Introduction
The prevalence of type 2 diabetes mellitus (T2DM or T2D) rose dramatically over the past decades, afflicting ∼8% adults globally (; ; ). The ever-rising T2D prevalence imposes profound sequelae on the cardiovascular system (; ; ; ). Cardiovascular disease (CVD) becomes the leading cause of death in T2D, with coronary artery disease (CAD) and ischemic cardiomyopathy being the main culprits (; ; ). In addition to CAD, small vessel disease and diminished cardiac capillary density also occur ().
T2D evokes unique changes in myocardium independent of classical risk factors, including CAD, hypertension, and valvular heart disease. This is a condition termed as diabetic cardiomyopathy (DCM) (; ). Early-stage DCM is usually characterized by structural and functional abnormalities, including myocardial hypertrophy, stiffening, interstitial fibrosis, and diastolic dysfunction (; ; ; ; ). Similar findings were observed in animal models of T2D, including compromised diastolic and systolic function accompanied by changes in cardiomyocyte function and intracellular Ca2+ (; ; ; ; ).
Patients with T2D exhibit an increased risk of heart failure (HF). Of note, 25% of T2D patients exhibit various types of HF, including HF with preserved, reduced, and midrange ejection fraction (HFpEF, HFrEF, and HFmrEF, respectively), with mortality increased by 30%–50% and worsened prognosis (). The two predominant types of HF in DCM are HFrEF and HFpEF, with concentric left ventricular (LV) remodeling and diastolic dysfunction. HFpEF accounts for approximately half of the HF incidence in T2D, characterized by a normal or near-normal LV ejection fraction (LVEF) ≥50% and exercise intolerance as the chief complaint (). Other than T2D, various complications, including obesity, hypertension, dyslipidemia, renal disease, and atrial fibrillation, also closely correlate with HFpEF (). Given the high prevalence and poor prognosis of HFpEF, intensive therapy is pertinent for the management of cardiovascular anomalies in patients with T2D (; ). Nonetheless, current treatment modality remains dismal largely due to the poorly defined pathophysiology of HFpEF in T2D. In this review, we discuss potential pathophysiological mechanisms and treatment options for HFpEF in T2D patients. We will also summarize recent clinical trials.
Epidemiology and prognosis of HFpEF in T2D
T2D plays an essential role in the onset of HFpEF (; ). Compared with non-diabetics, the incidence and mortality of CVD and HF are much higher in T2D patients (; ). Patients with T2D initially present with normal systolic but progressively impaired diastolic function, indicative of HFpEF. Approximately 45% of T2D patients develop HFpEF, and the prevalence of co-morbid T2D is more abruptly increased in T2D patients with new-onset HFpEF ().
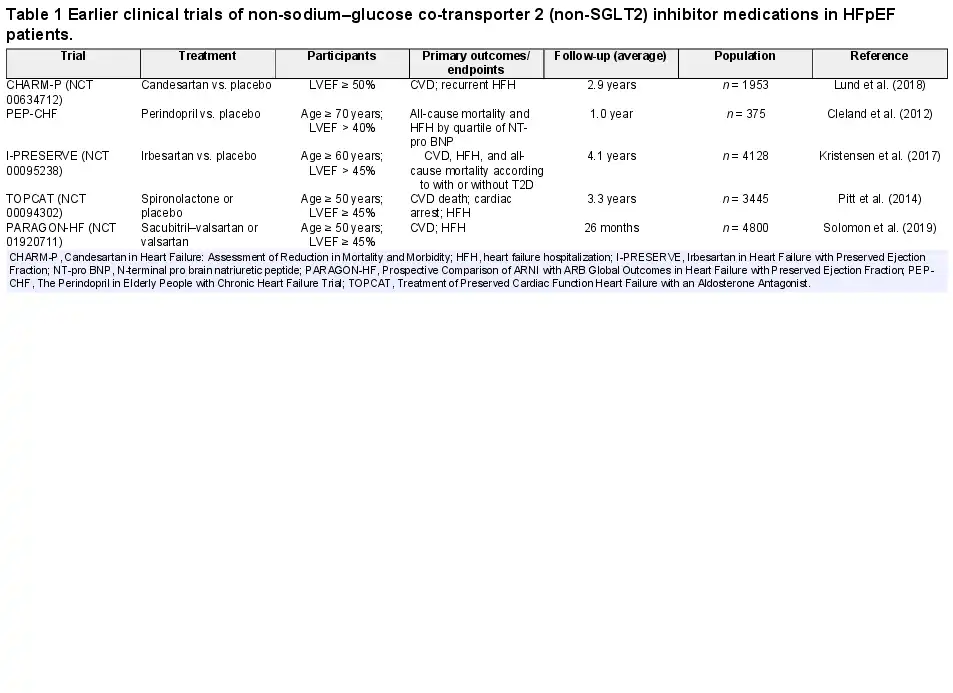
T2D in combination with HFpEF greatly enhances CVD risk and mortality (). TOPCAT (LVEF > 40%) is a multicenter, randomized, double-blind, placebo-controlled study of microvascular complications in T2D patients, including neuropathy, nephropathy, and retinopathy (; ). According to the CHARM Trial, 3023 patients were defined as LVEF > 40%, with an average follow-up of 37.7 months. Patients with T2D display greater volume overload, more severe myocardial injury, higher body mass index, hypertension (especially systolic pressure), coronary microvascular dysfunction, and renal abnormality (). The I-PRESERVE Study with LVEF ≥ 50% revealed that HFpEF patients with T2D had higher rates of cardiovascular mortality, hospitalization, and all-cause mortality (; ; Table 1). Overall, these clinical trials denote a close tie between HFpEF-associated mortality and T2D, favoring a positive correlation between T2D and HFpEF-associated morbidity and mortality. Possible contributions from T2D-induced cellular, molecular, and metabolic abnormalities are considered the major driving forces for the worsened HFpEF pathology.
Pathophysiology of HFpEF in T2D
Clinical reports favor a DCM phenotype distinct from dilated cardiomyopathy. T2D patients display myocardial dysfunction in the absence of coronary heart diseases (e.g. CAD) (), possibly due to metabolic abnormalities, e.g. advanced glycation end product (AGE) deposition, lipid toxicity, and microvascular rarefication (). These adverse effects of T2D were more pronounced in HFpEF (; ). T2D with HFpEF is featured by LV diastolic dysfunction (LVDD) and decreased LV cavity with elevated LV filling pressure, as well as endothelial and coronary microvascular dysfunction (). In T2D, hearts are exposed to hyperglycemic environments with abundant fatty acids and cytokines (; ). Hyperglycemia provokes adverse clinical outcomes in T2D through AGE production (), leading to accumulation of reactive oxygen species (ROS), inflammation, mitochondrial damage, and apoptosis (; ). Hyperglycemia prompts endothelial dysfunction and elevated serum cholesterol, resulting myocardial dysfunction and ventricular–vascular uncoupling (). In this context, subclinical variations develop, prompting cardiovascular events, including HFpEF (Figure 1; ). Rigorous glucose control is of enormous importance for the clinical outcome of HFpEF.
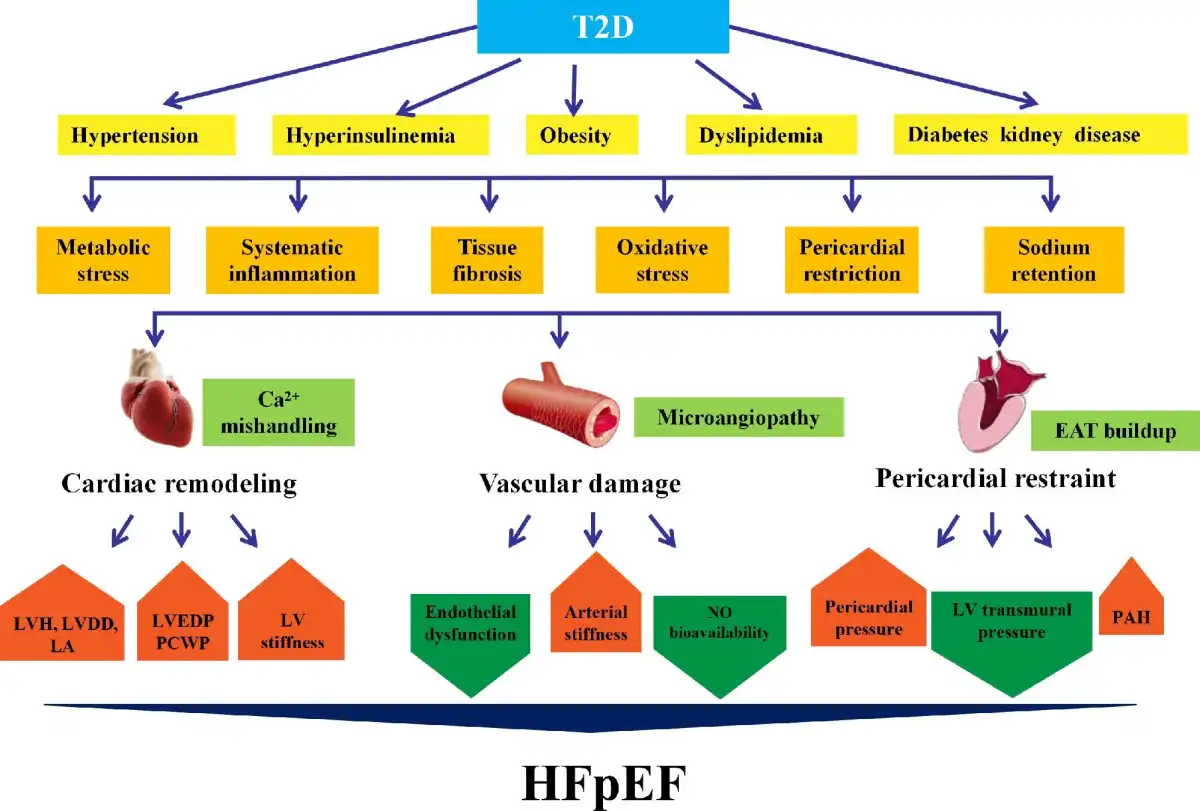
Figure 1
Pathology of HFpEF with T2D. Metabolic disturbances such as hyperglycemia, hypertension, hyperinsulinemia, obesity, and renal disease prompt the development of HFpEF in T2D. T2D patients with HFpEF often exhibit LVDD and small LV cavities with elevated LV filling pressures, as well as vascular damage, including endothelial and coronary microvascular dysfunction. T2D and obesity are intertwined and are accompanied by EAT buildup, deteriorating myocardial inflammation, and fibrosis. EAT leads to mechanical stress as a pericardial restraint for LV function. Abnormal hemodynamics between myocardium and pericardium increase pericardial pressure and LVEDP and reduce LV transmural pressure, resulting in higher pulmonary capillary pressure. LA, left atrium; PCWP, pulmonary capillary wedge pressure.
LVDD
HFpEF in T2D is difficult to diagnose due to relatively mild and atypical symptoms, including poor exercise capacity (). T2D can directly affect myocardial structure and function to prompt HFpEF (). T2D-induced specific cardiomyocyte anomalies include compromised Ca2+ handling, encompassing cytosolic and mitochondrial Ca2+ handling (). HFpEF is often manifested as diastolic dysfunction, with elevated LV filling pressure and plasma levels of BNP and NT-pro BNP (; ). The precise pathogenesis behind HFpEF is complex and multifactorial, with altered cardiac metabolism such as those seen in T2D being a critical contributing factor ().
It is noteworthy that diastolic dysfunction occurs in T2D patients without obvious CAD symptoms. Three quarters of T2D patients may present diastolic dysfunction (), usually defined as defects in the integral of active diastolic relaxation dependent upon myofilament dissociation and cytosolic Ca2+ reuptake in early diastolic phase. Passive stiffness is related to viscoelasticity controlled by mechanical changes from sarcomere to extracellular matrix. T2D also induces ROS generation and deposition of AGEs in endothelial and smooth muscle cells, prompting concentric LV remodeling and wall stiffness (). Elevated serum AGEs were associated with prolongation of LV relaxation in early stage of DCM (). In T2D, insulin resistant, and obese patients, circulating levels of glucose, free fatty acid (FFA), and proinflammatory cytokines were elevated, along with compromised angiogenesis, leading to pathological hypertrophy and diastolic dysfunction (). The severity and duration of hyperglycemia function as an important determinant for the onset of LVDD. High FFA intake, for example, leads to lipid toxicity and accumulation of triglyceride (TG) in the myocardium. Cardiac steatosis, as visualized using proton magnetic resonance spectroscopy, denotes high TG content in the heart muscles to prompt diastolic defect (). From these data, prominent changes in Ca2+ handling, deposition of AGEs, and insulin metabolic signaling may influence cardiac interstitial fibrosis/fiber stiffness and diastolic dysfunction in T2D patients with HFpEF.
Endothelial dysfunction
Endothelial defect usually encompasses impaired vasodilation, enhanced vasoconstriction, arterial stiffness, and overt atherogenesis. In addition, signs of dyspnea and fatigue are also noted in association with submaximal exercise and altered bioavailability of nitric oxide (NO). Impaired insulin metabolic sensitivity is a vital pathophysiological abnormality tied to DCM, which inhibits endothelial NO synthase (eNOS) and NO production (; ; ). The prevalence of endothelial dysfunction is high in HFpEF patients (). Endothelial dysfunction is linked to adverse outcomes in HFpEF patients, with a main role of poor NO availability (). Compared with HFpEF patients without endothelial dysfunction in coronary arteries, patients with comprised coronary endothelial function possess more severe clinical outcomes associated with T2D. The main driving force for myocardial dysfunction in T2D includes insulin resistance and impaired glucose tolerance, long before the onset of full-blown T2D (). In addition, increased arterial stiffness and LV end-systolic stiffness or elasticity result in altered preload and afterload, ultimately leading to a major fluctuation of stroke volume (). These changes help to explain the role of blood pressure instability in HFpEF patients. Exercise tolerance test suggests that patients with HFpEF may not get more benefits compared with those with HFrEF, courtesy of the loss of exercise benefit in the presence of endothelial dysfunction ().
Coronary microvascular dysfunction
The new focal point for HFpEF etiology has been shifted from LV volume overload to coronary microvascular inflammation. Coronary microvascular dysfunction is very difficult to define in HFpEF based on clinical indicators. In HFpEF, concentric LV remodeling is rooted from coronary microvascular endothelial inflammation in T2D (). In contrast, eccentric LV remodeling in HFrEF is driven by cardiomyocyte death due to ischemia, viral infection, and lipotoxicity. HFpEF patients with endothelial-dependent microvascular dysfunction exhibit more severe diastolic defects and poor prognosis (). In addition to compromised systolic reserve, insufficient vasodilation may be seen related to reduced LV end-systolic volume (LVESV) and elevated stroke volume in HFpEF. In HFpEF patients, the mean systemic vascular resistance and effective arterial elasticity usually drop, particularly during exercise (). Elevated AGEs and hyperglycemia may prompt vascular damage in various vascular beds (). Microvascular remodeling and angiogenesis are important regulatory mechanisms in coronary vessels, affecting vascular function (). It is perceived that decreased NO levels contribute to endothelial-dependent coronary micro-vasodilatation defects, causing vessel dilatory dysfunction in T2D.
Ventricular–vascular uncoupling
With concurrent T2D and HFpEF, endothelial dysfunction indecently disrupts ventricular–vascular uncoupling and contributes to pathological changes in T2D with HFpEF (; ). Systolic dysfunction often leads to subsequent impairment of LV diastolic function. Meanwhile, the interplay between vascular stiffness and diastolic reserve plays a major role in the etiology of HFpEF (). Ventricular load also seems to be important, with increased late systolic load exerting a greater adverse effect on LV diastolic function in comparison with increased early systolic pressure (). The inability to dilate blood vessels with poor systolic reserve results in a dynamic limitation of ventricular the artery coupled with exercise intolerance in HFpEF patients ().
Diabetic kidney disease
Chronic end-stage renal disease, defined as diabetic kidney disease (DKD), is seen in ∼50% of T2D patients (). DKD contributes to secondary hypertension and anemia (). In T2D, hyperglycemia leads to altered systemic metabolism. Cell stresses, including oxidative stress, endoplasmic reticulum (ER) stress, AGE buildup, inflammation, and histone and chromosomal abnormalities, all contribute to the etiology of DKD (). Chronic renal dysfunction is related to increased hospitalization and death in HFpEF (). Consequently, T2D, kidney dysfunction, and HF are suggested to form a ‘vicious circle’ (). Decreased renal perfusion and elevated central venous pressure are the most important hemodynamic factors for the heart–kidney interplay. Moreover, the renin–angiotensin–aldosterone system and sympathetic nervous system are overactivated in T2D, leading to lipolysis and the onset of insulin resistance (). Reduced sodium excretion promotes a proinflammatory state. This proinflammatory environment perpetuates a vicious cycle between the heart and kidney in HFpEF ().
Pericardial restriction
T2D is often concurrent with obesity to evoke metabolic changes, inflammation, fibrosis, and myocardial stiffness, all considered HFpEF phenotypes (; ). One of the hallmarks of T2D and obesity is accumulation of epicardial adipose tissue (EAT), a form of endocrine tissues capable of deteriorating myocardial inflammation and fibrosis via the release of paracrine and autocrine factors (). Furthermore, enlargement of EAT leads to mechanical stress on pericardial restriction (). In HFpEF, EAT thickness is closely associated with LV hypertrophy (LVH). Abnormal hemodynamics between myocardium and pericardium yields elevated pericardial pressure and LV end-diastolic pressure (LVEDP) and decreased LV transmural pressure that may elevate pulmonary capillary pressure. Finally, increased EAT volumes might also directly influence cardiac function and hemodynamics through mechanical interaction. Patients with HFpEF, particularly for obese HFpEF, exhibit an increased EAT thickness in collaboration with higher cardiac filling pressure and pericardial restraint, leading to pericardial constriction (; ; ). With the escalating pericardial constraint, intracavitary diastolic filling pressure needs to be higher to achieve an adequate preload volume, which further increases pulmonary capillary hydrostatic pressures in patients with HFpEF. These structural changes may probably be resulted from inflammation evoked by adipokines. Moreover, excessive collagen deposition, abnormal protein glycosylation, and abnormal collagen cross-linking in the myocardium lead to increased LV filling pressure, decreased diastolic compliance, and an increased risk for development of HFpEF (; ).
Molecular mechanisms behind HFpEF in T2D
In T2D patients, decreased diastolic function accompanied by altered protein expression governing relaxation was revealed. Metabolic abnormalities in T2D seem to play a vital role in conjunction with hyperglycemia, proinflammatory responses, and lipotoxicity (). Other confounding factors, including interstitial fibrosis, vascular damaging, dysregulated NO, and cyclic guanine monophosphate (cGMP), may also contribute to the etiology of HFpEF. For example, interruption in the cGMP–PKG signal transduction and the rise of protein kinase C alpha (PKCα) activity are deemed key regulatory factors of cardiomyocyte stiffness in DCM (Figure 2).
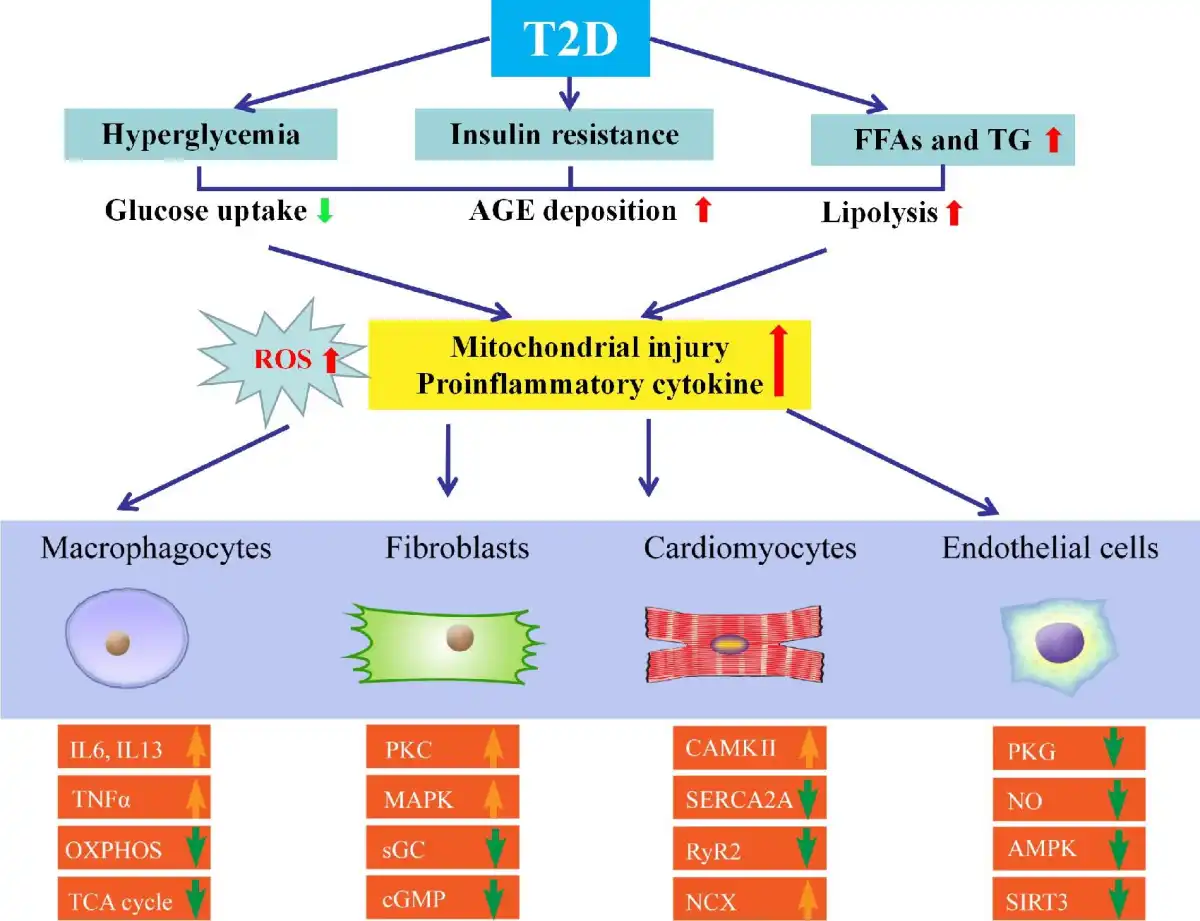
Figure 2
Molecular mechanisms behind HFpEF with T2D. Various metabolic abnormalities in T2D play an essential role in conjunction with the buildup of proinflammatory cytokines and ROS. Other confounding factors, including hyperglycemia, lipotoxicity, and increased levels of FFA, insulin, and AGEs, also contribute to HFpEF. Compromised NO bioavailability and sensitivity, oxidative stress and inflammation, and impaired angiogenesis are involved. Crucial molecular contributors to pathological hypertrophy include G protein-coupled receptors, stress hormone ligands, and signaling kinases in T2D with HFpEF. Relaxation period includes active and passive phases. Downregulated insulin signaling is a hallmark of T2D along with changes in other signaling cascades, including downregulated AMPK signaling and hyperactivated PKC and MAPK. IL6, interleukin 6; IL13, interleukin 13; OXPHOS, oxidative phosphorylation; TCA, tricarboxylic acid cycle; TNFα, tumor necrosis factor-α.
LVH is a cardinal feature of HFpEF and the major reason for elevated diastolic filling pressure and diastolic dysfunction. Myocardial fibrosis, impaired Ca2+ handling, oxidative stress, mitochondrial dysfunction, and metabolic reprogramming are all key components of pathological cardiac hypertrophy (; ; ; ). Crucial molecular contributors to this pathological hypertrophy in T2D with HFpEF include G protein-coupled receptors, stress hormone ligands, and signaling kinases (). The precise roles of these signaling machineries in HFpEF remain at large in humans. The ratio of NAD+ to NADH decreased in the hearts of patients with T2D, while NAD+ is required for metabolic oxidation (). These findings suggest the possible involvement of altered energy metabolism in pathological hypertrophy.
Interstitial fibrosis is commonly noted in DCM accompanied by changes in a wide variety of cell signaling cascades and extracellular matrix proteins (e.g. formation of insoluble AGEs) (; ). In addition, aberrant activation of protein kinases such as calcium/calmodulin-dependent protein kinase type II subunit beta (CAMKII) has been shown to contribute to excitation–contraction coupling defects in T2D. CAMKII may be turned on by the Ca2+–calmodulin complex, cAMP, and ROS. Overactivation of CAMKII phosphorylates substrate proteins governing Ca2+ handling and fosters LVH, mitochondrial injury, inflammation, and arrythmia (; ). It is believed that transient receptor potential channel 3 (TRPC3) and TRPC6 may serve as triggers for the hyperactivation of CAMKII (). This is supported by the observation that ablation of TRPC3 or TRPC6 mitigates pressure overload-evoked maladaptive hypertrophy (). Among various potential regulators for TRP channels, stimulation of calcineurin–NFAT is suggested to offer a direct link between gene expression and intracellular Ca2+ signaling (; ).
Diastolic function denotes myocardial relaxation capacity following a normal heartbeat (). The relaxation period includes active and passive phases at both cellular and tissue levels (). Upon excitation, intracellular Ca2+ is dumped from the intracellular sarcoplasmic reticulum (SR) store with the binding of ryanodine receptor (RyR) to troponin C. Relaxation initiates with the dissociation of Ca2+ ions from the troponin complex—prior to SR reuptake and efflux of Ca2+ through Na+–Ca2+ exchange protein (NCX). To lower cytosolic Ca2+ in diastole, phospholamban is phosphorylated to disinhibit sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA2), enabling SR Ca2+ reuptake (). This is supported by findings that cardiac overexpression of phospholamban inhibits SERCA2A and SR Ca2+ uptake to compromise cardiac function, while phospholamban knockout mice display better Ca2+ cycling and myocardial contractility. With the uptake of Ca2+ into SR during the diastolic period, reverse-mode NCX is increased at the sarcolemma. Decreased myocardial compliance (muscle stiffening) was due to suppressed cGMP-dependent PKG and hypo-phosphorylation of titin (). cGMP and its cognate kinase, PKG, are known for the maintenance of vascular and endothelial function. As such, the LV chamber becomes less compliant and fails to dilate properly courtesy of hypo-phosphorylation of titin in diastole. Activation of PKG or protein kinase A (PKA, cAMP-driven and possibly PKG-independent) may thus correct these irregularities and reinstate LV chamber compliance ().
Hyperglycemia, a hallmark of T2D, is one of the key causes of endothelial dysfunction (; ). Exposure of endothelial cells to high glucose leads to ROS production, while glycosylation inhibits eNOS activation, angiogenesis, and mitochondrial dysfunction. Quenching of NO by AGEs plays a vital role in vasodilatory impairment in T2D. Depletion of NADPH and excessive AGE production may result in elevated permeability of endothelial cells, inhibition of eNOS activity, influence of the coagulation system, and activation of NADPH oxidase and nuclear factor-κB (NF-κB) (). Downregulated insulin signaling is a hallmark of T2D, and other signaling cascade changes occur, including downregulated adenosine monophosphate activated protein kinase (AMPK) signaling and increased PKC and mitogen-activated protein kinase (MAPK) signaling with undesirable effects (; ). Downregulation of peroxisome-proliferator-activated receptor-γ coactivator 1α (PGC1α) evokes altered oxidative mitochondrial function in T2D. In plasma from HFpEF patients, metabolomic profiling exhibited altered levels of β-oxidation (). Reduced mitochondrial oxidative metabolism is accompanied by increased glycolysis, although such change may be translated to higher glucose uptake through glycolysis from pyruvate oxidation (). Impaired insulin signaling inhibits glucose oxidation by negative feedback regulation via the Randle cycle.
Microvascular disease is also a common feature of T2D (). Mechanisms include depressed NO signaling, increased oxidative stress and inflammation, impaired angiogenesis, and other abnormalities. For generality, the main factor is NAD-dependent protein deacetylase sirtuin-3 (SIRT3). SIRT3 knockdown of endothelial cells in mice impairs glycolysis and angiogenesis and is associated with diastolic dysfunction. However, a role for SIRT3 in HFpEF patients is lacking. Compared with healthy controls, patients with HFpEF display a greater product of end-systolic pressure and stroke volume, myocardial blood flow, and myocardial oxygen consumption (). These changes denote an imbalance between demand and supply with elevated cardiac work in HFpEF with β-adrenergic stimulation.
Nuclear factor erythroid 2-like factor 2 (Nrf2) is a master regulator of oxidative stress with a vital role in DKD (). Under unstressed conditions, Nrf2 is ubiquitinated and degraded by proteasomes through Kelch-like ECH-associated protein 1 (Keap1), while Keap1 undergoes various chemical modifications to reduce its affinity for Nrf2 and inhibit its degradation under oxidative stress (; ). Bardoxolone methyl is a synthetic triterpenoid, which not only upregulates Nrf2 but also retards inflammation, perhaps through additional actions on NF-κB. Currently, bardoxolone methyl is going through clinical trials for type 1 diabetic nephropathy and other renal diseases.
Treatment options for HFpEF in T2D
It is important to identify and treat potential risk factors and other comorbidities such as hypertension, CAD, atrial fibrillation, and valvular heart disease for HFpEF in T2D (Table 1). Therapeutically, mineralocorticoid receptor antagonists (MRA) were reported to reduce HFH in HFpEF, although the effects on mortality-related outcomes and quality of life remain unclear for MRA (). On the other hand, classical β-blockers display little effectiveness in treating obesity/T2D-associated HFpEF, although larger cohorts are needed. Likewise, angiotensin converting enzyme inhibitors exhibit little or no effect on cardiovascular mortality, all-cause mortality, and HFH (). In the PARAGON-HF Trial using angiotensin receptor/neprilysin inhibitor (ARNI, sacubitril/valsartan), there was little difference in primary endpoints of HFpEF. Nonetheless, significant benefits were noted in two subgroups (women and LVEF ≤ 57%). It is suggested that ARNI may be an effective option for certain HFpEF patients (). Comorbidities are common in HFpEF patients, including hypertension, thus necessitating intensive management for hypertension. The ACC/AHA guidelines recommend a target systolic blood pressure <130 mmHg in HFpEF patients. In addition, observational studies suggest that statins may also offer benefits in HFpEF ().
There are several mechanisms in T2D with HFpEF, including sodium retention and subsequent volume overload and elevated filling pressure, increased proinflammatory cytokines, impaired skeletal muscle function, and cardio–respiratory uncoupling (; ; ). Historically, treatment based on the cardiovascular system, with neurohormonal blockade, has been claimed unsuccessful. Specific subgroups of patients with HFpEF and those with combinations of clinical comorbidities, including T2D and metabolic dysfunction, might be more new remedies for T2D with HFpEF.
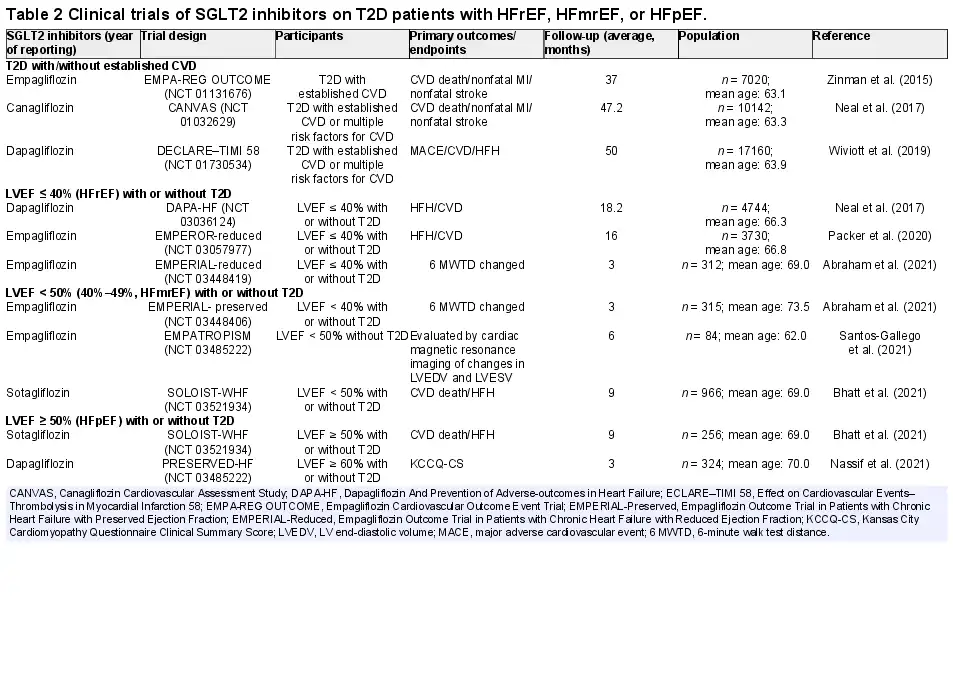
Of note, the use of new medications and combination therapies for glycemic control in T2D is rapidly evolving (; ; ; ; ; ). As patients with T2D often have other risk factors, including obesity, hypertension, dyslipidemia, and renal disease, current treatment strategies favor combination therapies, such as anti-hyperglycemic and anti-hypertensive therapy, to retard the progression of HFpEF. A multifactorial approach targeting multiple, if not all, risk factors has displayed better promises compared with glucose control alone. The most potent antidiabetic medications to prevent HF development in T2D include SGLT2 inhibitors, which retard the onset and progression of HF regardless of the presence of T2D or not (; Table 2).
Lifestyle modification
Favorable clinical outcome of exercise training in T2D with HFpEF is reminiscent of that seen in HFpEF individuals (; ). Several clinical trials have depicted that exercise training improves exercise capacity and quality of life in HFpEF patients (; ; ). On the other hand, calorie restriction and weight loss may be indicated for obese T2D patients with HFpEF. In general, the benefits of exercise training and lifestyle modification seem to reside outside the heart. In T2D and obesity, myocardial glucose is replaced by uptake of non-esterified FFA as the main energy source. Such substrate switch is closely associated with the onset of LVDD, the effect of which can be alleviated by exercise and lifestyle modification ().
Diuretics
Patients with HFpEF generally develop fluid retention and volume overload, dyspnea, and severe exertional incapacity, with a more pronounced symptomatic manifestation with T2D. Peripheral impairments, such as skeletal muscle dysfunction, impaired peripheral oxygen delivery, and time-varying dysfunction, may result in reduced exercise capacity in T2D patients with HFpEF. Meanwhile, diuretics are recommended for T2D patients with HFpEF and are usually required to treat symptoms and signs of fluid overload. Diuretics are commonly used in pulmonary arterial hypertension (PAH), supporting the utility of diuretics in reducing risks of hospitalization for HF (). Most clinical trials on the efficacy of treatments for HFpEF have produced neutral results, although strong evidence supports the benefits of diuretics as effective therapies.
Phosphodiesterase inhibitors
There is evidence for interference of beta-adrenergic receptor (beta-AR) microdomains in HFpEF, with T2D or obese heart exhibiting altered expression levels of beta-AR, coupled with elevated activity of phosphodiesterases (PDEs) as AMP-hydrolysing enzyme. PDE-5 inhibitor sildenafil is an established therapy for PAH. Compared with placebo, treatment with sildenafil did not lead to an improvement in exercise capacity or clinical status in HFpEF, with depressed myocardial oxygen supply particularly during exercise. The evidence of prominent endothelial dysfunction or deficient cGMP signaling could suggest that interventions targeting the NO–cGMP–PDE pathway may be promising for future treatment.
SGLT2 inhibitors
SGLT2 inhibitors impose benefits on glucose control, as well as renal and sodium–water-related metabolism and hemodynamics (; ), to alleviate clinical symptoms associated with HFpEF (; ). The main benefits of SGLT2 inhibitors are suggested to be improved arterial stiffness and coronary blood flow by way of natriuresis and glycosuria. Meanwhile, SGLT2 inhibitors may impact myocardium directly through alleviating cardiac afterload, interstitial fibrosis, and energy matrix displacement (; ; ). Beyond the glycemic control, SGLT2 inhibitors are deemed effective in the management of medium- to long-term T2D-associated complications (). Hyperglycemia impairs cardiac function, leading to compromised glucose uptake by T2D (; ). The increased level of β-hydroxybutyric acid in response to SGLT2 inhibitors leads to a shift of fuel supply from FFA and glucose to the more energy-efficient ketones. Thus, improvement of metabolic efficiency of the heart and kidney while reducing oxygen consumption may be a potential new direction in the future (). These SGLT2 inhibitor-induced biological effects may support the beneficial hemodynamic effects of SGLT2 inhibitors through increasing cardiac mitochondrial energy output. Empagliflozin, canagliflozin, and dapagliflozin have been shown to inhibit cardiac Na+/K+ exchanger (NHE), resulting in reduced cytoplasmic Na+ and Ca2+ and increased mitochondrial Ca2+ (). These effects may have a cardioprotective effect, as increased intracellular Na+ and NHE activities are associated with myocardial hypertrophy, exacerbation of HF, and arrhythmias ().
Metformin
In T2D patients with stable CAD, metformin improved LV diastolic function, although the mechanism of action remains undefined. Metformin was associated with lower all-cause mortality in the subgroup of patients with T2D and HFpEF and poor glycemic control (). The possible explanation of its anti-atherosclerotic effect may be its multiple effects on vascular endothelial cells, smooth muscle cells, lipids, and chronic systemic inflammation (), and results show that long-term metformin administration contributes to good lipid metabolism in T2D HFpEF patients.
Dipeptidylpeptidase-4 inhibitors
Due to the concern of blood glucose control, dipeptidylpeptidase-4 (DPP-4) inhibitors as the drug for the treatment of HFpEF with T2D attract considerable attention. DPP-4 inhibitors can inhibit the decomposition of glucagon-like peptide 1 (GLP-1) and correct glycogenesis of liver in order to improve blood glucose control. The Saxagliptin Assessment of Vascular Outcomes Recorded in Patients with Diabetes Mellitus Study showed that saxagliptin compared with the placebo group was neutral for the major adverse effect of MACE endpoints on cardiovascular death, nonfatal myocardial infarction, or stroke, but had an increased rate of HF rehospitalization (). At present, only animal studies have shown that DPP-4 inhibitors may promote myocardial fibrosis in elderly diabetic mice (; ). However, the relationship between the effects of DPP-4 inhibitors and HFpEF is not clear, which needs to be revealed in the future.
GLP-1 agonists
There was no significant difference in cardiovascular events and hospital stays between GLP-1 agonists and placebo in the ELIXA and ESXCEL Trials (; ). Importantly, the Evaluation of Cardiovascular Outcome Results (LEADER) as well as Semaglutide in Subjects with T2D (SUSTAIN-6) Trials showed superiority compared with placebo by decreasing the risk of major cardiovascular events (; ). It is noteworthy that the LEADER Trial showed that liralutide failed to improve the hospitalization rate of HF. The effect of this GLP-1 agonist on HF function showed that liralutide appeared to deteriorate HFrEF. However, the effects of GLP-1 agonists on T2D and HFpEF are not clear.
Conclusion and future perspectives
Clinical trial data show that the incidence rate and long-term mortality rate of T2D patients with HFpEF are higher than those of non-diabetic patients. Treatments for T2D patients with HFpEF are urgently needed. One of the major burning obstacles for clinical therapeutics of HFpEF is the poorly understood pathophysiology behind HFpEF, making drug development a perplexing task. Several potential therapeutic targets have been identified thus far. However, individual targeting of molecular signaling pathways defined in HFpEF might not effectively restore diastolic function due to the apparent drug toxicity and off-target effects. Therefore, future drug development requires a more comprehensive approach, not only for HFpEF comorbidities but also for classification and phenotypic identification in HFpEF.
Acknowledgements
We wish to thank Dr Aislinn O'Kane from the University of Wyoming College of Heath Sciences for her kind editing. The authors wish to sincerely apologize to those authors whose important work cannot be included due to space limitations.
References
- Abraham W.T., Lindenfeld J., Ponikowski P., et al (2021). Effect of empagliflozin on exercise ability and symptoms in heart failure patients with reduced and preserved ejection fraction, with and without type 2 diabetes. Eur. Heart J.42, 700–710.
- Akiyama E., Sugiyama S., Matsuzawa Y., et al (2012). Incremental prognostic significance of peripheral endothelial dysfunction in patients with heart failure with normal left ventricular ejection fraction. J. Am. Coll. Cardiol.60, 1778–1786.
- Ananthram M.G., Gottlieb S.S. (2021). Renal dysfunction and heart failure with preserved ejection fraction. Heart Fail. Clin.17, 357–367.
- Anker S.D., Butler J., Filippatos G.S., et al (2019). Evaluation of the effects of sodium–glucose co-transporter 2 inhibition with empagliflozin on morbidity and mortality in patients with chronic heart failure and a preserved ejection fraction: rationale for and design of the EMPEROR-Preserved Trial. Eur. J. Heart Fail.21, 1279–1287.
- Atila Uslu G., Uslu H. (2022). Evaluating the effects of Juglans regia L. extract on hyperglycaemia and insulin sensitivity in experimental type 2 diabetes in rat. Arch. Physiol. Biochem.128, 121–125.
- Ayton S.L., Gulsin G.S., McCann G.P., et al (2022). Epicardial adipose tissue in obesity-related cardiac dysfunction. Heart108, 339–344.
- Berthiaume J.M., Kurdys J.G., Muntean D.M., et al (2019). Mitochondrial NAD+/NADH redox state and diabetic cardiomyopathy. Antioxid. Redox Signal.30, 375–398.
- Bhatt D.L., Szarek M., Steg P.G., et al (2021). Sotagliflozin in patients with diabetes and recent worsening heart failure. N. Engl. J. Med.384, 117–128.
- Borlaug B.A. (2014). The pathophysiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol.11, 507–515.
- Borlaug B.A., Kass D.A. (2006). Mechanisms of diastolic dysfunction in heart failure. Trends Cardiovasc. Med.16, 273–279.
- Borlaug B.A., Olson T.P., Lam C.S., et al (2010). Global cardiovascular reserve dysfunction in heart failure with preserved ejection fraction. J. Am. Coll. Cardiol.56, 845–854.
- Bowen T.S., Herz C., Rolim N.P.L., et al (2018). Effects of endurance training on detrimental structural, cellular, and functional alterations in skeletal muscles of heart failure with preserved ejection fraction. J. Card. Fail.24, 603–613.
- Boyer J.K., Thanigaraj S., Schechtman K.B., et al (2004). Prevalence of ventricular diastolic dysfunction in asymptomatic, normotensive patients with diabetes mellitus. Am. J. Cardiol.93, 870–875.
- Braunwald E. (2019). Diabetes, heart failure, and renal dysfunction: the vicious circles. Prog. Cardiovasc. Dis.62, 298–302.
- Bullard K.M., Cowie C.C., Lessem S.E., et al (2018). Prevalence of diagnosed diabetes in adults by diabetes type—United States, 2016. MMWR Morb. Mortal. Wkly Rep.67, 359–361.
- Cavender M.A., Steg P.G., Smith S.C. Jr, et al (2015). Impact of diabetes mellitus on hospitalization for heart failure, cardiovascular events, and death: outcomes at 4 years from the reduction of atherothrombosis for continued health (REACH) registry. Circulation132, 923–931.
- Ceylan-Isik A.F., Fliethman R.M., Wold L.E., et al (2008). Herbal and traditional Chinese medicine for the treatment of cardiovascular complications in diabetes mellitus. Curr. Diabetes Rev.4, 320–328.
- Chirinos J.A., Segers P., Rietzschel E.R., et al (2013). Early and late systolic wall stress differentially relate to myocardial contraction and relaxation in middle-aged adults: the Asklepios study. Hypertension61, 296–303.
- Cleland J.G., Taylor J., Freemantle N., et al (2012). Relationship between plasma concentrations of N-terminal pro brain natriuretic peptide and the characteristics and outcome of patients with a clinical diagnosis of diastolic heart failure: a report from the PEP-CHF study. Eur. J. Heart Fail.14, 487–494.
- Cosentino F., Grant P.J., Aboyans V., et al (2020). 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J.41, 255–323.
- De Marco C., Claggett B.L., de Denus S., et al (2021). Impact of diabetes on serum biomarkers in heart failure with preserved ejection fraction: insights from the TOPCAT trial. ESC Heart Fail.8, 1130–1138.
- Dei Cas A., Fonarow G.C., Gheorghiade M., et al (2015). Concomitant diabetes mellitus and heart failure. Curr. Probl. Cardiol.40, 7–43.
- Del Buono M.G., Arena R., Borlaug B.A., et al (2019). Exercise intolerance in patients with heart failure: JACC state-of-the-art review. J. Am. Coll. Cardiol.73, 2209–2225.
- Dhulekar J., Simionescu A. (2018). Challenges in vascular tissue engineering for diabetic patients. Acta Biomater.70, 25–34.
- Dillmann W.H. (2019). Diabetic cardiomyopathy. Circ. Res.124, 1160–1162.
- Edelmann F., Gelbrich G., Dungen H.D., et al (2011). Exercise training improves exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction: results of the Ex-DHF (Exercise Training in Diastolic Heart Failure) pilot study. J. Am. Coll. Cardiol.58, 1780–1791.
- Ejiri K., Miyoshi T., Kihara H., et al (2020). Effect of luseogliflozin on heart failure with preserved ejection fraction in patients with diabetes mellitus. J. Am. Heart Assoc.9, e015103.
- Elsanhoury A., Nelki V., Kelle S., et al (2021). Epicardial fat expansion in diabetic and obese patients with heart failure and preserved ejection fraction—a specific HFpEF phenotype. Front. Cardiovasc. Med.8, 720690.
- Gaasch W.H., Zile M.R. (2004). Left ventricular diastolic dysfunction and diastolic heart failure. Annu. Rev. Med.55, 373–394.
- Ghosh S., Rodrigues B., Ren J. (2007). Rat models of cardiac insulin resistance. Methods Mol. Med.139, 113–143.
- Green J.B., Bethel M.A., Armstrong P.W., et al (2015). Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med.373, 232–242.
- Hegyi B., Bers D.M., Bossuyt J. (2019). CaMKII signaling in heart diseases: emerging role in diabetic cardiomyopathy. J. Mol. Cell. Cardiol.127, 246–259.
- Hogan P.G., Chen L., Nardone J., et al (2003). Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev.17, 2205–2232.
- Holman R.R., Bethel M.A., Mentz R.J., et al (2017). Effects of once-weekly exenatide on cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med.377, 1228–1239.
- Ipp E., Genter P., Childress K. (2017). Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med.376, 890–891.
- Jankauskas S.S., Kansakar U., Varzideh F., et al (2021). Heart failure in diabetes. Metabolism125, 154910.
- Jering K.S., Zannad F., Claggett B., et al (2021). Cardiovascular and renal outcomes of mineralocorticoid receptor antagonist use in PARAGON-HF. JACC: Heart Fail.9, 13–24.
- Jia G., Hill M.A., Sowers J.R. (2018). Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ. Res.122, 624–638.
- Kashiwagi A., Araki S., Maegawa H. (2021). Sodium–glucose cotransporter 2 inhibitors represent a paradigm shift in the prevention of heart failure in type 2 diabetes patients. J. Diabetes Investig.12, 6–20.
- Kessler E.L., Oerlemans M., van den Hoogen P., et al (2021). Immunomodulation in heart failure with preserved ejection fraction: current state and future perspectives. J. Cardiovasc. Transl. Res.14, 63–74.
- Kim A.H., Jang J.E., Han J. (2022). Current status on the therapeutic strategies for heart failure and diabetic cardiomyopathy. Biomed. Pharmacother.145, 112463.
- Kitzman D.W., Brubaker P.H., Herrington D.M., et al (2013). Effect of endurance exercise training on endothelial function and arterial stiffness in older patients with heart failure and preserved ejection fraction: a randomized, controlled, single-blind trial. J. Am. Coll. Cardiol.62, 584–592.
- Kitzman D.W., Brubaker P.H., Morgan T.M., et al (2010). Exercise training in older patients with heart failure and preserved ejection fraction: a randomized, controlled, single-blind trial. Circ. Heart Fail.3, 659–667.
- Klaiber M., Kruse M., Volker K., et al (2010). Novel insights into the mechanisms mediating the local antihypertrophic effects of cardiac atrial natriuretic peptide: role of cGMP-dependent protein kinase and RGS2. Basic Res. Cardiol.105, 583–595.
- Kobayashi E.H., Suzuki T., Funayama R., et al (2016). Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun.7, 11624.
- Kosmala W., Sanders P., Marwick T.H. (2017). Subclinical myocardial impairment in metabolic diseases. JACC Cardiovasc. Imag.10, 692–703.
- Kristensen S.L., Mogensen U.M., Jhund P.S., et al (2017). Clinical and echocardiographic characteristics and cardiovascular outcomes according to diabetes status in patients with heart failure and preserved ejection fraction. Circulation135, 724–735.
- Lalic N.M. (2021). Interdisciplinary assessment and diagnostic algorithm: the role of the diabetologist. Diabetes Res. Clin. Pract.176, 108850.
- Lam C.S., Brutsaert D.L. (2012). Endothelial dysfunction: a pathophysiologic factor in heart failure with preserved ejection fraction. J. Am. Coll. Cardiol.60, 1787–1789.
- Lascar N., Brown J., Pattison H., et al (2018). Type 2 diabetes in adolescents and young adults. Lancet Diabetes Endocrinol.6, 69–80.
- Lazo M., Halushka M.K., Shen L., et al (2015). Soluble receptor for advanced glycation end products and the risk for incident heart failure: the Atherosclerosis Risk in Communities Study. Am. Heart J.170, 961–967.
- Leite-Moreira A.F., Lourenco A.P., Roncon-Albuquerque R. Jr, et al (2012). Diastolic tolerance to systolic pressures closely reflects systolic performance in patients with coronary heart disease. Basic Res. Cardiol.107, 251.
- Li S.Y., Yang X., Ceylan-Isik A.F., et al (2006). Cardiac contractile dysfunction in Lep/Lep obesity is accompanied by NADPH oxidase activation, oxidative modification of sarco(endo)plasmic reticulum Ca2+-ATPase and myosin heavy chain isozyme switch. Diabetologia49, 1434–1446.
- Liang T., Gao F., Chen J. (2021). Role of PTEN-less in cardiac injury, hypertrophy and regeneration. Cell Regen.10, 25.
- Lin X., Wang Q., Sun S., et al (2020). Astragaloside IV promotes the eNOS/NO/cGMP pathway and improves left ventricular diastolic function in rats with metabolic syndrome. J. Int. Med. Res.48, doi: 10.1177/0300060519826848.
- Lopaschuk G.D., Verma S. (2016). Empagliflozin's fuel hypothesis: not so soon. Cell Metab.24, 200–202.
- Low Wang C.C., Hess C.N., Hiatt W.R., et al (2016). Clinical update: cardiovascular disease in diabetes mellitus: atherosclerotic cardiovascular disease and heart failure in type 2 diabetes mellitus—mechanisms, management, and clinical considerations. Circulation133, 2459–2502.
- Lund L.H., Claggett B., Liu J., et al (2018). Heart failure with mid-range ejection fraction in CHARM: characteristics, outcomes and effect of candesartan across the entire ejection fraction spectrum. Eur. J. Heart Fail.20, 1230–1239.
- Luo F., Das A., Chen J., et al (2019). Metformin in patients with and without diabetes: a paradigm shift in cardiovascular disease management. Cardiovasc. Diabetol.18, 54.
- MacDonald M.R., Petrie M.C., Varyani F., et al (2008). Impact of diabetes on outcomes in patients with low and preserved ejection fraction heart failure: an analysis of the Candesartan in Heart Failure: Assessment of Reduction in Mortality and Morbidity (CHARM) programme. Eur. Heart J.29, 1377–1385.
- Mahaffey K.W., Neal B., Perkovic V., et al (2018). Canagliflozin for primary and secondary prevention of cardiovascular events: results from the CANVAS program (Canagliflozin Cardiovascular Assessment Study). Circulation137, 323–334.
- Marso S.P., Daniels G.H., Brown-Frandsen K., et al (2016). Liraglutide and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med.375, 311–322.
- Martin N., Manoharan K., Thomas J., et al (2018). Beta-blockers and inhibitors of the renin–angiotensin aldosterone system for chronic heart failure with preserved ejection fraction. Cochrane Database Syst. Rev.6, CD012721.
- McDonagh T.A., Metra M., Adamo M., et al (2021). 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J.42, 3599–3726.
- McHugh K., DeVore A.D., Wu J., et al (2019). Heart failure with preserved ejection fraction and diabetes: JACC state-of-the-art review. J. Am. Coll. Cardiol.73, 602–611.
- McMurray J.J., Adamopoulos S., Anker S.D., et al (2012). ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012. Eur. J. Heart Fail.14, 803–869.
- Meza C.A., La Favor J.D., Kim D.H., et al (2019). Endothelial dysfunction: is there a hyperglycemia-induced imbalance of NOX and NOS? Int. J. Mol. Sci.20, 3775.
- Mihara K., Cao X.R., Yen A., et al (1989). Cell cycle-dependent regulation of phosphorylation of the human retinoblastoma gene product. Science246, 1300–1303.
- Molkentin J.D. (2004). Calcineurin–NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc. Res.63, 467–475.
- Mouton A.J., Li X., Hall M.E., et al (2020). Obesity, hypertension, and cardiac dysfunction: novel roles of immunometabolism in macrophage activation and inflammation. Circ. Res.126, 789–806.
- Mudaliar S., Alloju S., Henry R.R. (2016). Can a shift in fuel energetics explain the beneficial cardiorenal outcomes in the EMPA-REG OUTCOME study? A unifying hypothesis. Diabetes Care.39, 1115–1122.
- Mulvihill E.E., Varin E.M., Ussher J.R., et al (2016). Inhibition of dipeptidyl peptidase-4 impairs ventricular function and promotes cardiac fibrosis in high fat-fed diabetic mice. Diabetes65, 742–754.
- Muskiet M.H.A., Tonneijck L., Huang Y., et al (2018). Lixisenatide and renal outcomes in patients with type 2 diabetes and acute coronary syndrome: an exploratory analysis of the ELIXA randomised, placebo-controlled trial. Lancet Diabetes Endocrinol.6, 859–869.
- Nakagawa Y., Kuwahara K. (2020). Sodium–glucose cotransporter-2 inhibitors are potential therapeutic agents for treatment of non-diabetic heart failure patients. J. Cardiol.76, 123–131.
- Nakamura M., Sadoshima J. (2018). Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol.15, 387–407.
- Nakamura M., Sadoshima J. (2020). Cardiomyopathy in obesity, insulin resistance and diabetes. J. Physiol.598, 2977–2993.
- Nassif M.E., Windsor S.L., Borlaug B.A., et al (2021). The SGLT2 inhibitor dapagliflozin in heart failure with preserved ejection fraction: a multicenter randomized trial. Nat. Med.27, 1954–1960.
- Neal B., Perkovic V., Matthews D.R. (2017). Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med.377, 644.
- Obokata M., Reddy Y.N.V., Borlaug B.A. (2020). Diastolic dysfunction and heart failure with preserved ejection fraction: understanding mechanisms by using noninvasive methods. JACC Cardiovasc. Imag.13, 245–257.
- Oh J., Lee S.H., Lee C.J., et al (2021). Sodium–glucose co-transporter 2 inhibitors: a new path for heart failure treatment. Korean Circ. J.51, 399–408.
- Packer M., Anker S.D., Butler J., et al (2020). Cardiovascular and renal outcomes with empagliflozin in heart failure. N. Engl. J. Med.383, 1413–1424.
- Patel N., Ju C., Macon C., et al (2016). Temporal trends of digoxin use in patients hospitalized with heart failure: analysis from the American Heart Association Get With The Guidelines—Heart Failure Registry. JACC Heart Fail.4, 348–356.
- Paulus W.J., Dal Canto E. (2018). Distinct myocardial targets for diabetes therapy in heart failure with preserved or reduced ejection fraction. JACC Heart Fail.6, 1–7.
- Paulus W.J., Tschope C. (2013). A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol.62, 263–271.
- Phan T.T., Abozguia K., Nallur Shivu G., et al (2009). Heart failure with preserved ejection fraction is characterized by dynamic impairment of active relaxation and contraction of the left ventricle on exercise and associated with myocardial energy deficiency. J. Am. Coll. Cardiol.54, 402–409.
- Pillai V.B., Sundaresan N.R., Kim G., et al (2010). Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3–LKB1–AMP-activated kinase pathway. J. Biol. Chem.285, 3133–3144.
- Pitt B., