Introduction
The current systemic therapeutic agents against solid tumors comprise chiefly chemotherapy, targeted therapy, immune checkpoint inhibitors (ICIs). Although these therapies have improved outcomes, better ways of treatment are expected. Antibody-drug conjugates (ADCs) are rapidly emerging class of therapeutic agents combining cytotoxic drugs and targeted antibodies via a chemical linker (Fig. 1). The concept of targeted chemotherapy was first proposed by a German scientist, Paul Ehrlich 100 years ago, known as ‘magic bullet’.
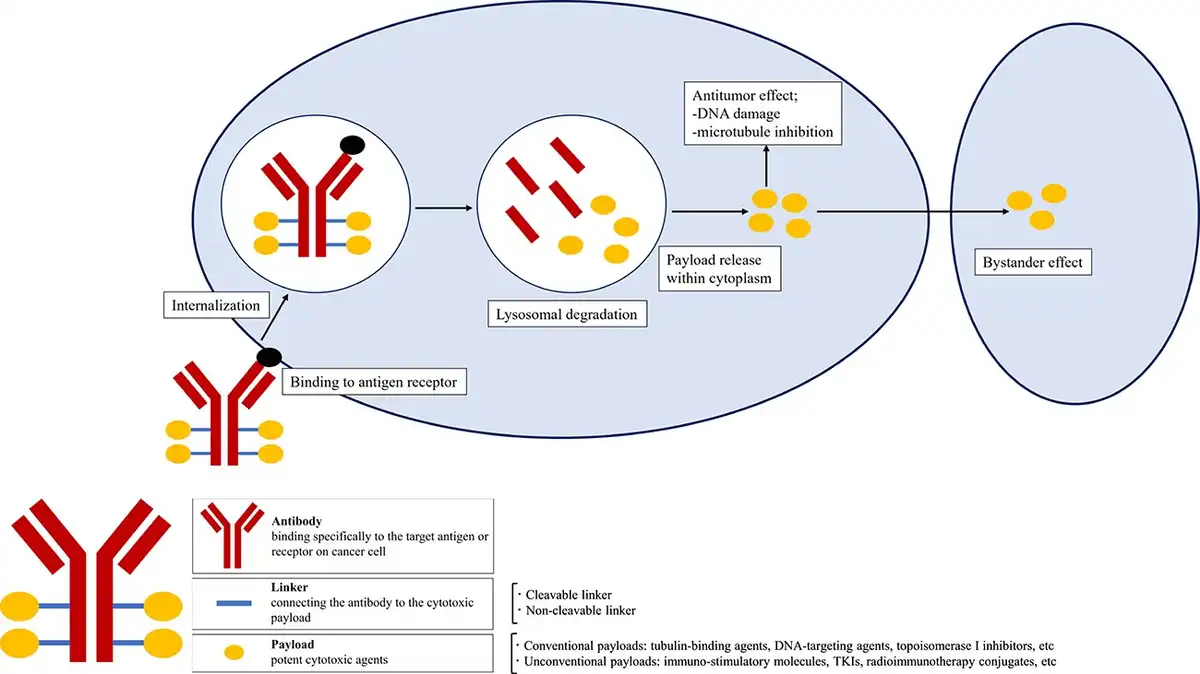
Figure 1
Structure of antibody-drug conjugates (ADCs). ADCs have three components: a monoclonal antibody that binds to an antigen or receptor on cancer cells, a linker that connects the antibody and payload, and a payload that is a potent cytotoxic agent. ADCs bind to target antigens on the surface of cancer cell followed by internalisation. After lysosomal degradation, payloads are released into the cytoplasm and induces cell death via DNA damage or microtubule inhibition. Moreover, payloads pass through the cell membrane and show antitumor effect on surrounding cancer cells, called bystander effect.
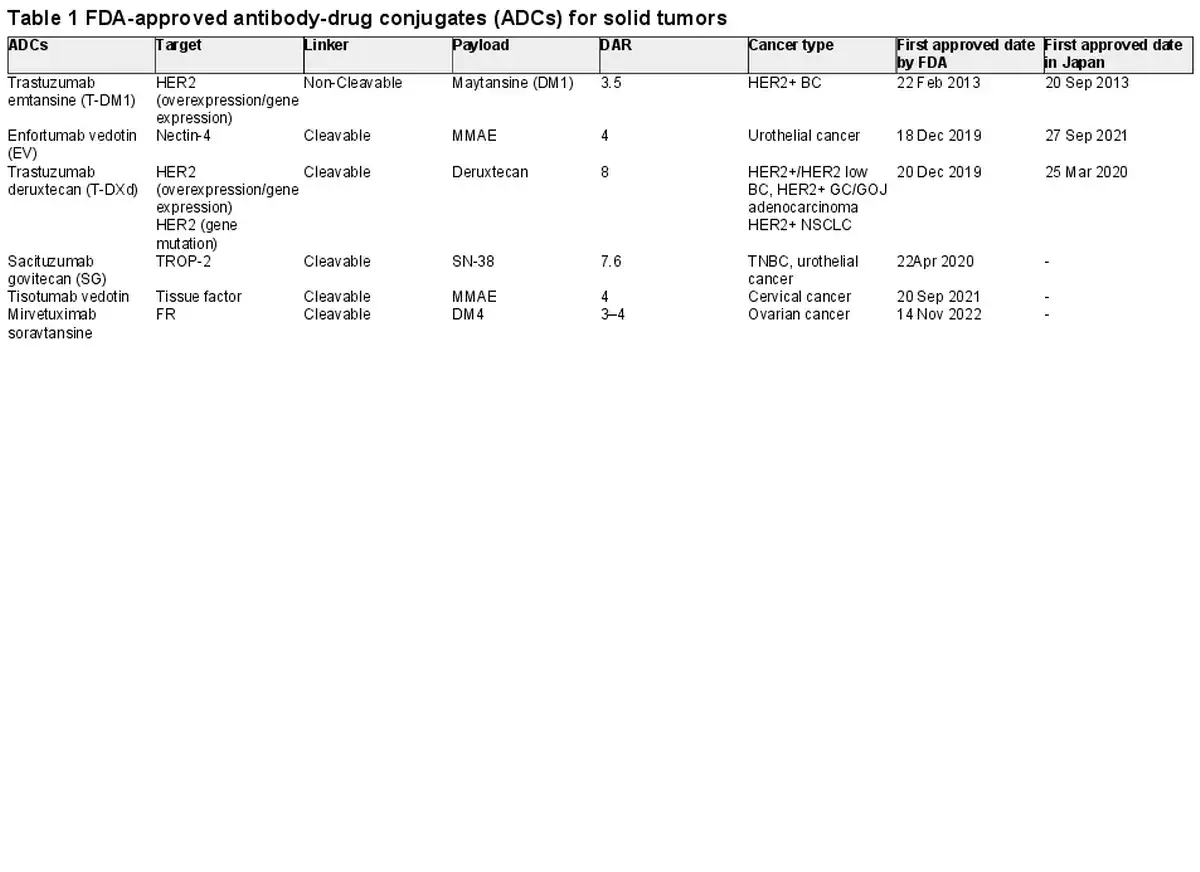
The US Food and Drug Administration (FDA) firstly approved Gemtuzumab ozogamicin (Mylotarg®) in 2000 for the treatment of adults with CD33-positive acute myeloid leukemia (). As for solid tumors, trastuzumab emtansine (T-DM1) was approved by FDA in 2013 for the treatment of metastatic breast cancer. As of 1 November 2023, the FDA has approved 13 ADCs for solid or hematologic cancers (6 for solid tumors) (Table 1, 2). In this review, we aim to provide a brief overview of the ADCs for solid tumors, their structure and mechanism of action, available clinical trial data.
