnicity (HR [95% CI] = 1.33 [1.05 to 1.68]), whereas sex, overall race, and insurance status had no notable effect (Table 3). After multivariable analysis, only adolescent age (12 to 17, HR [95% CI] = 2.11 [1.42 to 3.13]) was considered a notable predictor of worse 5-year survival (Figure 2).
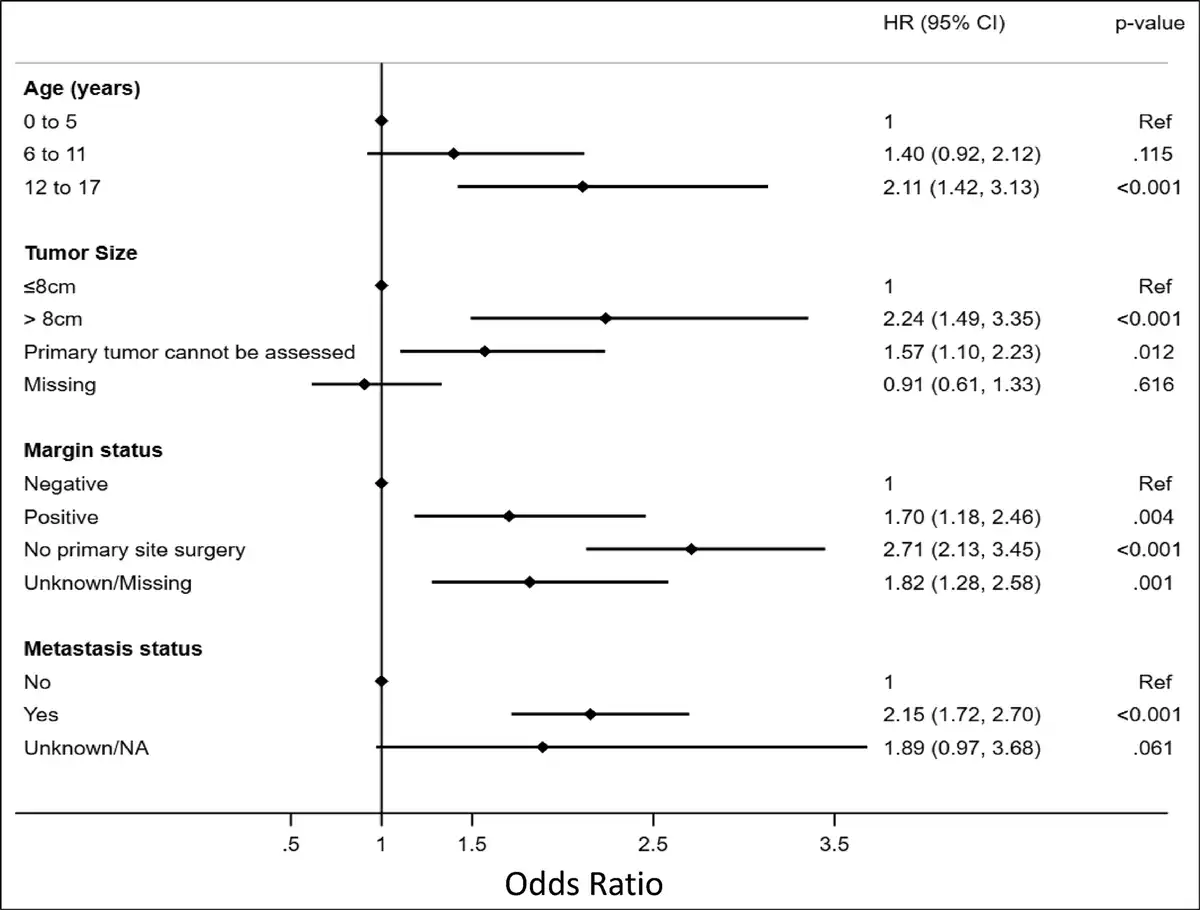
Figure 2
Graph/plot showing multivariable analysis of independent variables predictive of survival in pediatric patients diagnosed with Ewing sarcoma.
