(See the editorial commentary by Stratton on pages 668–70.)
The global increase in antibiotic resistance is reviving the need for alternative antimicrobial strategies, including phage therapy. This “forgotten cure” was developed in parallel to antibiotics during the first half of the 20th century and is still commonly used in countries of the former Soviet Union []. However, it was not developed on a large scale in Western countries, and information on phage pharmacokinetics/pharmacodynamics (PK/PD), drug interactions, in vivo efficacy, and emergence of resistance remains scarce. Phages have been administered by various routes, including inhalation for pneumonia (reviewed in []), surgical rinsing for chronic osteomyelitis [], and intravenous injection for severe systemic infections, such as typhoid fever (reviewed in []). However, only a few studies provide a comprehensive picture linking phage pharmacology to antibacterial efficacy [, ]. Moreover, with few exceptions [], the emergence of phage resistance is seldom addressed, even in recent clinical studies [].
Detailed understanding of bacterial resistance to phages is critical if phages were to be used more broadly in the clinical setting. Phage-resistant bacteria can result from several mechanisms, including modification of cell-surface receptors, restriction-modification of incoming (foreign) phage DNA, or interphage immunity []. Resistance mutations may arise spontaneously in vitro and are likely to be selected in vivo as well []. Some mutations may affect LPS (a common phage receptor) and impact bacterial fitness or virulence []. However, other kinds of mutations or further mutations restoring virulence cannot be excluded and must be scrutinized.
The intrinsic bactericidal properties of anti-infective compounds are most reliably studied in models of therapeutic sanctuaries, where natural host defenses are poorly involved. Experimental endocarditis (EE) and experimental meningitis are 2 such models. Experimental meningitis implicates a special anatomical setting where drug distribution depends on the blood–brain barrier. In contrast, EE mirrors the general situation encountered in many deep-seated infections. Moreover, endocarditis pathogens surround themselves with amorphous aggregates of platelet-fibrin clots, which cellular host defenses cannot penetrate (for review see []). Thus, the capability of antimicrobials to penetrate into vegetations is a critical issue.
In these experiments, we systematically explored the efficacy of a clinically used antipseudomonas phage cocktail (cocktail PP1131 currently investigated in the multicentric Phagoburn clinical trial for treatment of burn-wound infections; http://www.phagoburn.eu) in a dual in vitro and in vivo model of experimental endocarditis due to Pseudomonas aeruginosa.
Phage therapy alone was active both in vitro and in animals with EE. Moreover, it was highly synergistic with antibiotics. Although phage-resistant bacteria emerged in vitro, they were not detectable in vivo due to fitness alteration in the animal milieu. These results suggest that phage therapy alone or combined with antibiotics (in this case ciprofloxacin) deserves further consideration for future clinical application.
METHODS
Strains, Phage Cocktail, and Antibiotics
A total of 33 P. aeruginosa reference strains and clinical isolates from our own clinical strain collection were used in in vitro susceptibility determinations (Supplementary Table 1). These isolates were not meant to cover the diversity of contemporary pseudomonas from our hospitals but rather to select strains with extreme phenotypes to be tested in our experiments. Growth conditions and reagents are given in the Supplementary Materials.
In Vitro Studies
The host range of the individual phages composing the PP1131 cocktail was determined using spot or plaque assays []. Experiments were done in triplicates. Plasma clots were prepared in 96-well microplates, as previously described []. Phage titers were measured by plaque assays []. The activity of the phage cocktail was considered bactericidal when it killed ≥3 log colony-forming units (CFUs) of the starting inoculum in the clot. Details are provided in the Supplementary Materials.
Animal Studies
Experiments were approved by and in adherence with the guidelines of the Swiss Animal Protection Law, Veterinary office, State of Vaud. Experiments were performed under license number 879.9 and in accordance with the regulations of the cantonal committee on animal experimentation of the State of Vaud, Switzerland. The production of catheter-induced aortic vegetations and the installation of an intravenous line into the superior vena cava, connected to an infusion pump to deliver the phage cocktail, were performed in female Wistar rats as previously described []. Details are provided in the Supplementary Materials.
Histology
Semithin and ultrathin sections were prepared as previously described [] and cut with a Leica Ultracut microtome. Sections were stained for light microscopy using the modified Brown and Brenn method, as previously described []. Ultrathin sections of 50 nm were stained as previously described []. For phage observation, phage cocktail was adsorbed on copper 200-mesh grid coated with Formvar-carbon and stained with uranyl acetate. Micrographs were taken with a transmission electron microscope FEI CM100 at an acceleration voltage of 80 kV with a TVIPS TemCam-F416 digital camera (EMF, UNIL).
Cytokine Measurements
The serum inflammatory cytokines interleukin 1β (IL-1β), interleukin 6 (IL-6), and tumor necrosis factor α (TNFα) were quantified prior to infection (24 hours after surgery), prior to therapy (18 hours after bacterial inoculation), and at sacrifice (6 hours after onset of therapy). Plasma was kept at −80°C, and cytokines titers were measured using the Biorad Bio-Plex Rat Cytokine Kit in a Luminex 200 system and following manufacturer’s recommendations (MEF platform, UNIL).
Detection and Characterization of Phage Resistance
Phage-resistant bacteria were isolated from broth cultures, in vitro fibrin clots, and rat vegetations by plating on blood agar plates containing 1010 PFU (plaque forming unit) preabsorbed phages. Resistance rates were determined as the number of bacteria growing on plates preadsorbed with phages divided by the number of bacteria growing on plain plates. In vivo infectivity of phage resistance was assessed by inoculating rats with catheter-induced aortic vegetations as previously described. Full genomic DNA sequencing was performed by the Center for Integrative Genomics (University of Lausanne, Switzerland) using the Pacific Biosciences RSII platform. Total DNA was extracted using the DNeasy Blood and Tissue kit according to manufacturer’s instructions. GenDB [], BLASTn [], and BRIG [] were used for genomic analysis. Subsurface twitching motility tests were performed as described []. LPS was extracted using the iNTRON LPS extraction kit, resolved on NuPAGE 4%–12% BisTris gels, and silver stained as previously described []. Details are provided in the Supplementary Materials.
RESULTS
Pseudomonas aeruginosa Host Range of Individual Phages of the PP1131 Cocktail
The 12 phages contained in the PP1131 cocktail were evaluated for their ability to lyse a panel of 33 laboratory and clinical isolates of P. aeruginosa from our own collection using spot assays (Supplementary Tables 1 and 2). This test showed that 31 of 33 (94%) of the P. aeruginosa isolates were lysed by at least 2 phages, whereas only strains PA7 and PA10 were resistant to the 12 phages. The relatedness of the 12 phages regarding strain specificity was further evaluated by hierarchical cluster analysis based on a binary matrix derived from the spot assay. Supplementary Figure 1 indicates that the phages of the cocktail were different from each other regarding their host specificity profiles, thus ensuring coverage of multiple strains.
Of note, spot assays indicate both lysis from within (due to productive phage infection) and lysis from without (due to phage attachment only). Therefore, although they assess specificity of phage–bacteria interactions, they may underestimate genuine productive infectivity. In the present experiments, the PP1331 phage cocktail achieved productive infection in 28 of 33 (84%) of the tested strains (Supplementary Figure 2).
Activity of the Phage Cocktail in the In Vitro Fibrin-Clot Model
The efficacy of the cocktail was tested in fibrin clots simulating endocarditis vegetations []. Two P. aeruginosa strains were used: namely, strain P7, which was not infected by any of the cocktail’s phages, and strain CHA which was susceptible to all of them. For resistant strain PA7, no phage-induced killing and no in situ phage amplification were observed (Figure 1A). Moreover, physical disintegration of the clots, possibly due to the action of bacterial proteases, was observed after 24 hours (Figure 1C).
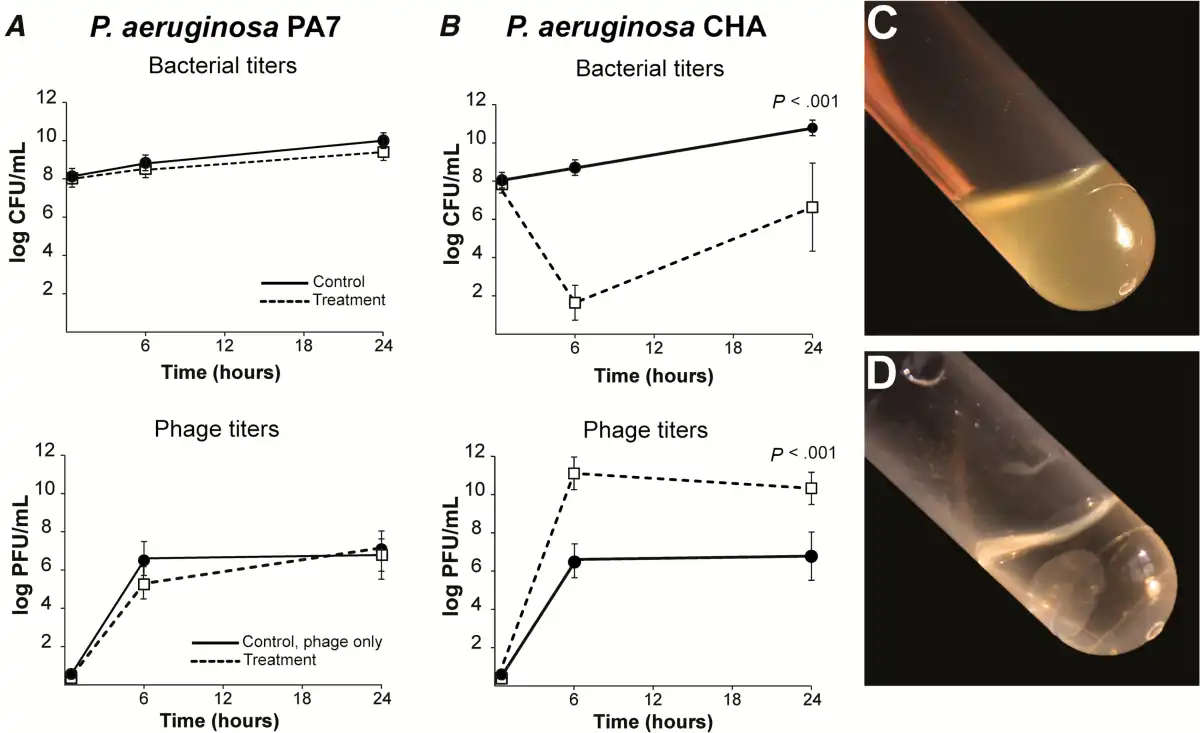
Figure 1
Activity of phage cocktail PP1131 in in vitro fibrin clots. Clots were produced from rat plasma and infected with 108 colony-forming units (CFUs)/mL of either phage-resistant Pseudomonas aeruginosa strain PA7 (A) or phage-susceptible strain CHA (B). Clots were left untreated (solid lines) or exposed to 108 PFUs/mL of PP1131 for 24 hours at 37°C (dashed lines). Phage titers in noninfected clots (solid lines) and in PA7-infected or CHA-infected clots (dashed lines) were determined 6 hours and 24 hours after exposure to the phage cocktail. Clots infected with strain PA7 lysed in spite of phage treatment (C), whereas those infected with phage-susceptible CHA and treated with phages remained intact (D). P values were determined using the Mann–Whitney test.
In contrast, a significant loss of bacterial viability of approximately 6 log CFUs/g was observed after 6 hours when CHA-infected clots were challenged with the cocktail (Figure 1I (P = .0001 compared with the initial bacterial load). Phage-induced killing was accompanied by a phage amplification of 5 log PFUs/g of clots (P = .0001 compared with noninfected clots). In addition, phage-induced bacterial killing prevented bacterial-induced clot disintegration (Figure 1D), indicating that phages diffused through the fibrin meshwork and killed bacteria located inside the clots. Yet, the sharp initial phage-induced killing was followed by bacterial regrowth after 24 hours (Figure 1B). This regrowth was caused by the development of phage-resistant bacterial mutants and was not accompanied by further phage amplification.
Frequency of Phage Resistance in Broth Cultures and Fibrin Clots
In the absence of phages, spontaneous resistant mutants occurred at an average frequency of ca 10–7 and at ca 10–8 against the whole cocktail (result not shown), both in batch cultures and in fibrin clots after 6 hours and 24 hours (Supplementary Table 3, column 2). In contrast, the proportion of phage-resistant bacteria increased sharply after exposure to phages due to the replacement of phage-susceptible bacteria by resistant subpopulations, (ie, 10–4 at 6 hours and 10–2 at 24 hours) (Supplementary Table 3, columns 5 and 6).
Prevention of Phage Resistance Using Phage/Antibiotic Combinations
To prevent the development of phage-resistant subpopulations, we attempted to combine phages with antibiotics. Combining the phage cocktail with 2.5 times the minimum inhibitory concentration (MIC) of ciprofloxacin or meropenem inhibited the regrowth of phage-resistant mutants after 24 hours (Figure 2A and 2B, dashed dotted lines, respectively). Moreover, the 2 antibiotics were highly synergistic with phages at 24 hours, as defined by a ≥3 log CFUs/mL decrease in bacterial viable counts when compared with single phage or antibiotic therapy alone.
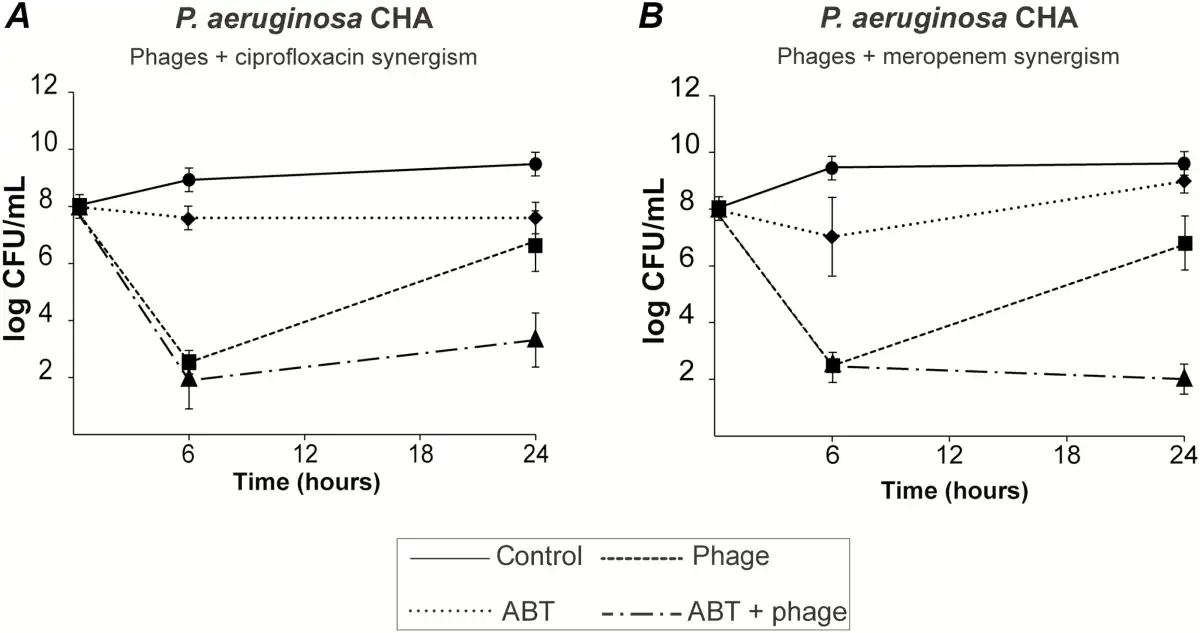
Figure 2
Bactericidal synergism between phages and selected antibiotics. Bacterial killing by phage-antibiotic combinations was tested by using 108 PFUs/mL of PP1131 with 2.5-times the minimum inhibitory concentration (MIC) of ciprofloxacin (A) or meropenem (B) (MIC of 0.19 µg/mL and 0.125 µg/mL, respectively). Each value represents the mean ± SD of 4–16 independent clots. Abbreviation: ABT, antibiotic.
Phage Pharmacokinetics
The pharmacokinetics of the phage cocktail were assessed in the plasma and tissues of noninfected rats with catheter-induced aortic vegetations following a single intravenous bolus injection or during continuous infusion (Figure 3A, solid and dashed line, respectively). A high plasma titer (8.5 ± 1.5 log PFUs/mL) was measured 1 hour after bolus injection, followed by a continuous decrease to 4.9 ± 0.6 log PFUs/mL at 24 hours (elimination half-life = ca 2.3 hours). In parallel, the concentrations of phages in vegetations decreased from 7.5 ± 0.3 to 6.2 ± 0.9 log PFUs/g between 6 hours and 24 hours after administration. A similar tendency was observed in the spleen, kidney, liver, lung, and brain (elimination half-life = ca 9 hours) (Supplementary Figure 3). This demonstrated phage diffusion into the organs and delayed phage clearance compared with plasma. During continuous infusion, phage titers in plasma progressively increased to reach a plateau of 8.5 ± 0.2 log PFUs/mL after 6 hours, which is comparable with to the concentration achieved after 1 hour following bolus injection. Phage concentrations in the vegetations and organs remained high after 24 hours (7.4 ± 0.9 log PFUs/mL) (Supplementary Figure 3).
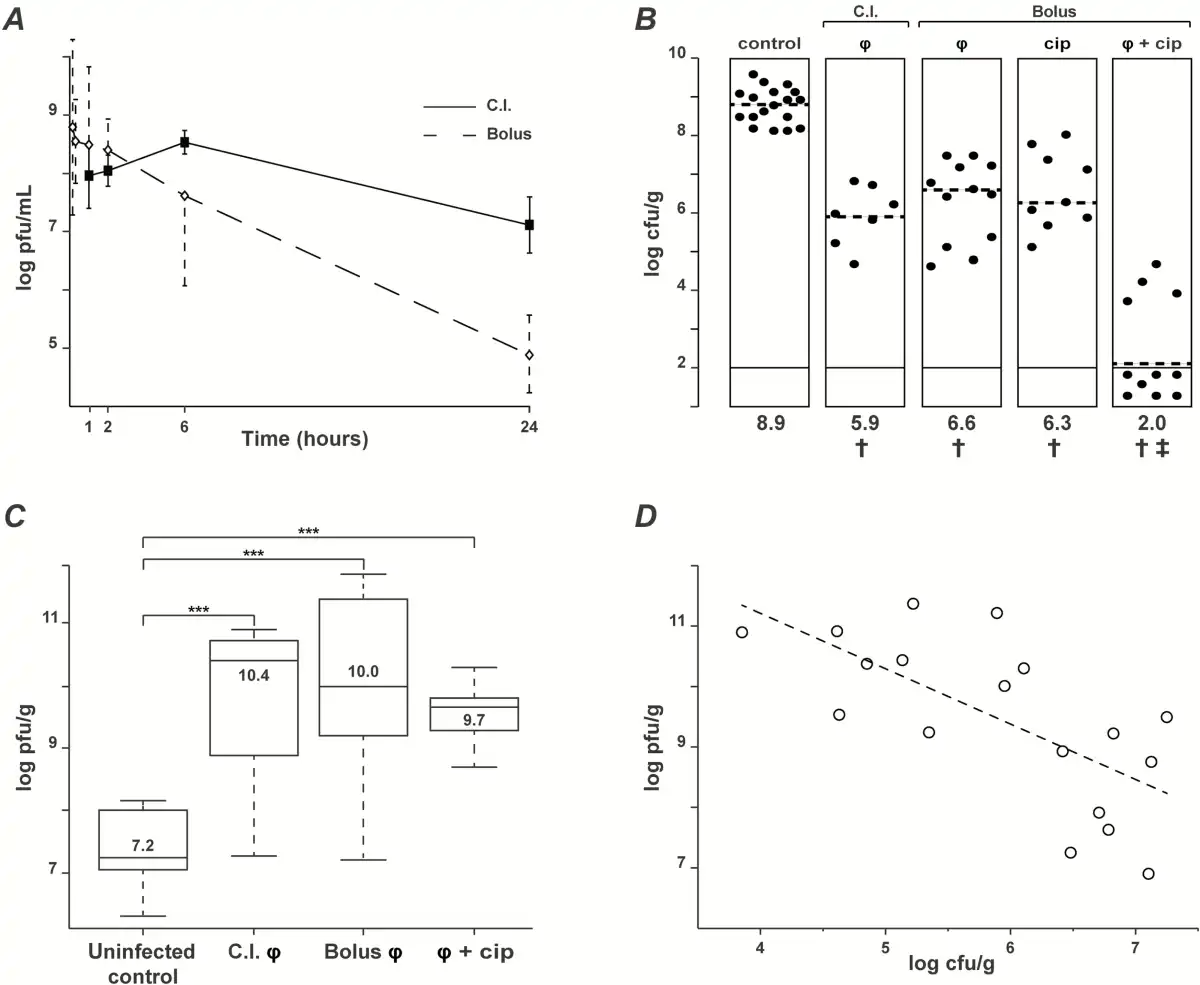
Figure 3
Pharmacokinetics/pharmacodynamics (PK/PD) and therapeutic efficacy of phage cocktail PP1131 and ciprofloxacin in rats with experimental endocarditis (EE) due to Pseudomonas aeruginosa CHA. A, Pharmacokinetics in the absence of infection of phage cocktail PP1131 in rat plasma after either a single intravenous bolus injection (1 mL of 1010 PFUs/mL in 1 minute, dashed line, Bolus) or the same amount of phages administered through continuous infusion (0.1 mL/h of 1010 PFUs/mL over 24 hours, solid line: continuous infusion). Each value represents the mean ± SD from 8–10 individual animals. B, Bacterial loads of infected vegetations after 6 hours of phage treatment administered through continuous infusion or in single bolus and treatment with ciprofloxacin alone or combined with phages. Each dot represents the vegetation of a single animal. The mode of injection and type of treatment are indicated at the top of columns (C.I.: continuous infusion; φ: phages). Statistical results are indicated at the bottom of columns (†: control vs all types of treatment, P < .0005; ‡: combination vs control and all other types of treatments, P < .0001; results were compared by the Mann–Whitney test). C, Phage titers in vegetations measured in uninfected rats treated with bolus phage injection (control) or infected rats treated with the same regimens as in Figure 2B (***, P < .0001 using the Mann–Whitney test). D, Correlation between the decrease in vegetation bacterial loads and the vegetation phage titers resulting from in situ phage amplification (continuous infusion and bolus injection pooled together; correlation value [r] = −0.66; Pearson 2-tailed correlation test: P = .003). Abbreviation: cip, ciprofloxacin.
Treatment of Experimental Endocarditis
Inoculation with 108 CFUs of P. aeruginosa CHA induced EE with bacterial loads in vegetations of control rats of >8 log CFUs/g 18 hours after bacterial challenge (Figure 3B). Administration of the phage cocktail 18 hours after bacterial challenge by either continuous or bolus injection decreased median vegetation bacterial titers by 3.0 and 2.3 log CFUs/g, respectively, within 6 hours of therapy (Figure 3B) (P < .0001 compared with treatment onset). No significant difference between the 2 routes of administration was observed. Notably, a single bolus injection of ciprofloxacin simulating a single oral dose of 750 mg in adults [] decreased median vegetation bacterial titers by 2.6 log CFUs/g (Figure 3B), which was comparable with phage therapy. Moreover, the combination of phages with ciprofloxacin was highly synergistic, resulting in negative vegetation cultures in 7 of 11 rats receiving combined therapy after 6 hours, as compared with 0 of 28 in phage-alone or ciprofloxacin-alone treated groups (Figure 3B) (P < .005). Likewise, the residual median vegetation bacterial titers of ≤2 log CFUs/g in the combined therapy group were significantly lower than the ≥6 log CFUs/g titers detected in the monotherapy groups (P < .0001).
In Situ Penetration of Phages in Valve Tissues
Optical microscopy and transmission electron microscopy were performed on vegetation samples to visualize the presence and activity of phages inside the vegetations. Figure 4 presents relevant pictures of these experiments, including intravegetation P. aeruginosa in untreated rats (Figure 4A and 4B), negatively stained phages from the cocktail with a capsid size of ca 70 nm (Figure 4C), and intravegetation lysed bacteria in phage-treated rats with bacterial ghosts containing phage particles (Figure 4D).
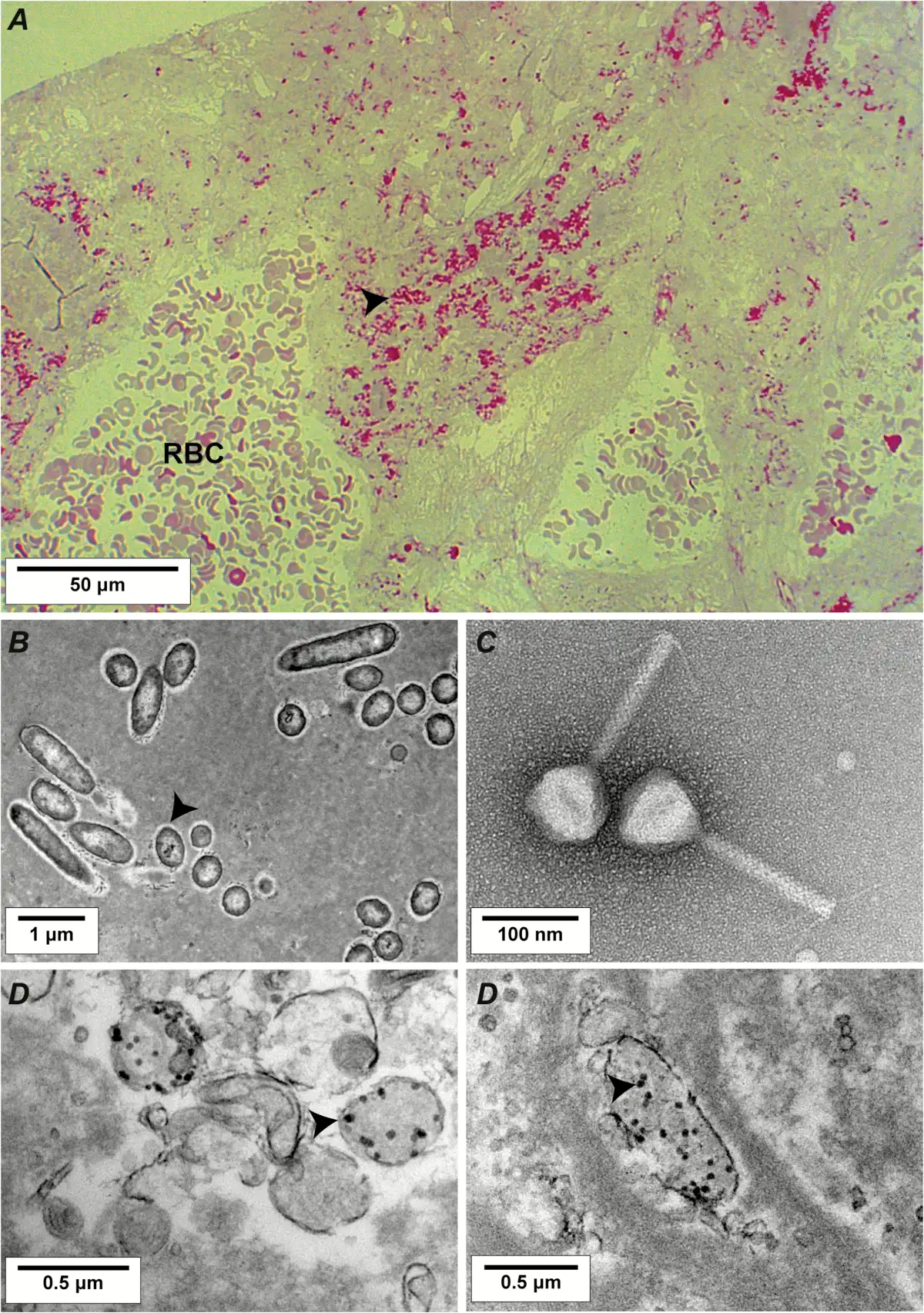
Figure 4
Light microscopy and transmission electron microscopy of rat vegetations after 6 hours of phage therapy. A, Semithin section (200 nm) of control vegetation infected with Pseudomonas aeruginosa CHA stained using the modified Braun Brenn staining protocol []. Arrows indicate bacteria, stained in red. B, Transmission electron microscopy of ultrathin sections (50 nm) of control vegetation infected with P. aeruginosa CHA positive-stained using uranium acetate and lead citrate. Arrows indicate bacteria. C, Example of a negative staining of a myoviridae phage present in the cocktail; other phages morphotypes including podoviridae were also present. D, Transmission electron microscopy of vegetation infected by P. aeruginosa CHA and treated with the phage cocktail. Arrows indicate phage capsids inside lysed bacteria. Abbreviation: RBC, red blood cell.
Correlation Between Phage Multiplication, Antimicrobial Activity, and Cytokine Production
In vivo antibacterial activity correlated with phage amplification (ca 3 log PFUs/g of vegetations) after both continuous and bolus injection (Figure 3C). This relation was further underlined by the scatter plot presented in Figure 3D, which showed a significant inverse correlation between phage and bacterial concentrations in the vegetations (correlation value r = −0.66, Pearson 2-tailed correlation test; P = .003).
Successful phage therapy also correlated with the production of specific inflammatory cytokines (Figure 5). Concentrations of IL-1β, IL-6, and TNFα in plasma were quantified by Luminex first before inoculation, second before treatment onset, and lastly after 6 hours of phage therapy in uninfected and infected rats (Figure 5A). Several observations were drawn from these measurements. First, although there were trends in cytokine-induction by bacteria and phages alone, these were not statistically significant. Second, TNFα levels remained essentially unaltered in all of the groups throughout the experiment. Finally, only phage treatment, but not ciprofloxacin, significantly increased plasma levels of IL-1β and IL-6 (Figure 5B). Because ciprofloxacin is not bacteriolytic, the increase in IL-1β and IL-6 levels was likely related to phage-induced bacterial lysis.
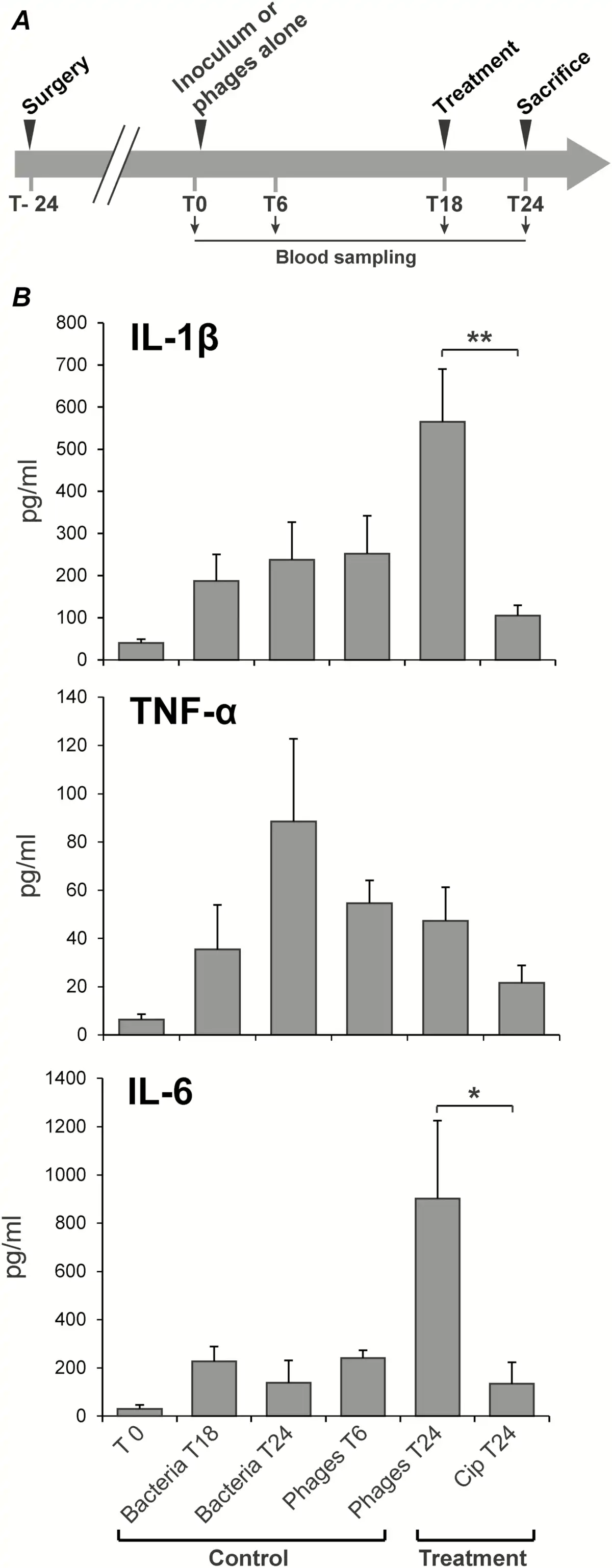
Figure 5
Cytokine quantification in rat plasma during experimental endocarditis. A, Protocol of blood sampling for cytokine quantification. B, Levels of interleukin 1β (IL-1β), interleukin 6 (IL-6), and tumor necrosis factor α (TNF-α) measured after 6 hours of phage or antibiotherapy (treatment). Controls included rats 24 hours after surgery (inoculum, T0), untreated but infected rats for 18 hours or 24 hours (bacteria T18 and T24), and uninfected rats receiving phage for 6 hours (Phages T6). Each value represents the mean ± SEM from 4–10 individual animals (*, P = .03; **, P = .005 using the Mann–Whitney test). Abbreviation: cip, ciprofloxacin.
Phage Resistance in Experimental Endocarditis
Because spontaneous phage-resistant mutants occurred at a rate of 10–7 in batch cultures and fibrin clots, they were also expected to emerge in infected vegetations. To test this assumption, phage-resistant mutants were sought in infected valves by plating vegetation extracts directly on agar plates containing preadsorbed phages. Unexpectedly, no phage-resistant mutants could be isolated from the infected vegetations either before or after therapy, therefore suggesting that phage resistance might result in altered virulence or fitness and interfere with successful bacterial survival or growth in vivo. Two resistant isolates recovered from fibrin clots, 19/2 and 24/2, which showed different phage resistance patterns, were further characterized to verify this hypothesis. Figure 6A shows that these resistant isolates had markedly different phage susceptibility profiles when compared with the parent strain. Mutant 24/2 conferred turbid plaques, whereas mutant 19/2 was resistant to 10 of the 12 phages and displayed a melanized (dark colonies) phenotype.
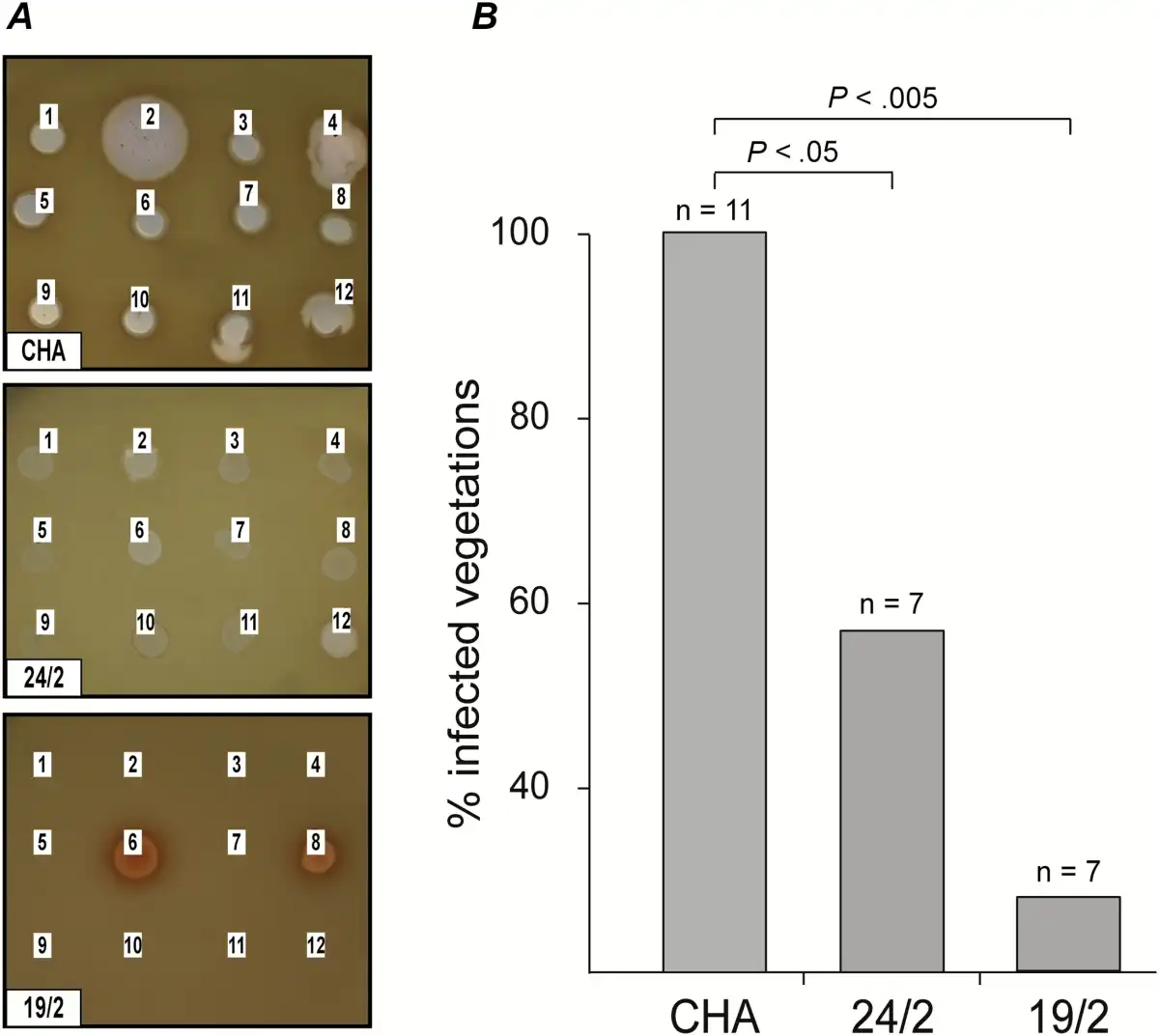
Figure 6
Infectivity of phage-resistant Pseudomonas aeruginosa mutants in rats with catheter-induced vegetations. A, Two phage resistant P. aeruginosa mutants, 24/2 and 19/2, were isolated in vitro from fibrin clots and showed different phage resistance patterns. Lysis zones at the site of the phage deposits indicated phage-sensitivity. The absence of lysis indicated phage resistance. B, The isolates’ infectivity was tested in rats with catheter-induced vegetations. Both variants showed a loss of infectivity (>40% for 24/2 and >70% for 19/2) as compared with the parent strain CHA. P values were determined using Fisher’s exact test.
To determine whether these mutants had altered virulence, they were tested for their capability to infect sterile vegetations. A single bolus of 108 CFUs (ie, infective dose 90% of parent strain) led to the infection of <60% (P < .05) and <30% (P < .01) of vegetations for mutants 19/2 and 24/2, respectively (Figure 6B). Moreover, the median bacterial densities in the infected vegetations were significantly lower than in rats infected with the parent strain (CHA: 8.9 CFUs/g; 24/2: 7.3 CFUs/g; and 19/2: 6.8 CFUs/g; P < .05), and blood cultures were negative.
To further investigate the origin of this impaired in vivo infectivity, the genomic DNA of the parent strain CHA and the 2 mutants was sequenced, assembled, and compared (Figure 7A). Mutant 24/2 displayed a 15-basepair deletion at the 3’ end of the pilT gene (Supplementary Figure 4), which encodes an ATPase involved in motility []. As a result, mutant 24/2 exhibited loss of twitching motility (Figure 7B). Mutant 19/2 had a 362-kb genomic deletion encompassing 342 genes. These included the galU gene, which is involved in P. aeruginosa LPS core synthesis. galU deletion was shown to be responsible for phage resistance []. The resulting absence of O antigen and LPS core truncation was confirmed by LPS extraction followed by gel electrophoresis (Figure 7C). Mutant 19/2 also showed a 3-fold decrease in its MIC of ciprofloxacin (0.064 vs 0.192 µg/mL for mutant 19/2 and parent CHA, respectively).
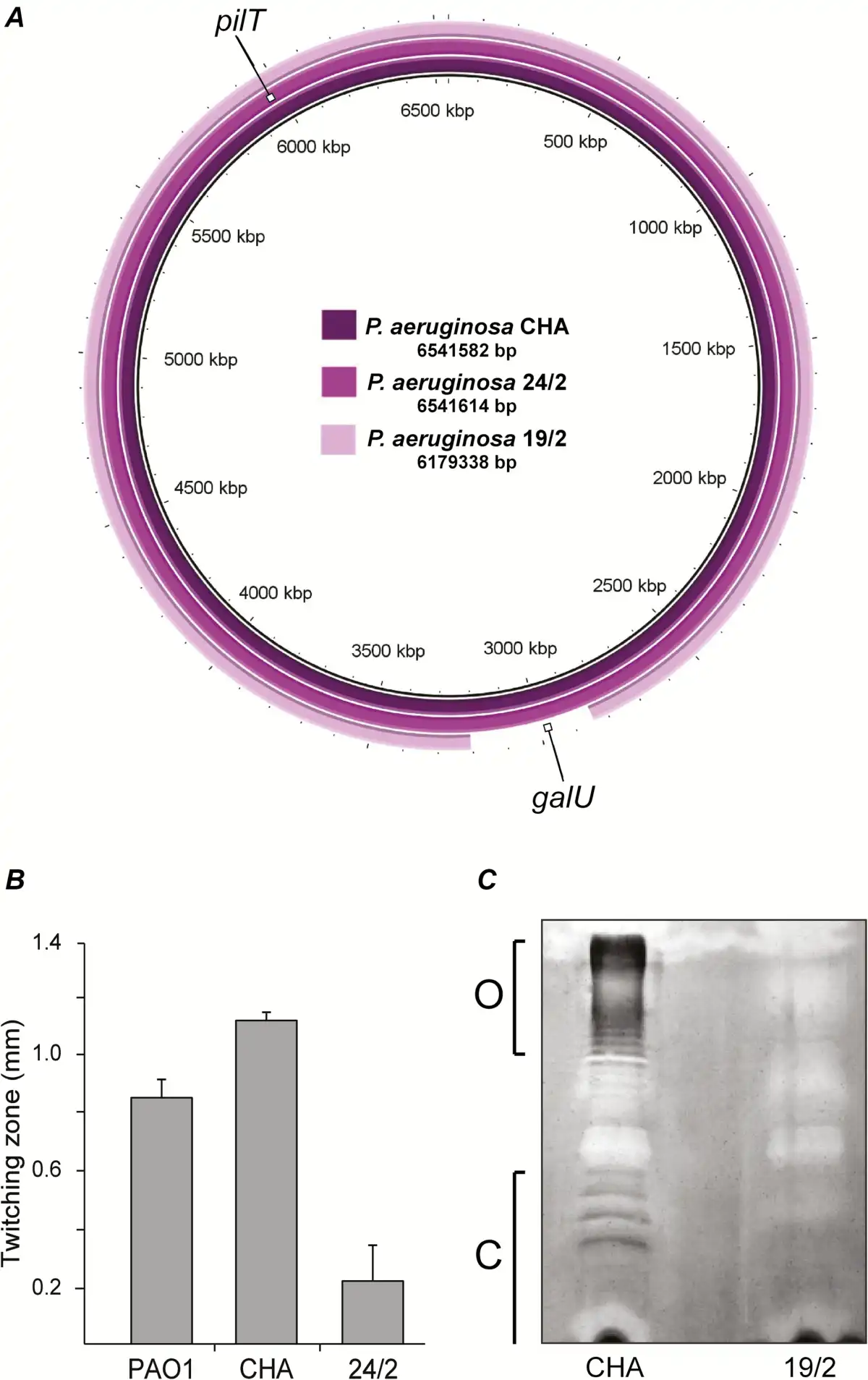
Figure 7
Characterization of phage-resistant Pseudomonas aeruginosa mutants. The 2 phage-resistant P. aeruginosa mutants, 24/2 and 19/2, isolated in vitro were further characterized regarding to genomic content and phenotypes. A, Variant 24/2 displayed a 15-basepair deletion in the pilT gene, and variant 19/2 displayed a 362-kb chromosomal deletion that encompassed the galU gene. B, Impaired twitching motility resulting from the pilT alteration in variant 24/2, as compared with wild-type PAO1 and parent CHA. C, Impaired LPS synthesis resulting from the galU deletion in variant 19/2 with absence of O-antigen (O) and LPS core (C), as compared with parent CHA.
DISCUSSION
Although phage therapy is a promising alternative to antibiotics against specific and difficult-to-treat infections, careful appraisal of its therapeutic potential is an absolute prerequisite before clinical application.
Here we evaluated the in vitro activity of the whole phage cocktail and its individual phage components against a panel of independent P. aeruginosa isolates in test tubes and in fibrin clots. This revealed the presence of bacterial strains with opposite susceptibility profiles, either resistant to all of the cocktail’s phages (eg, strain PA7) or susceptible to all of them (eg, strain CHA). The frequency of spontaneous phage resistance mutations of the susceptible strain CHA was found to be ca 10–7. This predicted that phage resistance would emerge in infected fibrin clots, which contained ≥108 CFUs/g. This was indeed the case. However, these approach experiments highlighted 2 additional important facts. First, phages could readily diffuse into clots, kill the overwhelming majority of phage-susceptible bacteria in situ, and protect the fibrin matrix from bacterial-induced disintegration. Second, combining phages with low concentrations of ciprofloxacin or meropenem (2.5 × the MIC) inhibited the regrowth of phage-resistant mutants, suggesting potential success of in vivo therapy.
The in vivo experiments provided further critical information. Regarding PK/PD parameters, phages were relatively stable in plasma (elimination half-life of ca 2.3 hours following bolus administration) and persisted longer in organs (half-life up to 9 hours). These values were consistent with those observed in previous work [] and confirmed that phages, whose sizes vary from ca 50 nm to 200 nm, can diffuse into various body compartments [, , ]. As a result, phages were able to kill bacteria inside valve vegetations and multiply locally by up to 3 log PFUs/g within 6 hours. Phage-induced killing correlated with a burst of IL-1β and IL-6. Because cytokine levels were measured only at a single time point, the data did not permit extrapolating the dynamics of cytokine responses over time. However, the significant increase in IL-1β and IL-6 levels in rats treated with phages—as compared with rats treated with ciprofloxacin—most likely reflected the release of cell debris by phage-mediated lysis. Accordingly, it is known that both cytokines are inducible by LPS [] and that similar results were obtained in EE using a bactericidal phage lysin [].
From a therapeutic point of view, the combination of phages with ciprofloxacin exhibited a highly synergistic effect and resulted in 7 of 11 (64%) negative valve cultures within 6 hours. This remarkable result has no precedent in antimicrobial therapy of EE with virtually any drug or pathogen, especially not with P. aeruginosa [].
Phage-antibiotic (or PAS) synergism has been formerly reported []. The mechanism of PAS is proposed to result from antibiotic-induced bacterial elongation, which may facilitate the access of phages to their bacterial target []. However, none of the previous PAS studies were done in vivo, and none of them demonstrated the extensive killing of >7 orders of magnitude in 6 hours reported herein []. Interestingly, phage-resistant subpopulations were readily detected in broth cultures and infected clots, but not in vegetations. This suggests that preexisting phage-resistant mutants might be hampered in vivo, as proposed by others [, , , , ].
In fact, the mutations that conferred resistance to the phage cocktail came at a great physiological cost. The present experiments provide 2 examples supporting this notion. Mutant 24/2 exhibited defective cell motility due to disruption of the pilT gene, whose product is an ATPase providing energy for type IV pilus contraction and bacterial twitching []. Type IV pilus is also a phage receptor, and pilus contraction is believed to bring the phages closer to the bacterial envelope []. Mutant 19/2 had a truncated LPS resulting from a large deletion encompassing the galU gene involved in the synthesis of the LPS core [, ]. LPS can also act as a phage receptor, and its alteration can confer phage resistance []. Both type IV pili and LPS are major virulence factors in P. aeruginosa, and their mutation can result in attenuated virulence [, ]. Thus, while these mutations confer resistance to phages, they may also decrease fitness in animals while preserving normal growth in less stringent in vitro conditions.
Although these observations are promising regarding phage antibacterial activity, caution should be raised as to the use of phage cocktails versus single-phage preparations. Although phage cocktails provide broader strain coverage than single phages, they also carry greater risks of unwanted gene transfer and interphage interference. Ideally phages should be tailored against their target pathogen, and specificity should be preferred to exhaustiveness, just like for antibiotic therapy.
Altogether our results provide a strong proof of concept for phage therapy of deep-seated and systemic infections. Moreover they raise the provocative possibility that certain infections might be cured by a single phage injection most efficiently combined with synergistic antibiotics. Indeed, once locally established, phages can multiply in situ and do not necessitate further administration.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Notes
Acknowledgments. We are indebted to Marlyse Giddey, Matthew Parkan, Sara Mitri, Glib Mazepa, Shawna McCallin, Jean Daraspe, and Gilles Willemen for their invaluable technical support and fruitful discussions.
Financial support. This work was supported in part by the Swiss National Research Foundation (CR31I3_166124 to Y. A. Q., 310030-143799 to J. M. E.) and the European Commission research grant (FP7-PHAGOBURN to Y. A. Q.). This work was also supported by a nonrestricted grant from the Loterie Suisse Romande (to Y. A. Q.). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Potential conflict of interest. J. G. is employed by the commercial company Pherecydes Pharma as COO. The bacteriophage cocktail PP1131 used in this study is produced under license by the company Pherecydes Pharma and used in a human clinical trial (Phagoburn, NCT02116010, http://www.phagoburn.eu/). Y. A. Q. is coinvestigator in the clinical trial Phagoburn, which uses the PP1131 bacteriophage cocktail. All authors have submitted the ICMJE Form for Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. Chanishvili N. Phage therapy—history from Twort and d’Herelle through Soviet experience to current approaches. Adv Virus Res2012; 83:3–40.
- 2. Abedon ST. Phage therapy of pulmonary infections. Bacteriophage2015; 5:e1020260.
- 3. Lang G, Kehr P, Mathevon H, Clavert JM, Séjourne P, Pointu J. Bacteriophage therapy of septic complications of orthopaedic surgery. Rev Chir Orthop Reparatrice Appar Mot1979; 65:33–7.
- 4. Speck P, Smithyman A. Safety and efficacy of phage therapy via the intravenous route. FEMS Microbiol Lett2016; 363.
- 5. Chhibber S, Kaur S, Kumari S. Therapeutic potential of bacteriophage in treating Klebsiella pneumoniae B5055-mediated lobar pneumonia in mice. J Med Microbiol2008; 57:1508–13.
- 6. Pouillot F, Chomton M, Blois H, et al. Efficacy of bacteriophage therapy in experimental sepsis and meningitis caused by a clone O25b:H4-ST131 Escherichia coli strain producing CTX-M-15. Antimicrob Agents Chemother2012; 56:3568–75.
- 7. Marza JA, Soothill JS, Boydell P, Collyns TA. Multiplication of therapeutically administered bacteriophages in Pseudomonas aeruginosa infected patients. Burns2006; 32:644–6.
- 8. Wright A, Hawkins CH, Anggård EE, Harper DR. A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa; a preliminary report of efficacy. Clin Otolaryngol2009; 34:349–57.
- 9. Rhoads DD, Wolcott RD, Kuskowski MA, Wolcott BM, Ward LS, Sulakvelidze A. Bacteriophage therapy of venous leg ulcers in humans: results of a phase I safety trial. J Wound Care2009; 18:237–8, 40–3.
- 10. Labrie SJ, Samson JE, Moineau S. Bacteriophage resistance mechanisms. Nat Rev Microbiol2010; 8:317–27.
- 11. Lenski RE. Experimental studies of pleiotropy and epistasis in Escherichia coli variation in competitive fitness among mutants resistant to virus T4. Evolution1988; 42:425–32.
- 12. Scanlan PD, Buckling A, Hall AR. Experimental evolution and bacterial resistance: (co)evolutionary costs and trade-offs as opportunities in phage therapy research. Bacteriophage2015; 5:e1050153.
- 13. Le S, Yao X, Lu S, et al. Chromosomal DNA deletion confers phage resistance to Pseudomonas aeruginosa. Sci Rep2014; 4:4738.
- 14. Que YA, Moreillon P. Infective endocarditis. Nat Rev Cardiol2011; 8:322–36.
- 15. Clokie MRJ, Kropinski AM. Bacteriophages. Methods and protocols. Volume 1: Isolation, characterization, and interactions. New York: Humana Press, 2009.
- 16. Entenza JM, Haldimann A, Giddey M, Lociuro S, Hawser S, Moreillon P. Efficacy of iclaprim against wild-type and thymidine kinase-deficient methicillin-resistant Staphylococcus aureus isolates in an in vitro fibrin clot model. Antimicrob Agents Chemother2009; 53:3635–41.
- 17. Entenza JM, Vouillamoz J, Glauser MP, Moreillon P. Levofloxacin versus ciprofloxacin, flucloxacillin, or vancomycin for treatment of experimental endocarditis due to methicillin-susceptible or -resistant Staphylococcus aureus. Antimicrob Agents Chemother1997; 41:1662–7.
- 18. Broskey NT, Daraspe J, Humbel BM, Amati F. Skeletal muscle mitochondrial and lipid droplet content assessed with standardized grid sizes for stereology. J App Physio2013; 115:765–70.
- 19. Taylor RD. Modification of the Brown and Brenn gram stain for the differential staining of gram-positive and gram-negative bacteria in tissue sections. Am J Clin Pathol1966; 46:472–4.
- 20. Meyer F, Goesmann A, McHardy AC, et al. GenDB—an open source genome annotation system for prokaryote genomes. Nucleic Acids Res2003; 31:2187–95.
- 21. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol1990; 215:403–10.
- 22. Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics2011; 12:402.
- 23. Semmler AB, Whitchurch CB, Mattick JS. A re-examination of twitching motility in Pseudomonas aeruginosa. Microbiology1999; 145:2863–73.
- 24. Zhu ZX, Cong WT, Ni MW, et al. An improved silver stain for the visualization of lipopolysaccharides on polyacrylamide gels. Electrophoresis2012; 33:1220–3.
- 25. Mattick JS. Type IV pili and twitching motility. Annu Rev Microbiol2002; 56:289–314.
- 26. Dean CR, Goldberg JB. Pseudomonas aeruginosa galU is required for a complete lipopolysaccharide core and repairs a secondary mutation in a PA103 (serogroup O11) wbpM mutant. FEMS Microbiol Lett2002; 210:277–83.
- 27. Merril CR, Biswas B, Carlton R, et al. Long-circulating bacteriophage as antibacterial agents. Proc Natl Acad Sci U S A1996; 93:3188–92.
- 28. Górski A, Wazna E, Dabrowska BW, Dabrowska K, Switała-Jeleń K, Miedzybrodzki R. Bacteriophage translocation. FEMS Immunol Med Microbiol2006; 46:313–9.
- 29. de Bont N, Netea MG, Rovers C, et al. LPS-induced release of IL-1 beta, IL-1Ra, IL-6, and TNF-alpha in whole blood from patients with familial hypercholesterolemia: no effect of cholesterol-lowering treatment. J Interferon Cytokine Res2006; 26:101–7.
- 30. Entenza JM, Loeffler JM, Grandgirard D, Fischetti VA, Moreillon P. Therapeutic effects of bacteriophage Cpl-1 lysin against Streptococcus pneumoniae endocarditis in rats. Antimicrob Agents Chemother2005; 49:4789–92.
- 31. Papadakis JA, Samonis G, Maraki S, Boutsikakis J, Petrocheilou V, Saroglou G. Efficacy of amikacin, ofloxacin, pefloxacin, ciprofloxacin, enoxacin and fleroxacin in experimental left-sided Pseudomonas aeruginosa endocarditis. Chemotherapy2000; 46:116–21.
- 32. Bayer AS, Norman D, Kim KS. Efficacy of amikacin and ceftazidime in experimental aortic valve endocarditis due to Pseudomonas aeruginosa. Antimicrob Agents Chemother1985; 28:781–5.
- 33. Robaux MA, Dube L, Caillon J, et al. In vivo efficacy of continuous infusion versus intermittent dosing of ceftazidime alone or in combination with amikacin relative to human kinetic profiles in a Pseudomonas aeruginosa rabbit endocarditis model. J Antimicrob Chemother2001; 47:617–22.
- 34. Ryan EM, Alkawareek MY, Donnelly RF, Gilmore BF. Synergistic phage-antibiotic combinations for the control of Escherichia coli biofilms in vitro. FEMS Immunol Med Microbiol2012; 65:395–8.
- 35. Knezevic P, Curcin S, Aleksic V, Petrusic M, Vlaski L. Phage-antibiotic synergism: a possible approach to combatting Pseudomonas aeruginosa. Res Microbiol2013; 164:55–60.
- 36. Kamal F, Dennis JJ. Burkholderia cepacia complex phage-antibiotic synergy (PAS): antibiotics stimulate lytic phage activity. Appl Environ Microbiol2015; 81:1132–8.
- 37. Comeau AM, Tétart F, Trojet SN, Prère MF, Krisch HM. Phage-antibiotic synergy (PAS): beta-lactam and quinolone antibiotics stimulate virulent phage growth. PLoS One2007; 2:e799.
- 38. Scanlan PD, Hall AR, Blackshields G, et al. Coevolution with bacteriophages drives genome-wide host evolution and constrains the acquisition of abiotic-beneficial mutations. Mol Biol Evol2015; 32:1425–35.
- 39. Smith HW, Huggins MB, Shaw KM. The control of experimental Escherichia coli diarrhoea in calves by means of bacteriophages. J Gen Microbiol1987; 133:1111–26.
- 40. Bradley DE. Shortening of Pseudomonas aeruginosa pili after RNA-phage adsorption. J Gen Microbiol1972; 72:303–19.
- 41. Rodríguez-Rojas A, Mena A, Martín S, Borrell N, Oliver A, Blázquez J. Inactivation of the hmgA gene of Pseudomonas aeruginosa leads to pyomelanin hyperproduction, stress resistance and increased persistence in chronic lung infection. Microbiology2009; 155:1050–7.
- 42. Comolli JC, Hauser AR, Waite L, Whitchurch CB, Mattick JS, Engel JN. Pseudomonas aeruginosa gene products PilT and PilU are required for cytotoxicity in vitro and virulence in a mouse model of acute pneumonia. Infect Immun1999; 67:3625–30.