Structure and mechanism
Antibody
Monoclonal antibodies are the components that allow the ADCs to bind specifically to the target antigen or receptor on cancer cell. The antibodies for ADCs require an adequate binding affinity, efficient internalisation, low immunogenicity and a long half-life. Murine antibodies were often used in the early days of ADCs development, but their high immunogenicity made them likely to cause serious side effects. Currently, development is increasingly used chimeric, humanised and fully humanised antibodies with low immunogenicity. In the five types of Immunoglobulin (M, A, D, E, G), Immunoglobulin G (IgG) is the most commonly used in ADCs. IgGs are classified into IgG1–4, but IgG3 is usually not used because of its short half-life, and IgG1 is widely used due to its immunogenic functions such as antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cell-mediated phagocytosis (ADCP) and complement-dependent cytotoxicity (CDC) ().
Payload
Potent cytotoxic agents are the ‘payloads’ of ADCs. Many of the cytotoxic agents in ADCs are very low IC50 (the drug-concentration to inhibit the growth of 50% of cells), so that they cannot be used as drugs on their own because they exhibit strong toxicity. The payloads chiefly classified into tubulin-binding agents (auristatins or maytansinoids), DNA-targeting agents (calicheamicins, duocarmycin), topoisomerase I inhibitors (deruxctecan, SN-38). In these cytotoxic agents, auristatins are the largest family of payloads so far, due to their favorable biochemical characteristics. Auristatins include two types of derivatives: monomethyl auristatin E (MMAE) and monomethyl auristatin F (MMAF). While the MMAF shows low membrane permeability, the MMAE has favorable permeability to cell membrane and can diffuse into nearby cancer cells and kill them, called the bystander effects. Currently, in approved ADCs for solid tumors, MMAE is loaded in enfortumab vedotin (EV) and tisotumab vedotin ().
Furthermore, several types of alternative payloads are under development, such as radionucleotides, immune modulators, tyrosine kinase inhibitors and dual-distinct payloads. Moreover, the conjugation of payloads to bispecific antibodies is another strategy (). The drug-antibody ratio (DAR) is the amount of payloads loaded to the antibody and is a critical factor to determine the efficacy of the ADCs. ADCs with higher DAR values increase potency in vitro but are usually less favorable in vivo due to the unstable structure and low binding capacity, resulting in faster plasma clearance. Therefore, in most of ADCs, their DAR values are in the range of 2–4. However, several ADCs have emerged with favorable pharmacokinetics profiles even in DAR values of around 8, such as trastuzumab deruxtecan (T-DXd) and sacituzumab govitecan (SG) (,).
Linker
Linkers connect the antibody to the cytotoxic payload and define several characteristics of ADCs. Ideal linkers should ensure ADCs stability in the bloodstream but release payloads at the tumor site. The conjugation site has a significant impact on the stability and pharmacokinetics of ADCs. Site-specific binding produces homogeneous ADCs whose properties can be tailored to maximise therapeutic range ().
Linkers can be categorised into cleavable and non-cleavable. Cleavable linkers release the payload based on some factors of cancer cell such as pH, reducing agents and lysosomal protease. While cleavable linkers have the advantage of providing a bystander effect, they have the disadvantage of increased toxicity in normal tissues. Conversely, non-cleavable linkers are more stable in plasm, so that they release payload only after complete lysosomal degradation of the antibody and limit premature release of payload. Whereas these features of non-cleavable linkers offer less toxicity, bystander effect is not expected because of the extremely low cell membrane permeability. Among the 13 approved ADCs, cleavable linkers are the most common (11 of 13) and non-cleavable linkers are only two (trastuzumab emtansine, belantamab mafodotin).
Mechanism of action
The action of ADCs begins with the recognition and binding of target antigens on the surface of cancer cell. After that, the ADCs are internalised through endocytosis with formation of early endosomes. The early endosomes mature into late endosomes, which then fuse with lysosomes. Whereas ADCs with cleavable linkers release their payload depending on the localisation of enzymes and other factors, the ADCs with non-cleavable linkers release their payloads when antibodies are degraded in lysosomes. The payloads are released into the cytosol and induces cell death via DNA damage or microtubule inhibition. In addition, after the payloads are released intracellularly, they may pass through the cell membrane and exert an antitumor effect on surrounding cancer cells, referred to as the bystander effect. The bystander effect is important because tumors are heterogeneous populations of cancer cells and target antigens are not expressed by all cancer cells. This bystander effect may explain the efficacy of T-DXd in patients with ‘HER2-low’ breast cancer. Furthermore, the ADCs display anticancer effect through immunogenic reactions including ADCC, ADCP and CDC.
ADCs for solid tumors
Trastuzumab emtansine (T-DM1)
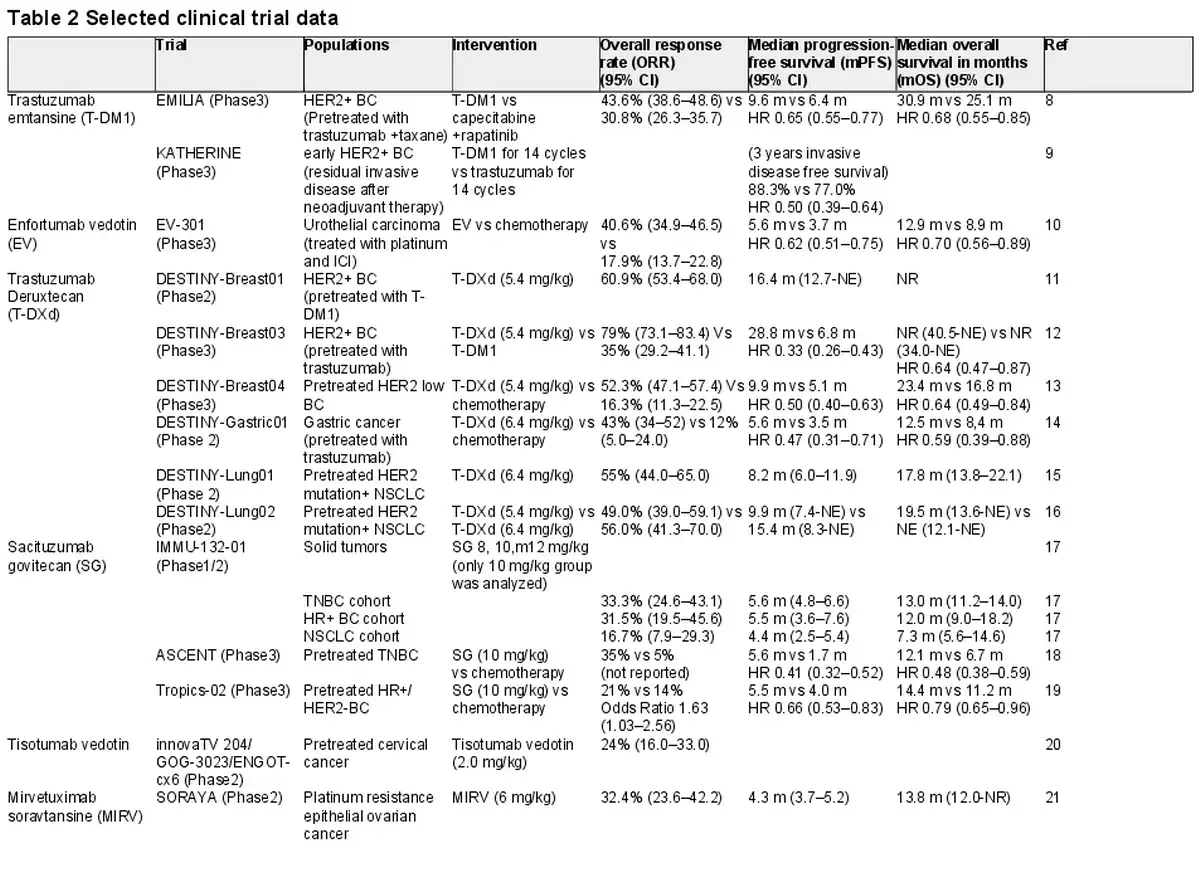
T-DM1 is the first ADC approved for solid tumors (). It combines the anti-HER2 IgG1 monoclonal antibody to the cytotoxic payload maytansine (DM1), using a non-cleavable linker. FDA approved T-DM1 for the treatment of HER2-positive metastatic breast cancer (BC) based on the results of EMILIA trial, a global phase III trial. The overall response rate (ORR) was 43.6 vs 30.8%, median progression free survival (mPFS) was 9.6 m (HR 0.65, 95%CI 0.55–0.77), median overall survival (mOS) was 30.9 m (HR 0.68, 95% CI 0.55–0.85). The most frequent grade ≥ 3 treatment-emergent adverse events (TEAEs) were thrombocytopenia (12.9%) and elevated serum concentrations of aspartate aminotransferase (4.3%) and alanine aminotransferase (2.9%).
In addition, it is used for an adjuvant therapy for residual disease in early-stage HER2-positive BC after neoadjuvant therapy, on the basis of KATHERINE trial (). In this phase III trial, T-DM1 (for 14 cycles) was compared with trastuzumab for patients with HER2-positive early breast cancer who were found to have residual invasive disease at surgery after neoadjuvant therapy including a trastuzumab and a taxane (with or without anthracycline). 3 years invasive disease free survival was 88.3% (HR 0.50, 95%CI 0.39–0.64). Grade ≥ 3 TEAEs were reported that a thrombocytopenia (5.7%) and hypertension (2.0%). Assigned therapy were completed in 71.4 (T-DM1 group) and 81.0% of patients (trastuzumab group). T-DM1 was discontinued due to thrombocytopenia (4.2%), elevated blood bilirubin level (2.6%), elevated aspartate aminotransferase level (1.6%), elevated alanine aminotransferase level (1.5%), peripheral sensory neuropathy (1.5%) and decreased ejection fraction (1.2%).
Enfortumab vedotin
Nectin-4 is a cell-adhesion molecule that may associated with cancer cell growth and proliferation and overexpressed in urothelial cancer and breast cancer. EV is an ADC consisting of a fully human monoclonal nectin-4 antibody and monomethyl auristatin E (MMAE) (). A phase III trial EV-301 was conducted to evaluated EV as compared with investigator’s choice chemotherapy (docetaxel, paclitaxel or vinflunine) in patients with locally advanced or metastatic urothelial cancer who had previously treated with a platinum-based chemotherapy and PD-1/PD-L1 inhibitor. The mPFS was 5.6 months (HR 0.62, 95% CI 0.51–0.75) and the mOS was 12.9 months (HR 0.70, 95% CI 0.56–0.89). The most common grade ≥ 3 treatment-related adverse events (TRAEs) were maculopapular rash (7.4%), fatigue (6.4%) and decreased neutrophil count (6.1%).
Trastuzumab deruxtecan
T-DXd consists of a humanised HER2 antibody (trastuzumab) and topoisomerase I inhibitor (deruxtecan) (). The FDA approved T-DXd for the treatment of pretreated HER2-positive metastatic BC based on the data of a phase II trial, DESTINY-Breast01. Then, in DESTINY-Breast03, a phase III trial comparing T-DXd with T-DM1 in metastatic HER2-positive BC, T- DXd demonstrated clinical advantage (). The mPFS was 28.8 months (HR 0.33, 95% CI 0.26–0.43) and mOS was NR (HR 0.64, 95% CI 0.47–0.87). The most common grade ≥ 3 TEAEs where neutrophil count decreased (16%), anemia (9%), platelet count decreased (8%), nausea (7%). Drug-related interstitial lung disease (ILD) or pneumonitis occurred in 15% of patients, including grade 1 (4%), grade 2 (10%), grade 3 (<1.0%) and grade ≥ 4 (0%).
Furthermore, DESTINY-Breast04, a phase III trial, was conducted to investigate the efficacy of T-DXd in the treatment of HER2-low metastatic BC (). This study revealed a meaningful improvement in mPFS (HR 0.50, 95% CI 0.40–0.63) and mOS (HR 0.64, 95% CI, 0.49–0.84). The most frequent grade ≥ 3 AEs were neutropenia (13.7%), anemia (8.1%) and fatigue (7.5%). Drug-related ILD or pneumonitis occurred in 12.1% of patients, with grade 1 (3.5%), grade 2 (3.5%), grade 3 (1.3%) and grade 5 (0.8%).
In DESTINY-Gastric01, randomised phase II trial, T-DXd showed significant response as compared with chemotherapy in HER2-positive advanced gastric or gastroesophageal junction cancer patients who have previously treated with trastuzumab (). The PFS was 5.6 months (HR 0.47, 95% CI 0.31–0.71) and the mOS was 12.5 months (HR 0.59, 95% CI 0.39–0.88). The most common grade ≥ 3 AEs were with decreased neutrophil count (51%), anemia (38%), a decreased white-cell count (21%), and decreased appetite (17%). A total of 12 patients (10%) had drug-related ILD or pneumonitis, with three events of grade 1, six events of grade 2, two events of grade 3, one event of grade 4 and no grade 5 events.
In phase II, DESTINY-Lung01 trial evaluated the efficacy of T-DXd (6.4 mg/kg) in the treatment of pretreated metastatic HER2-mutant NSCLC patients (). The ORR was 55% (95% CI, 44–65), the mPFS was 8.2 months (95% CI, 6.0–11.9) and the mOS was 17.8 months (95% CI, 13.8–22.1). Grade ≥ 3 TRAEs occurred in 46% of patients, including neutropenia (19%) and anemia (10%). Drug-related ILD or pneumonitis were reported in 24 patients (26%), with 3 events of grade 1, 15 events of grade 2, 4 events of grade 3 and two events of grade 5. The median time to the onset of ILD was 141 days (range, 14–462).
Additionally, the clinical benefit of T-DXd (5.4 mg/kg) compared with T-DXd (6.4 mg/kg) was reported in phase II DESTINY-Lung02 trial (). Both doses of T-DXd showed clinical activity; however, the safety profile was tolerable favoring T-DXd 5.4 mg/kg. Grade ≥ 3 drug-related TEAEs occurred in 38.6 (in 5.4 mg/kg group) and 58.0% patients (in 6.4 mg/kg group). Drug-related ILD were 12.9 and 28.0% of patients (grade ≥ 3 were 2.0% in both group), respectively.
Sacituzumab govitecan
TROP2 (trophoblast antigen 2) is a transmembrane glycoprotein coded by the gene TACSTD2, which primarily acts as intracellular calcium signal transducer (). While TROP2 is overexpressed in many human epithelial tissues, the expression increases in tumor tissue compared to normal tissues and has been associated with poor prognosis. This overexpression occurs in various tumor types including breast cancer, NSCLC, colon cancer, esophaegeal squamous cancer, thyroid cancer and hepatobiliary cancer.
SG, formerly IMMU-132, is composed of a humanised TROP2 antibody and the active metabolite of the topoisomerase I inhibitor irinotecan (SN-38).
ASCENT trial, a phase III trial comparing SG with a physician’s choice chemotherapy (eribulin, vinorelbine, capecitabine or gemcitabine) in TNBC patients, revealed a survival benefit of SG (). The mPFS was 5.6 months (HR 0.41, 95% CI 0.32–0.52) and the mOS was 12.1 months (HR 0.48, 95% CI, 0.38–0.59). The most common grade ≥ 3 AEs were neutropenia (51%), leukopenia (10%), diarrhea (10%), anemia (8%) and febrile neutropenia (6%).
In the Tropics-02 study, a randomised phase III trial, SG was compared with physician’s choice chemotherapy (eribulin, vinorelbine, capecitabine or gemcitabine) in the HR+/HER2– breast cancer patients with endocrine-resistant, chemotherapy-treated (). The mPFS was 5.5 months (HR 0.66, 95% CI 0.53–0.83) and the mOS was14.4 months (HR 0.79, 95% CI, 0.65–0.96). The most frequent grade ≥ 3 TEAEs were neutropenia (51%) and diarrhea (10%).
Tisotumab vedotin
Tisotumab vedotin is an ADC consisting of a monoclonal antibody against tissue factor (TF) and MMAE (). TF is a transmembrane glycoprotein physiologically expressed on fibroblasts and subendothelial cells and serves as an initiator of the extrinsic coagulation cascade. Conversely, TF is frequently overexpressed on several solid tumors, including cervical cancer and related to tumor growth, metastasis and angiogenesis.
A phase II trial, innovaTV 204/GOG-3023/ENGOT-cx6, reported that the sclinical activity of Tisotumab vedotin in patients with pretreated recurrent or metastatic cervical cancer. The confirmed ORR was 24% (95% CI, 16.0–33.0), including 7% of CR and 17% of PR. The most common grade ≥ 3 TRAEs included neutropenia (3%), fatigue (2%), ulcerative keratitis (2%) and peripheral neuropathies (2% each with sensory, motor, sensorimotor and neuropathy peripheral).
Mirvetuximab soravtansine
Mirvetuximab soravtansine (MIRV) is an ADC with folate receptor an (Fra) antibody conjugated with the maytansinoid (DM4) as payload (). In the SORAYA study, a single arm, phase II study investigated a clinical activity of MIRV in patients with FRa positive, platinum resistant epithelial ovarian cancer. The ORR was 32.4% (95% CI, 23.6–42.2), the mPFS was 4.3 months (95% CI, 3.7–5.2) and the mOS was 13.8 months (95% CI, 12.0-NR). The most common grade ≥ 3 TRAEs were blurred vision (6%), keratopathy (9%), dry eye (2%), diarrhea (2%) and neutropenia (2%).
Datopotamab deruxtecan
Datopotamab deruxtecan (Dato-DXd) is a novel ADC composed of humanised anti TROP2 IgG1 monoclonal antibody, a cleavable linker, a topoisomerase I inhibitor payload and has a DAR of 4 (). Although both of Dato-DXd and SG are the TROP2 ADCs, the two ADCs are different in some points. Whereas 90% of the payload of SG is released in 3 days, just 5% of the payload of Dato-DXd is released after 21 days. Because of this pharmacokinetic feature, Dato-DXd is administered every 3 weeks in contrast to SG dosed on Days 1 and 8.
TROPION-Pan Tumor01, a phase I basket trial, is investigating the safety and antitumor activity of Dato-DXd at dose levels of 4–8 mg/kg in unresectable advanced solid tumors. In patients treated at 6 mg/kg of NSCLC cohort, the ORR was 26% (95% CI, 14.6–40.3), the mPFS was 6.9 months (95% CI, 2.7–8.8) and the mOS was 11.4 months (95% CI, 7.1–20.6). The most frequent grade ≥ 3 TEAEs were pneumonia, anemia and lymphocytopenia. The potential ILD occurred in 28 patients across expansion doses (13 ≥ grade 3, 15 ≤ grade2).
Furthermore, some phase III trials of Dato-DXd are ongoing. TROPION-Lung 01, a phase III study evaluating Dato-DXd as compared with docetaxel (DTX) in pretreated NSCLC patients, demonstrated greater efficacy with manageable safety profile (). The confirmed ORR was 26.4 (Dato-DXd) and 12.8% (DTX), the median duration of response (DOR) was 7.1 and 5.6 months, and the mPFS in non-squamous histology subgroup was 5.6 and 3.7 months (HR 0.63, 95% CI 0.51–0.78). The most common TRAEs in Dato-DXd group were stomatitis (49.2%, mostly grade 1–2) and nausea (37%). Drug-related ILD grade ≥ 3 occurred in 3.4% in Dato-DXd group and 1.4% in DTX group.
The combination therapies of Dato-DXd with other anticancer agents for NSCLC patients are investigated in TROPION-Lung07 (ClinicalTrials.gov identifier: NCT05555732), TROPION-Lung08 (ClinicalTrials.gov identifier: NCT05215340), AVANZAR (ClinicalTrials.gov identifier: NCT05687266). TROPION-Lung07, a phase III trial, is evaluating Dato-DXd plus pembrolizumab with or without a platinum agent in NSCLC with a PD-L1 tumor proportion score (TPS) of <50%.
TROPION-Lung08 is a phase III trial to evaluate Dato-DXd plus pembrolizumab comparing with pembrolizumab as first-line therapy for patients with a PD-L1 TPS of ≥50%. AVANZAR, a phase III study, examined the survival benefit of Dato-DXd plus durvalumab plus carboplatin against pembrolizumab plus platinum-based chemotherapy as first-line treatment.
In phase III TROPION-Breast01 trial, Dato-DXd was investigated in hormone (HR) positive/HER2 negative BC patients priory treated with endocrine therapy and cytotoxic chemotherapy. Dato-DXd showed clinical benefit (). The confirmed response was 36.4% (Dato-DXd) and 22.9% (investigator’s choice of chemotherapy), and the mPFS was 6.9 months and 4.9 months (HR 0.63, 95% CI 0.52–0.76). OS data were not mature. The grade ≥ 3 TRAEs were less likely in Dato-DXd group.
Patritumab deruxtecan (HER3-DXd)
Human epidermal growth factor receptor 3 (HER3) is a tyrosine kinase receptor that expressed in various solid tumors, including breast cancer and NSCLC (,). It is reported that high expression of HER3 is associated with poor prognoses and resistance mechanism of EGFR TKI.
HER3-DXd is a novel ADC composed of a humanised HER3 antibody and a topoisomerase I inhibitor payload. It has a high DAR value of 8. HERTHENA-Lung01 is a phase II trial to evaluate the efficacy and safety of HER3-DXd in EGFR mutated NSCLC patients previously treated with EGFR TKI and platinum-based chemotherapy (). In this study, HER3-DXd demonstrated significant efficacy. The confirmed ORR was 29.8% (95CI, 23.9–36.2), the mPFS was 5.5 months (95% CI, 5.1–5.9) and the mOS was 11.9 months (95% CI, 11.2–13.1). Grade ≥ 3 TEAEs occurred in 64.9% of patients, including thrombocytopenia (20.9%), neutropenia (19.1%), anemia (14%) and leukopenia (10%). A phase III trial in EGFR mutated NSCLC patients pretreated with EGFR TKI is ongoing (HERTHENA-Lung02; ClinicalTrials.gov identifier: NCT05338970).
The U31402-A-J101 study is a phase I/II trial to assess the maximum tolerated dose (in the dose-escalation part) and the safety and efficacy (in the dose expansion part) of HER3-DXd in patients with previously treated HER3 expressing BC (). In patients with HR+/HER2- BC, the confirmed ORR was 30.1% (95% CI, 21.8–39.4), the mPFS was 7.4 months (95% CI, 4.7–8.4) and the mOS was 14.6 months (95% CI, 11.3–19.5). In patients with TNBC, the confirmed ORR was 22.6% (95% CI, 12.3–36.2), the mPFS was 5.5 months (95% CI, 3.9–6.8) and the mOS was 14.6 months (95% CI, 11.2–17.2). In patients with HER2+ BC, the confirmed ORR was 42.9% (95% CI, 17.1–71.1), mPFS was 11.0 months (95% CI, 4.4–16.4), mOS was 19.5 months (95% CI, 12.2- NE). Grade ≥ 3 TEAEs occurred in 71.4% of all patients: 64.6 (4.8 mg/kg group) and 81.6% of patients (6.4 mg/kg group). In all patients, the most common grade ≥ 3 TEAEs were decreased neutrophil count (39.5%), decreased platelet count (30.8%), anemia (18.6%) and decreased white blood cell count (18.1%). Several studies to evaluate HER3-DXd are ongoing, such as ICARUS-Breast (ClinicalTrials.gov identifier: NCT04965766), SOLTI TOT-HER3 (ClinicalTrials.gov identifier: NCT04610528) and SOLTI-2103 VALENTINE (ClinicalTrials.gov identifier: NCT05569811).
Future perspective
The development of ADCs has the following directions; stabilisation in vivo (increase hydrophilicity, site specific conjugation), enhanced efficacy (increase DAR, dual payload, bystander effect, combination therapy) and reduction of toxicity (tumor specific antigen, tumor site specific payload release, dose-optimisation). Improvements in antibodies, linkers, payloads and conjugation are underway to achieve these goals.
Efficacy enhancement
In general, increasing hydrophilicity of ADCs result in improving stability in circulating plasma and anticancer effect. One of the such strategy is that the incorporation of polyethylene glycol (PEG) as a side chain in a linker contributes to increase hydrophilicity and decrease plasma clearance (,). Although the PEGylate linker has been a valid approach to improve the pharmacokinetics properties of ADCs, there are some limitations such as hypersensitivity, non-biodegradablity and accelerated blood clearance.
Currently, polysarcosine (PSR), an intermediate and byproduct of glycine synthesis and degradation, is considered a promising alternative to PEG in terms of biodegradablility and ease of synthesis and versatility (,).
While payload masking linkers are impressive methods for ADCs hydrophilicity, hydrophilic payloads are another approach to ADCs stabilisation and enables high DAR. For example, β-D-glucuronyl-monomethylauristatin E (MMAU) is the novel payload which enable high DAR = 8 ADCs with stability and efficacy due to its hydrophilicity ().
The therapeutic effect will be enhanced by increasing the number of payloads in the ADCs. Most ADCs linkers used so far have linear structures, allowing only a single payload attachment. However, branched linkers enable the loading of two or more payload molecules while maintaining the ADCs’ stability (,).
The use of dual payload is another approach. Since in conventional cancer chemotherapy, combining two or more different anticancer agents have enhanced the therapeutic effect, it is expected that loading multiple payloads in ADCs would also enhance the antitumor effect. Several studies have been reported about dual payloads ADCs with antitumor activity, such as MMAE and MMAF (), MMAE and pyrrolobenzodiazepine (), MMAF and PNU-159682 ().
Bispecific antibodies provide another possibility to increase clinical benefit. Their selectivity of binding to two different tumor targets enhances the antitumor effect and reduces the off-target toxicity (). Several ADCs with bispecific antibodies are currently under development including Integrin/HER2 bispecific ADCs (), HER2/CD63 bispecific ADC () and HER2/prolactin receptor (PRLR) bispecific ADC ().
Moreover, the combination therapy of ADCs with other antitumor agents, including cytotoxic drugs, antiangiogenic agents, targeted antibodies and ICIs, is currently investigated ().
Optimising the safety of ADCs
The toxicity of ADCs varies widely among individuals due to organ functions, comorbidities, pharmacogenomics and so on. Several methods have been considered to optimise the dose of ADCs.
Antibody modifications
When target antigens are expressed in both tumor cells and normal tissues, on-target adverse events are increased. Probody-drug conjugates (PDC) are antibody prodrugs designed to remain intact in normal tissue by masking antigen-binding regions, while they are activated by protease in the tumor microenvironment. As a result, this approach enables safer binding to target antigens expressed in both cancer and normal cell ().
First-in-Human studies of CX-2029 and Praluzatamab ravtansine (CX-2009) revealed relevant improvements in the safety profiles. CX-2029 is a PDC comprising an anti-CD71 antibody conjugated to MMAE. CD71 is previously considered as undruggable ADC target because of broad expression in both of tumor and normal tissue. However, the phase I study of CX-2029 revealed the tolerability of targeting therapy for CD71 ().
Praluzatamab ravtansine (CX-2009) is a PDC consisting of an anti-CD166 antibody and DM4. Although CD166 is also widely expressed in tumor and normal tissues, the results of phase I study of praluzatamab ravtansine showed tolerable safety profiles ().
In addition, bispecific antibodies, binding to two different antigens, increase, selectivity resulting in the reduction of the off-target toxicity ().
Modifying conjugation technology or drug/linker chemistry
Tandem linker
One drawback associated with cleavable linkers is the potential to release payloads in plasma circulation before tumor targeting. If the payloads are released systemically prematurely, the efficacy of the remaining circulating ADCs may be reduced, leading to off-target toxicity. Tandem cleavable linkers are novel linkers that incorporate a β-glucuronide moiety and require tandem enzymic cleavage events (). These linkers reduce payloads release during circulation and off-target toxicity.
Silencing the Fc portion of the ADCs
Whereas the Fc domain of the antibody induces immunogenicity, the internalisation of ADCs into non-targeted cells via Fcγ receptors on immune cells may provoke off-target toxicities. By silencing the Fc domain, Fc-mediated off-target cytotoxicity was reduced ().
Novel site specific conjugation technologies
Conventional antibody conjugates have been constructed through cysteine or lysine residue side chains. These approaches were stochastic, non-specific conjugation and generated heterogenous ADCs. To overcome the drawback of non-specific conjugation, intense research to realise site specific conjugation has been conducted. Chemo-enzymatic methods include transglutaminase and glycan-mediated conjugation, and chemical methods include selective reduction of disulfides and N-terminal amine modifications ().
Dose-optimisation strategy
Five dose-optimisation strategies are known to minimise toxicity and maximise efficacy. These include body weight-based dose capping, treatment duration capping, dose schedule, response-guided dosing and randomised dose-finding ().
Body weight-based dose-capping
Body weight has been generally considered as significant factor to adjust drug dosage. Body weight-based dose-capping was adapted for EV, ADCs used in urothelial cancer patients (). The recommended dose of EV was 1.25 mg/kg, but the maximum dose was decided as 125 mg.
Treatment duration capping
Limiting the treatment duration is widely adopted when using taxanes or platinum salts to minimise chronic toxicities such as peripheral neuropathy (,).
Polatuzumab vedotin, ADCs for diffuse large B cell lymphoma patients, is used up to six cycles. Predicted risk of developing grade ≥ 2 peripheral neuropathy with polatuzumab vedotin increases by ≥50% when eight versus six treatment cycles are administered. These data supported the approval of polatuzumab vedotin for patients with relapsed and/or refractory diffuse large B cell lymphoma at a dose of 1.8 mg/kg every 3 weeks for a maximum of six cycles ().
Dose schedule
Dose frequency is fundamental for the safety profile of a drug. Dose fractionation offers the almost same cumulative dose with a lower peak plasma concentration (Cmax) compared with a single dose and reduce toxicity related to the Cmax. Within the FDA approved ADCs, 4 ADCs (gemtuzumab ozogamicin, inotuzumab ozogamicin, SG and EV) are administered weekly dosing (,).
Response-guided dosing
Response-guided dosing is a flexible strategy to modify the dose of drug based on initial response of individual patient. This method is used in inotuzumab ozogamicin, consisting of anti-CD22 antibody and calicheamicin. The subsequent dose is decreased in patients with complete response after an initial dose of inotuzumab ozogamicin ().
Randomised dose-finding study
Although conventional Phase II/III studies have been arranged to evaluate primary clinical efficacy and safety, these studies often lack data on multiple dose levels. Randomised dose-finding study is conducted to reveal an optimal dose level based on data of multiple dose levels. DESTINY-Lung02 is an example in which the optimal dose of T-DXd (5.4 vs 6.4 mg/kg) for NSCLC was assessed (). The confirmed ORR was 49.0% (95%CI, 39.0–59.1) and 56.0% (95% CI, 41.3–70.0), median DOR was 16.8 months (95% CI, 6.4- not estimable [NE]) and NE (95% CI, 8.3-NE) with 5.4 and 6.4 mg/kg, respectively. Grade ≥ 3 TEAEs were observed in 38.6 and 58.0% of patients with 5.4 and 6.4 mg/kg, respectively. ILD occurred in 12.9 and 28.0% of patients with 5.4 and 6.4 mg/kg, respectively. Based on this result, the approval dose of T-Dxd was 5.4 mg/kg for NSCLC.
Randomised dose-finding studies might be reasonable for other ADCs and indications, particularly for those in which fatal adverse events like as ILD and/or considerable grade ≥ 3 toxicities are observed either in trials or clinical practice.
Inverse targeting strategy
The inverse targeting concept is another strategy to reduce off-target toxicity by payload-binding selectivity enhancers (PBSE). PBSE is payload-binding fragments which is co-administered with ADCs to bind unconjugated payload and decrease distribution of payload into non-targeted cells. Preclinical study showed that anti-MMAE antibody fragment and anti-maytansinoid single domain antibody increased the therapeutic window of MMAE-based ADCs and DM4-based ADCs, respectively (,).
Pharmacogenomics
Pharmacogenomics is the term that has been used to mean the study of how a person’s genetic characteristics alter their response to drug. Such genomic technologies are expected to clarify in which patients the toxicity is enhanced ().
As an example of pharmacogenomics, UGT1A1 polymorphisms is known to cause delayed metabolism of SN-38. SN-38, the active metabolite of irinotecan, is glucuronosylated and excreted by UGT1A1 (). UGT1A1 has a genetic polymorphism, and patients homozygous or heterozygous for either UGT1A1*6 or UGT11*28 are known to have a delayed metabolism of SN-38 resulting in higher incidence of toxicity.
This correlation was observed as well in the study of ADCs. SG, loading SN-38 as payload, reported that the toxicity was increased in patients with deleterious UGT1A1 polymorphisms ().
Conclusions
In summary, we have reviewed the structure and mechanism of ADCs, clinical data of approved ADCs for solid tumors. Based on the ability to deliver novel payloads selectively to targeting cancer cells, they have enabled more effective treatment with fewer side effects. Some ADCs show efficacy in multiple types of solid tumors, which suggests that ADCs may enable cancer treatment based on not only tumor types but also cell surface biomarkers. Further development on ADCs will bring new strategies for cancer treatment.
Funding
This research received no commercial or financial incentives.
Conflict of interest statement
Dr Takakura has no conflict of interest. Dr Shimizu has received research funding from AbbVie, Eli Lilly, LOXO Oncology, Novartis, Daiichi-Sankyo, Takeda Oncology, Bristol-Myers Squibb, Eisai, Incyte, AstraZeneca, Pfizer, Chordia Therapeutics, Astellas, Symbio Pharmaceuticals, 3D-Medicine, PharmaMar, Parexel, IQVIA; personal honoraria for lectures, speakers bureaus from Chugai, Taiho, MSD, IQVIA; support for attending meetings and travel from Roche; participation on a Data Safety Monitoring Board or Advisory Board of AbbVie, Chordia Therapeutics, Daiichi-Sankyo, Kyowa Kirin, Chugai; leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid of ESMO Targeted Anticancer Therapies (TAT), Asia Pacific Oncology Drug Development Consortium (APODDC), External IRB Member of Phase 1 Trials in Hong Kong, HKSAR, China. Dr Yamamoto has received research funding from AstraZeneca, Chugai Pharma, MSD, Taiho Pharmaceutical, Boehringer Ingelheim, Novartis, AbbVie, Amgen, Asahi Kasei, Janssen, Bristol-Myers Squibb Japan, IQVIA, EPS Corporation, Amgen, A2 Healthcare, Mebix, Ono Pharmaceutical; personal consulting fees from AstraZeneca, Chugai Pharma, MSD, Lilly Japan, Amgen, Novartis, Ono Pharmaceutical; honoraria for speakers bureaus, manuscript writing or educational events from AstraZeneca, Chugai Pharma, MSD, Takeda, Acuuray, AbbVie, Amgen, Ono Pharmaceutical, Guardant Health, Kyorin, Daiichi Sankyo, Taiho Pharmaceutical, Tsumura & Co., TERUMO, Lilly Japan, Boehringer Ingelheim Japan, Novartis, Pfizer, Miyarisan pharmaceutical, Merck biopharma, Janssen; participation on a Data Safety Monitoring Board or Advisory Board of AstraZeneca.
References
- 1. Sievers EL, Larson RA, Stadtmauer EA, et al Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol 2001;19:3244–54. https://doi.org/10.1200/JCO.2001.19.13.3244.
- 2. Beck A, Goetsch L, Dumontet C, Corvaïa N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat Rev Drug Discov 2017;16:315–37. https://doi.org/10.1038/nrd.2016.268.
- 3. Chen H, Lin Z, Arnst KE, Miller D, Li W. Tubulin inhibitor-based antibody-drug conjugates for cancer therapy. Molecules 2017;22:1281. https://doi.org/10.3390/molecules22081281.
- 4. Tarantino P, Pestana RC, Corti C, et al Antibody-drug conjugates: smart chemotherapy delivery across tumor histologies. CA Cancer J Clin 2021;72:165–82. https://doi.org/10.3322/caac.21705.
- 5. Lyon RP, Bovee TD, Doronina SO, et al Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat Biotechnol 2015;33:733–5. https://doi.org/10.1038/nbt.3212.
- 6. Tang Y, Tang F, Yang Y, et al Real-time analysis on drug-antibody ratio of antibody-drug conjugates for synthesis, process optimization, and quality control. Sci Rep 2017;7:7763. https://doi.org/10.1038/s41598-017-08151-2.
- 7. Strop P, Liu SH, Dorywalska M, et al Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol 2013;20:161–7. https://doi.org/10.1016/j.chembiol.2013.01.010.
- 8. Verma S, Miles D, Gianni L, et al Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med 2012;367:1783–91. https://doi.org/10.1056/NEJMoa1209124.
- 9. von Minckwitz G, Huang CS, Mano MS, et al Trastuzumab emtansine for residual invasive HER2-positive breast cancer. N Engl J Med 2019;380:617–28. https://doi.org/10.1056/NEJMoa1814017.
- 10. Powles T, Rosenberg JE, Sonpavde GP, et al Enfortumab vedotin in previously treated advanced urothelial carcinoma. N Engl J Med 2021;384:1125–35. https://doi.org/10.1056/NEJMoa2035807.
- 11. Modi S, Saura C, Yamashita T, et al Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N Engl J Med 2020;382:610–21. https://doi.org/10.1056/NEJMoa1914510.
- 12. Hurvitz SA, Hegg R, Chung WP, et al Trastuzumab deruxtecan versus trastuzumab emtansine in patients with HER2-positive metastatic breast cancer: updated results from DESTINY-Breast03, a randomised, open-label, phase 3 trial. Lancet 2023;401:105–17. https://doi.org/10.1016/S0140-6736(22)02420-5.
- 13. Modi S, Jacot W, Yamashita T, et al Trastuzumab deruxtecan in previously treated HER2-LowAdvanced breast cancer. N Engl J Med 2022;387:9–20. https://doi.org/10.1056/NEJMoa2203690.
- 14. Shitara K, Bang YJ, Iwasa S, et al Trastuzumab deruxtecan in previously treated HER2-positive gastric cancer. N Engl J Med 2020;382:2419–30. https://doi.org/10.1056/NEJMoa2004413.
- 15. Li BT, Smit EF, Goto Y, et al Trastuzumab deruxtecan in HER2-mutant non-small-cell lung cancer. N Engl J Med 2022;386:241–51. https://doi.org/10.1056/NEJMoa2112431.
- 16. Goto K, Goto Y, Kubo T, et al Trastuzumab deruxtecan in patients with HER2-mutant metastatic non-small-cell lung cancer: primary results from the randomized, phase II DESTINY-lung02 trial. J Clin Oncol 2023;41:4852–63. https://doi.org/10.1200/JCO.23.01361.
- 17. Bardia A, Messersmith WA, Kio EA, et al Sacituzumab govitecan, a Trop-2-directed antibody-drug conjugate, for patients with epithelial cancer: final safety and efficacy results from the phase I/II IMMU-132-01 basket trial. Ann Oncol 2021;32:746–56. https://doi.org/10.1016/j.annonc.2021.03.005.
- 18. Bardia A, Hurvitz SA, Tolaney SM, et al Sacituzumab govitecan in metastatic triple-negative breast cancer. N Engl J Med 2021;384:1529–41. https://doi.org/10.1056/NEJMoa2028485.
- 19. Rugo HS, Bardia A, Marmé F, et al Overall survival with sacituzumab govitecan in hormone receptor-positive and human epidermal growth factor receptor 2-negative metastatic breast cancer (TROPiCS-02): a randomised, open-label, multicentre, phase 3 trial. Lancet 2023;402:1423–33. https://doi.org/10.1016/S0140-6736(23)01245-X.
- 20. Coleman RL, Lorusso D, Gennigens C, et al Efficacy and safety of tisotumab vedotin in previously treated recurrent or metastatic cervical cancer (innovaTV 204/GOG-3023/ENGOT-cx6): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol 2021;22:609–19. https://doi.org/10.1016/S1470-2045(21)00056-5.
- 21. Matulonis UA, Lorusso D, Oaknin A, et al Efficacy and safety of mirvetuximab soravtansine in patients with platinum-resistant ovarian cancer with high folate receptor alpha expression: results from the SORAYA study. J Clin Oncol 2023;41:2436–45. https://doi.org/10.1200/JCO.22.01900.
- 22. Shimizu T, Sands J, Yoh K, et al First-in-human, phase I dose-escalation and dose-expansion study of trophoblast cell-surface antigen 2–directed antibody-drug conjugate datopotamab deruxtecan in non-small-cell lung cancer: TROPION-PanTumor01. J Clin Oncol 2023;41:4678–87. https://doi.org/10.1200/JCO.23.00059.
- 23. Ahn MJ, Lisberg A, Paz-Ares L, et al Datopotamab deruxtecan (Dato-DXd) vs docetaxel in previously treated advanced/metastatic (adv/met) non-small cell lung cancer (NSCLC): results of the randomized phase 3 study TROPION-Lung01. ESMO Congress 2023;34:S1305–6. https://doi.org/10.1016/j.annonc.2023.10.061.
- 24. Bardia A, Jhaveri K, Im SA, et al Datopotamab deruxtecan (Dato-DXd) vs chemotherapy in previously-treated inoperable or metastatic hormone receptor-positive, HER2-negative (HR+/HER2e) breast cancer (BC): primary results from the randomised phase III TROPION-Breast01 trial. ESMO Congress 2023;34:S1264–5. https://doi.org/10.1016/j.annonc.2023.10.015.
- 25. Ocana A, Vera-Badillo F, Seruga B, Templeton A, Pandiella A, Amir E. HER3 overexpression and survival in solid tumors: a meta-analysis. J Natl Cancer Inst 2013;105:266–73. https://doi.org/10.1093/jnci/djs501.
- 26. Mishra R, Patel H, Alanazi S, Yuan L, Garrett JT. HER3 signaling and targeted therapy in cancer. Oncol Rev 2018;12:355. https://doi.org/10.4081/oncol.2018.355.
- 27. Yu HA, Goto Y, Hayashi H, et al HERTHENA-Lung01, a phase II trial of patritumab deruxtecan (HER3-DXd) in epidermal growth factor receptor–mutated non–small-cell lung cancer after epidermal growth factor receptor tyrosine kinase inhibitor therapy and platinum-based chemotherapy. J Clin Oncol 2023;41:5363–75. https://doi.org/10.1200/JCO.23.01476.
- 28. Krop IE, Masuda N, Mukohara T, et al Patritumab deruxtecan (HER3-DXd), a human epidermal growth factor receptor 3-directed antibody-drug conjugate, in patients with previously treated human epidermal growth factor receptor 3-expressing metastatic breast cancer: a multicenter, phase I/II trial. J Clin Oncol 2023;41:5550–60. https://doi.org/10.1200/JCO.23.00882.
- 29. Pelegri-O’Day EM, Lin EW, Maynard HD. Therapeutic protein−polymer conjugates: advancing beyond PEGylation. J Am Chem Soc 2014;136:14323–32. https://doi.org/10.1021/ja504390x.
- 30. Burke PJ, Hamilton JZ, Jeffrey SC, et al Optimization of a PEGylated glucuronide-monomethylaur-istatin E linker for antibody-drug conjugates. Mol Cancer Ther 2017;16:116–23. https://doi.org/10.1158/1535-7163.MCT-16-0343.
- 31. Viricel W, Fournet G, Beaumel S, et al Monodisperse polysarcosine-based highly-loaded antibody-drug conjugates. Chem Sci 2019;10:4048–53. https://doi.org/10.1039/C9SC00285E.
- 32. Evans N, Grygorash R, Williams P, et al Incorporation of hydrophilic macrocycles into drug-linker reagents produces antibody-drug conjugates with enhanced in vivo performance. Front Pharmacol 2022;13:764540, 1–9. https://doi.org/10.3389/fphar.2022.764540.
- 33. Satomaa T, Pynnonen H, Vilkman A, et al Hydrophilic auristatin glycoside payload enables improved antibody-drug conjugate efficacy and biocompatibility. Antibodies 2018;7:15. https://doi.org/10.3390/antib7020015.
- 34. Kumar A, Mao S, Dimasi N, Gao C. Design and validation of linkers for site-specific preparation of antibody-drug conjugates carrying multiple drug copies per cysteine conjugation site. Int J Mol Sci 2020;21:6882. https://doi.org/10.3390/ijms21186882.
- 35. Lyon RP, Bovee TD, Doronina SO, et al Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat Biotechnol 2015;33:733–5. https://doi.org/10.1038/nbt.3212.
- 36. Levengood MR, Zhang X, Hunter JH, et al Orthogonal cysteine protection enables homogeneous multi-drug antibody-drug conjugates. Angew Chem Int Ed Engl 2017;56:733–7. https://doi.org/10.1002/anie.201608292.
- 37. Kumar A, Kinneer K, Masterson L, et al Synthesis of a heterotrifunctional linker for the site-specific preparation of antibody-drug conjugates with two distinct warheads. Bioorg Med Chem Lett 2018;28:3617–21. https://doi.org/10.1016/j.bmcl.2018.10.043.
- 38. Nilchan N, Li X, Pedzisa L, Nanna AR, Roush WR, Rader C. Dualmechanistic antibody-drug conjugate via site-specific selenocysteine/cysteine conjugation. Antib Ther 2019;2:71–8. https://doi.org/10.1093/abt/tbz009.
- 39. Ma J, Mo Y, Tang M, et al Bispecific antibodies: from research to clinical application. Front Immunol 2021;12:626616, 1–19. https://doi.org/10.3389/fimmu.2021.626616.
- 40. Yamaguchi A, Anami Y, Ha SYY, et al Chemical generation of small molecule-based bispecific antibody-drug conjugates for broadening the target scope. Bioorganic Med Chem 2021;32:116013. https://doi.org/10.1016/j.bmc.2021.116013.
- 41. De Goeij BECG, Vink T, Napel HT, et al Efficient payload delivery by a bispecific antibody–drug conjugate targeting HER2 and CD63. Mol Cancer Ther 2016;15:2688–97. https://doi.org/10.1158/1535-7163.MCT-16-0364.
- 42. Andreev J, Thambi N, Perez Bay AE, et al Bispecific antibodies and antibody–drug conjugates (ADCs) bridging HER2 and prolactin receptor improve efficacy of HER2 ADCs. Mol Cancer Ther 2017;16:681–93. https://doi.org/10.1158/1535-7163.MCT-16-0658.
- 43. Sasso JM, Tenchov R, Bird R, et al The evolving landscape of antibody–drug conjugates: In depth analysis of recent research progress. Bioconjug Chem 2023;34:1951–2000. https://doi.org/10.1021/acs.bioconjchem.3c00374.
- 44. Autio KA, Boni V, Humphrey RW, Naing A. Probody therapeutics: an emerging class of therapies designed to enhance on-target effects with reduced of-tumor toxicity for use in immuno-oncology. Clin Cancer Res 2020;26:984–9. https://doi.org/10.1158/1078-0432.CCR-19-1457.
- 45. Johnson M, El-Khoueiry A, Hafez N, et al Phase I, first-in-human study of the probody therapeutic CX-2029 in adults with advanced solid tumor malignancies. Clin Cancer Res 2021;27:4521–30. https://doi.org/10.1158/1078-0432.CCR-21-0194.
- 46. Boni V, Fildler MJ, Arkenau HT, et al Praluzatamab ravtansine, a CD166-targeting antibody–drug conjugate, in patients with advanced solid tumors: an open-label phase I/II trial. Clin Cancer Res 2022;28:2020–9. https://doi.org/10.1158/1078-0432.CCR-21-3656.
- 47. Chuprakov S, Ogunkoya AO, Barfield RM, et al Tandem-cleavage linkers improve the in vivo stability and tolerability of antibodydrug conjugates. Bioconjug Chem 2021;32:746–54. https://doi.org/10.1021/acs.bioconjchem.1c00029.
- 48. Aoyama M, Tada M, Yokoo H, Demizu Y, Ishii-Watabe A. Fcγ receptor-dependent internalization and of-target cytotoxicity of antibody-drug conjugate aggregates. Pharm Res 2022;39:89–103. https://doi.org/10.1007/s11095-021-03158-x.
- 49. Sadiki A, Vaidya S, Abdollahi M, et al Site-specific conjugation of native antibody. Antib Ther 2020;3:271–84. https://doi.org/10.1093/abt/tbaa027.
- 50. Liao MZ, Lu D, Kagedal M, et al Model-informed therapeutic dose optimization strategies for antibody-drug conjugates in oncology: what can we learn from US Food and Drug Administration-approved antibody-drug conjugates? Clin Pharmacol Ther 2021;110:1216–30.
- 51. Hanna KS. Clinical overview of enfortumab vedotin in the management of locally advanced or metastatic urothelial carcinoma. Drugs 2020;80:1–7. https://doi.org/10.1007/s40265-019-01241-7.
- 52. Rivera E, Cianfrocca M. Overview of neuropathy associated with taxanes for the treatment of metastatic breast cancer. Cancer Chemother Pharmacol 2015;75:659–70. https://doi.org/10.1007/s00280-014-2607-5.
- 53. Iveson T, Sobrero AF, Yoshino T, et al Prospective pooled analysis of four randomized trials investigating duration of adjuvant (adj) oxaliplatin-based therapy (3 vs 6 months {m}) for patients (pts) with high-risk stage II colorectal cancer (CC). J Clin Oncol 2019;37:3501–1. https://doi.org/10.1200/JCO.2019.37.15_suppl.3501.
- 54. Lu D, Gillespie WR, Girish S, et al Time-to-event analysis of polatuzumab vedotin-induced peripheral neuropathy to assist in the comparison of clinical dosing regimens. CPT Pharmacometrics Syst Pharmacol 2017;6:401–8. https://doi.org/10.1002/psp4.12192.
- 55. Norsworthy KJ, Ko CW, Lee JE, et al FDA approval summary: mylotarg for treatment of patients with relapsed or refractory CD33-positive acute myeloid leukemia. Oncologist 2018;23:1103–8. https://doi.org/10.1634/theoncologist.2017-0604.
- 56. Garrett M, Ruiz-Garcia A, Parivar K, Hee B, Boni J. Population pharmacokinetics of inotuzumab ozogamicin in relapsed/refractory acute lymphoblastic leukemia and nonHodgkin lymphoma. J Pharmacokinet Pharmacodyn 2019;46:211–22. https://doi.org/10.1007/s10928-018-9614-9.
- 57. Starodub AN, Ocean AJ, Shah MA, et al First-in-human trial of a novel anti-trop-2 antibody-sn-38 conjugate, sacituzumab govitecan, for the treatment of diverse metastatic solid tumors. Clin Cancer Res 2015;21:3870–8. https://doi.org/10.1158/1078-0432.CCR-14-3321.
- 58. Advani A, Coiffier B, Czuczman MS, et al Safety, pharmacokinetics, and preliminary clinical activity of inotuzumab ozogamicin, a novel immunoconjugate for the treatment of B-cell non-Hodgkin’s lymphoma: results of a phase I study. J Clin Oncol 2010;28:2085–93. https://doi.org/10.1200/JCO.2009.25.1900.
- 59. Bordeau BM, Nguyen TD, Polli JR, et al Payload-binding Fab fragments increase the therapeutic index of MMAE antibody–drug conjugates. Mol Cancer Ther 2023;22:459–70.
- 60. Nguyen TD, Bordeau BM, Balthasar JP. Use of payload binding selectivity enhancers to improve therapeutic index of maytansinoid-antibody-drug conjugates. Mol Cancer Ther 2023;22:1332–42. https://doi.org/10.1158/1535-7163.MCT-22-0804.
- 61. Pirmohamed M. Pharmacogenomics: current status and future perspectives. Nat Rev Genet 2023;24:350–62. https://doi.org/10.1038/s41576-022-00572-8.
- 62. Liu X, Cheng D, Kuang Q, Liu G, Xu W. Association of UGT1A1*28 polymorphisms with irinotecan-induced toxicities in colorectal cancer: a meta-analysis in Caucasians. Pharmacogenomics J 2014;14:120–9. https://doi.org/10.1038/tpj.2013.10.
- 63. Rugo HS, Tolaney S, Loirat D, et al Safety analyses from the phase 3 ASCENT trial of sacituzumab govitecan in metastatic triple-negative breast cancer. NPJ Breast Cancer 2022;8:98. https://doi.org/10.1038/s41523-022-00467-1.