Tumor-Specific Prognostic Factors
The most common primary tumor locations were reported within the lower extremities (32.62%) or pelvis (25.57%; Table 1). Approximately 12% of patients were found to have clinically identified macrometastatic disease on presentation. Of the limited cases with the reported largest tumor dimension, most were tumor sizes of <8 cm (AJCC T1 classification). When separated into alive vs. deceased groups, tumor-specific characteristics more commonly reported in the deceased group included pelvic tumor location (37.37% vs. 21.35), larger tumor size (>8 cm: 11.76% vs. 8.89%), and metastasis on presentation (20.69% vs. 10.21%; Table 2). Unadjusted tumor-specific variables associated with worse survival rates included primary tumor location (pelvis, HR [95% CI] = 1.96 [1.57 to 2.46]), larger tumor size (>8 cm, HR [95% CI] = 2.51 [1.68 to 3.75]), and presence of metastasis at the time of diagnosis (HR [95% CI] = 2.33 [1.87 to 2.91]; Table 3), whereas grade had no notable effect. Primary tumor locations involving facial and skull or mandible-specific locations had lower mortality rates. After multivariable analysis, larger tumor size (>8 cm, HR [95% CI] = 2.24 [1.49 to 3.35]) and positive metastases on presentation (HR [95% CI] = 2.15 [1.72 to 2.70]) were associated with considerable significance in predicting worse 5-year survival (Figure 2).
Treatment-Specific Prognostic Factors
Most patients underwent treatment with a sequenced chemotherapy and surgical resection; approximately 20% of patients had received some degree of radiation treatment (Figure 1). When separated into alive vs deceased groups, treatment-specific characteristics more commonly reported in the deceased group included no primary surgery being performed, whereas no differences were observed in chemotherapy alone or radiation alone treatments (Table 2). Alive patients were reported to undergo combined chemotherapy and surgery at a markedly higher rate than deceased patients (60.76% versus 39.35%, respectively), who more often underwent nonsurgical management. Unadjusted treatment-specific variables associated with worse survival rates were positive margins after surgical resection (HR [95% CI] = 1.71 [1.19 to 2.47]), tumors in which no primary site surgery was performed (HR [95% CI] = 2.66 [2.13 to 3.30]), and patients treated without chemotherapy or surgery (HR [95% CI] = 3.28 [2.13 to 5.05]; Table 3). After multivariable analysis, only positive margin status (HR [95% CI] = 1.70 [1.18 to 2.46]) was considered significant in predicting worse 5-year survival (Figure 2).
Discussion
ES remains the second most common pediatric primary malignant bone tumor reported in the United States. Although the advent of combined chemotherapy and wide resection protocols have markedly improved outcomes, novel panacea treatments remain elusive. This study demonstrated a 74.5% overall 5-year survival rate, with improved survival in localized tumors (84.7%) compared with patients presenting with metastatic disease (50.4%). Collectively, these results imply optimistically improving short-midterm survival, particularly for patients presenting with metastases. Patient demographic-, tumor-, and treatment-specific variables demonstrated an effect on survival. The multivariable predictors of worse mortality were found to include older age, larger tumor size (>8 cm), macrometastatic disease on presentation, and positive surgical margins.
Survival Rates
Overall, an aggregated 5-year survival rate of 74.5% was found among all included patients. Patients with localized disease had a comparatively improved 5-year survival rate of 84.70% as opposed to patients with macrometastatic disease on presentation with a survival rate of 50.4% (Figure 1). These findings are consistent, albeit slightly higher, compared with similar epidemiology-based and review studies for localized (approximately 70% to 80%) and metastatic (approximately 30% to 40%) disease. However, many of these larger population-driven and hospital database–driven reports are 1 to 2 decades old. Therefore, the authors believe that the improved survival rates reported in this study are likely a result of gradual improvement/emphasis on multidisciplinary care provided at larger tertiary referral centers with greater access to randomized control trials.
Patient-Specific Prognostic Factors
Increasing age was associated with overall worse survival rates, with adolescents demonstrating worse 5-year survival rates compared with younger age groups. This is consistent with previous findings that have shown the survival rate of ES to be inversely proportional to age among pediatric and young adult populations. Ewing tumor size and location have some purported age dependence with adolescents having markedly larger tumors and higher incidence of occurrence within the pelvis and axial skeleton as compared with preadolescent patients. Given the worse prognosis associated with axial tumors, this was likely a contributor to worse outcomes with increasing age in this analysis.
Diagnosis of ES primarily occurred in non-Hispanic White males, which is consistent with ES's historical predilection for European ancestries and strikingly low incidence in populations of African descent. This is likely secondary to genetic germline variants that are protective for African Americans. However, there is a tumor dedifferentiation risk of ES in people of African descent, which may be a result of a specific genetic alteration within the EGR2 gene. Regardless, patient sex and race did not account for notable differences in mortality status.
Tumor-Specific Prognostic Factors
As expected, larger tumor size and metastatic disease on presentation were both associated with worse overall survival rates. These findings are consistent with tumor staging through the AJCC updated staging guidelines for primary bone tumors. Importantly, the most updated AJCC staging system (eighth edition) separates pelvis and spinal primary bone tumors to better reflect the overall difference in survival or treatments, respectively, as compared with appendicular tumors. It should also be noted that this study's analysis showed notable differences in tumor location among deceased and nondeceased patients diagnosed with ES, with deceased patients having notably higher percentages of pelvis locations (Tables 2 and 3). Axial primary bone malignancies, particularly within the pelvis, have been convincingly proven to have worse long-term survival rates compared with appendicular tumors, hence the updates to the AJCC staging system. It is uncertain as to why pelvic tumor location did not remain a notable predictor of worse mortality after the adjusted multivariable analysis (Table 3 versus Figure 2); however, this may reflect improvements in targeted radiation and chemotherapy treatments. Overall, improvements in targeted adjuvant treatments and the emphasis on multidisciplinary care are likely the two drivers behind the improved metastatic disease and/or pelvic tumor location survival rates.
The updated AJCC also lumps the appendicular skeleton with trunk, skull, and facial bone tumors for classification purposes of primary bone tumors. This study found that facial and mandible-specific primary Ewing tumor locations have markedly improved survival compared with appendicular tumors. These facial and mandible-specific primary tumors may be more easily noticed by patients and parents and therefore diagnosed earlier and treated in a timelier manner. However, these findings appear to contrast with similar survival rates to contemporary appendicular tumors. Although unclear for the causality, facial and mandible-specific tumors may need to be considered for separate categorization in newer editions of the AJCC Cancer Staging Manual.
Treatment-Specific Prognostic Factors
Before the 1970s, amputation was the only believed methodology for managing local tumor burden. The shift to combined chemotherapy with limb-preservation surgery revolutionized outcomes and patient functionality. As expected, mortality was found to be markedly affected by treatment type, with chemotherapy and surgery combination treatment associated with improved 5-year survival rates in this analysis. Of note, limb-preservation surgery is contingent on the tumor's candidacy for resection and the patient/parents' desire for that surgery. For example, large and invasive pelvic tumors or metastatic burdens may preclude patients from undergoing combined chemotherapy and surgical management. These patients may be managed with isolated radiation treatments or combined with chemotherapy for palliative treatment. In this investigation, ES patients treated with likely isolated palliative radiation showed a trend toward improved survival compared with no treatment. We believe that this was likely unresectable tumors in central locations such as the previously mentioned pelvic location subgroup.
Among patients who undergo tumor resection, survival rates have been predicated on margin status and tumor response to chemotherapy. Similarly, this study showed that positive margins after surgical resection were associated with lower 5-year survival rates. Unfortunately, there is considerable variability in the methodology of reporting margins. For example, standard teaching stressed the importance of a 3-cm bony margin and soft-tissue layer surrounding the tumor. However, less stringent 1.5-cm margins have demonstrated equivalent oncologic outcomes in osteosarcoma. Despite these controversies, the basic principle of reducing tumor burden and local recurrence through negative margins upon wide resection persists. Histologic response to chemotherapy is important in ES; however, this variable is not reported in the NCDB. Providers should bear in mind that even with successful treatment and remission past the 5-year mark, patients may require continued monitoring from their regular oncologist with referral to the musculoskeletal oncology surgeon as needed. Even with curative treatment without recurrence, there are notable short- and long-term treatment-related complications as a result of the chemotherapy and resection of these malignant bone tumors.
Limitations
There are several limitations to the use of large databases. Although the NCDB captures most new cancer patients in the United States and displays a high rate of 5-year follow-up, it is ultimately a hospital-based data set and not population based (such as Surveillance, Epidemiology, and End Results data). Therefore, patient inclusion is specific only to patients treated by hospitals accredited by the American College of Surgeons Committee on Cancer. Furthermore, analysis is limited to variables provided, which often results in the lack of specific data granularity. This includes exact tumor location, lack of EWSR1 status for diagnostic confirmation, histologic response to chemotherapy, radiation dosing, type of chemotherapy received, or patients' involvement in clinical trials. The NCDB seeks to be a comprehensive database consistent in reporting variables among tumors. However, this may lead to errors in reporting or erroneous variables such as reporting various grades of ES of bone, which is generally universally considered undifferentiated.
In addition, the NCDB is unable to link separate or associated cancer diagnoses for the same patient, which may serve pertinent in patients with genetic predispositions. It reports each individual cancer diagnosis as a different case. However, researchers can exclude patients indicated to have a history of other cancers, thereby minimizing the effect of prior malignant neoplasms, as was done in this analysis. Furthermore, we were unable to report on survival rates regarding patients who initially presented with localized disease with later progression to metastatic disease despite having received treatment. Finally, this analysis was only able to report overall survival rather than event-free survival rates as recurrence and specific complications are not explicitly provided by the NCDB.
Looking Forward—Treatments on the Horizon
Compressed chemotherapy induction with vincristine, doxorubicin, and cyclophosphamide remains the standard for ES within the United States. Although various trials continue, major changes in chemotherapy toward either malignancy have not been implemented in the United States in a decade. Current efforts in novel treatments include investigations into stereotactic radiosurgery, proton beam therapy, and even some targeted immunotherapies. Low incidence rates and difficulty obtaining adequately powered clinical trials remain burdensome for these investigations.
Conclusion
ES remains the second most common primary bone tumor in pediatric and adolescent patients in the United States with reportedly stagnant survival rates across the past few decades. Overall, this study's ES cohort exhibited an 84.7% 5-year survival rate in patients with localized disease compared with 50.4% for patients presenting with metastatic disease (73.9% overall). The improved survival for metastatic patients compared with prior studies is likely a reflection of improvements in adjuvant treatments and the establishment of multidisciplinary care teams. Multivariable regression showed that older age, larger tumor size, macrometastases, and positive margins all result in worse 5-year survival rates. This analysis serves to establish updated survival rates of pediatric ES treated within the United States to set standards for comparison among future studies. Continued multi-institutional and international collaboration is needed to optimize current treatment results and develop novel targeted therapies.
References
1.
Siegel DA, Richardson LC, Henley SJ, et al.: Pediatric cancer mortality and survival in the United States, 2001-2016. Cancer 2020;126:4379-4389.2.
Close AG, Dreyzin A, Miller KD, Seynnaeve BKN, Rapkin LB: Adolescent and young adult oncology-past, present, and future. CA Cancer J Clin 2019;69:485-496.3.
Smith MA, Altekruse SF, Adamson PC, Reaman GH, Seibel NL: Declining childhood and adolescent cancer mortality. Cancer 2014;120:2497-2506.4.
Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: Challenges for the twenty-first century. J Clin Oncol 2010;28:2625-2634.5.
Ward E, DeSantis C, Robbins A, Kohler B, Jemal A: Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 2014;64:83-103.6.
Grünewald TG, Bernard V, Gilardi-Hebenstreit P, et al.: Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat Genet 2015;47:1073-1078.7.
Mirabello L, Zhu B, Koster R, et al.: Frequency of pathogenic germline variants in cancer-susceptibility genes in patients with osteosarcoma. JAMA Oncol 2020;6:724-734.8.
Postel-Vinay S, Véron AS, Tirode F, et al.: Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat Genet 2012;44:323-327.9.
Allison DC, Carney SC, Ahlmann ER, et al.: A meta-analysis of osteosarcoma outcomes in the modern medical era. Sarcoma 2012;2012:704872.10.
Grünewald TGP, Cidre-Aranaz F, Surdez D, et al.: Ewing sarcoma. Nat Rev Dis Primers 2018;4:5.11.
Angervall L, Enzinger FM: Extraskeletal neoplasm resembling Ewing's sarcoma. Cancer 1975;36:240-251.12.
Askin FB, Rosai J, Sibley RK, Dehner LP, McAlister WH: Malignant small cell tumor of the thoracopulmonary region in childhood: A distinctive clinicopathologic entity of uncertain histogenesis. Cancer 1979;43:2438-2451.13.
Jaffe R, Santamaria M, Yunis EJ, et al.: The neuroectodermal tumor of bone. Am J Surg Pathol 1984;8:885-898.14.
Board WCoTE: Soft Tissue and Bone Tumours. Lyon, France. International Agency for Research on Cancer, 2020.15.
Jawad MU, Cheung MC, Min ES, Schneiderbauer MM, Koniaris LG, Scully SP: Ewing sarcoma demonstrates racial disparities in incidence-related and sex-related differences in outcome: An analysis of 1631 cases from the SEER database, 1973-2005. Cancer 2009;115:3526-3536.16.
Eaton BR, Claude L, Indelicato DJ, et al.: Ewing sarcoma. Pediatr Blood Cancer 2021;68(suppl 2):e28355.17.
Gaspar N, Hawkins DS, Dirksen U, et al.: Ewing sarcoma: Current management and future approaches through collaboration. J Clin Oncol 2015;33:3036-3046.18.
Maheshwari AV, Cheng EY: Ewing sarcoma family of tumors. J Am Acad Orthop Surg 2010;18:94-107.19.
Bacci G, Ferrari S, Longhi A, et al.: Role of surgery in local treatment of Ewing's sarcoma of the extremities in patients undergoing adjuvant and neoadjuvant chemotherapy. Oncol Rep 2004;11:111-120.20.
Esiashvili N, Goodman M, Marcus RB Jr: Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol 2008;30:425-430.21.
Grier HE, Krailo MD, Tarbell NJ, et al.: Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 2003;348:694-701.22.
Rodríguez-Galindo C, Navid F, Liu T, Billups CA, Rao BN, Krasin MJ: Prognostic factors for local and distant control in Ewing sarcoma family of tumors. Ann Oncol 2008;19:814-820.23.
Hesla AC, Papakonstantinou A, Tsagkozis P: Current status of management and outcome for patients with Ewing sarcoma. Cancers (Basel) 2021;13:1202.24.
Pappo AS, Dirksen U: Rhabdomyosarcoma, Ewing sarcoma, and other round cell sarcomas. J Clin Oncol 2018;36:168-179.25.
Stahl M, Ranft A, Paulussen M, et al.: Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatr Blood Cancer 2011;57:549-553.26.
Boffa DJ, Rosen JE, Mallin K, et al.: Using the National Cancer Database for outcomes research: A review. JAMA Oncol 2017;3:1722-1728.27.
Bilimoria KY, Bentrem DJ, Stewart AK, Winchester DP, Ko CY: Comparison of commission on cancer-approved and -nonapproved hospitals in the United States: Implications for studies that use the National Cancer Data Base. J Clin Oncol 2009;27:4177-4181.28.
Mohanty S, Bilimoria KY: Comparing national cancer registries: The National Cancer Data Base (NCDB) and the Surveillance, Epidemiology, and End Results (SEER) program. J Surg Oncol 2014;109:629-630.29.
Ahrens S, Hoffmann C, Jabar S, et al.: Evaluation of prognostic factors in a tumor volume-adapted treatment strategy for localized Ewing sarcoma of bone: The CESS 86 experience. Cooperative Ewing Sarcoma Study. Med Pediatr Oncol 1999;32:186-195.30.
Bosma SE, Ayu O, Fiocco M, Gelderblom H, Dijkstra PDS: Prognostic factors for survival in Ewing sarcoma: A systematic review. Surg Oncol 2018;27:603-610.31.
Cotterill SJ, Ahrens S, Paulussen M, et al.: Prognostic factors in Ewing's tumor of bone: Analysis of 975 patients from the European Intergroup Cooperative Ewing's Sarcoma Study Group. J Clin Oncol 2000;18:3108-3114.32.
Damron TA, Ward WG, Stewart A: Osteosarcoma, chondrosarcoma, and Ewing's sarcoma: National Cancer Data Base Report. Clin Orthop Relat Res 2007;459:40-47.33.
Werier J, Yao X, Caudrelier JM, et al.: A systematic review of optimal treatment strategies for localized Ewing's sarcoma of bone after neo-adjuvant chemotherapy. Surg Oncol 2016;25:16-23.34.
Bleyer A, Montello M, Budd T, Saxman S: National survival trends of young adults with sarcoma: Lack of progress is associated with lack of clinical trial participation. Cancer 2005;103:1891-1897.35.
Kreyer J, Ranft A, Timmermann B, et al.: Impact of the Interdisciplinary Tumor Board of the Cooperative Ewing Sarcoma Study Group on local therapy and overall survival of Ewing sarcoma patients after induction therapy. Pediatr Blood Cancer 2018;65:e27384.36.
Reed DR, Naghavi A, Binitie O: Sarcoma as a model for adolescent and young adult care. J Oncol Pract 2019;15:239-247.37.
Strönisch A, Märdian S, Flörcken A: Centralized and interdisciplinary therapy management in the treatment of sarcomas. Life (Basel) 2023;13:979.38.
Friedman DN, Chastain K, Chou JF, et al. Morbidity and mortality after treatment of Ewing sarcoma: A single-institution experience. Pediatr Blood Cancer. 2017;64. doi:10.1002/pbc.26562.39.
Jenkin RD, Al-Fawaz I, Al-Shabanah M, et al.: Localised Ewing sarcoma/PNET of bone—Prognostic factors and international data comparison. Med Pediatr Oncol 2002;39:586-593.40.
Koohbanani B, Han G, Reed D, et al.: Ethnicity and age disparities in Ewing sarcoma outcome. Fetal Pediatr Pathol 2013;32:246-252.41.
Worch J, Ranft A, DuBois SG, Paulussen M, Juergens H, Dirksen U: Age dependency of primary tumor sites and metastases in patients with Ewing sarcoma. Pediatr Blood Cancer 2018;65:e27251.42.
Amin MB, Greene FL, Edge SB, et al.: The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J Clin 2017;67:93-99.43.
Worch J, Cyrus J, Goldsby R, Matthay KK, Neuhaus J, DuBois SG: Racial differences in the incidence of mesenchymal tumors associated with EWSR1 translocation. Cancer Epidemiol Biomarkers Prev 2011;20:449-453.44.
Beck R, Monument MJ, Watkins WS, et al.: EWS/FLI-responsive GGAA microsatellites exhibit polymorphic differences between European and African populations. Cancer Genet 2012;205:304-312.45.
Amin MB; American Joint Committee on Cancer, American Cancer Society: AJCC cancer staging manual, in American Joint Committee on Cancer. Chicago, IL, Springer, 2017.46.
Albergo JI, Gaston CL, Laitinen M, et al.: Ewing's sarcoma: Only patients with 100% of necrosis after chemotherapy should be classified as having a good response. Bone Joint J 2016;98-b:1138-1144.47.
Biswas B, Rastogi S, Khan SA, et al.: Developing a prognostic model for localized Ewing sarcoma family of tumors: A single institutional experience of 224 cases treated with uniform chemotherapy protocol. J Surg Oncol 2015;111:683-689.48.
Fizazi K, Dohollou N, Blay JY, et al.: Ewing's family of tumors in adults: Multivariate analysis of survival and long-term results of multimodality therapy in 182 patients. J Clin Oncol 1998;16:3736-3743.49.
Lee J, Hoang BH, Ziogas A, Zell JA: Analysis of prognostic factors in Ewing sarcoma using a population-based cancer registry. Cancer 2010;116:1964-1973.50.
Miller BJ, Gao Y, Duchman KR: Does surgery or radiation provide the best overall survival in Ewing's sarcoma? A review of the National Cancer Data Base. J Surg Oncol 2017;116:384-390.51.
Smeland S, Bielack SS, Whelan J, et al.: Survival and prognosis with osteosarcoma: Outcomes in more than 2000 patients in the EURAMOS-1 (European and American Osteosarcoma Study) cohort. Eur J Cancer 2019;109:36-50.52.
Verma V, Denniston KA, Lin CJ, Lin C: A comparison of pediatric vs. adult patients with the Ewing sarcoma family of tumors. Front Oncol 2017;7:82.53.
Wells ME, Eckhoff MD, Kafchinski LA, Polfer EM, Potter BK. Conventional cartilaginous tumors: Evaluation and treatment. JBJS Rev. 2021;9. doi:10.2106/JBJS.RVW.20.00159.54.
Berger M, Fagioli F, Abate M, et al.: Unusual sites of Ewing sarcoma (ES): A retrospective multicenter 30-year experience of the Italian Association of Pediatric Hematology and Oncology (AIEOP) and Italian Sarcoma Group (ISG). Eur J Cancer 2013;49:3658-3665.55.
Martin E, Radomski S, Harley EH: Pediatric Ewing sarcoma of the head and neck: A retrospective survival analysis. Int J Pediatr Otorhinolaryngol 2019;117:138-142.56.
Rehman R, Osto M, Parry N, et al.: Ewing sarcoma of the craniofacial bones: A qualitative systematic review. Otolaryngol Head Neck Surg 2022;166:608-614.57.
Friedman MA, Carter SK: The therapy of osteogenic sarcoma: Current status and thoughts for the future. J Surg Oncol 1972;4:482-510.58.
Eilber FR, Eckhardt J, Morton DL: Advances in the treatment of sarcomas of the extremity. Current status of limb salvage. Cancer 1984;54(11 suppl):2695-2701.59.
Link MP, Goorin AM, Miser AW, et al.: The effect of adjuvant chemotherapy on relapse-free survival in patients with osteosarcoma of the extremity. N Engl J Med 1986;314:1600-1606.60.
Hoang K, Gao Y, Miller BJ: The variability in surgical margin reporting in limb salvage surgery for sarcoma. Iowa Orthop J 2015;35:181-186.61.
Biermann JS, Siegel G. Orthopedic Knowledge Update: Musculoskeletal Tumors. Illinois: American Academy of Orthopaedic Surgeons; 2020:280–289.62.
Loh AH, Wu H, Bahrami A, et al.: Influence of bony resection margins and surgicopathological factors on outcomes in limb-sparing surgery for extremity osteosarcoma. Pediatr Blood Cancer 2015;62:246-251.63.
Arpaci E, Yetisyigit T, Seker M, et al.: Prognostic factors and clinical outcome of patients with Ewing's sarcoma family of tumors in adults: Multicentric study of the Anatolian Society of Medical Oncology. Med Oncol 2013;30:469.64.
He F, Zhang W, Shen Y, et al.: Effects of resection margins on local recurrence of osteosarcoma in extremity and pelvis: Systematic review and meta-analysis. Int J Surg 2016;36:283-292.65.
Brouwer CA, Gietema JA, van den Berg MP, et al.: Long-term cardiac follow-up in survivors of a malignant bone tumour. Ann Oncol 2006;17:1586-1591.66.
Marina NM, Liu Q, Donaldson SS, et al.: Longitudinal follow-up of adult survivors of Ewing sarcoma: A report from the Childhood Cancer Survivor Study. Cancer 2017;123:2551-2560.67.
Juergens C, Weston C, Lewis I, et al.: Safety assessment of intensive induction with vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) in the treatment of Ewing tumors in the EURO-E.W.I.N.G. 99 clinical trial. Pediatr Blood Cancer 2006;47:22-29.68.
Womer RB, West DC, Krailo MD, et al.: Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: A report from the Children's Oncology Group. J Clin Oncol 2012;30:4148-4154.69.
Team ACSMaEC. What's New in Ewing Tumor Research and Treatment? https://www.cancer.org/cancer/ewing-tumor/about/new-research.html#references (2021). Accessed December 11, 2023.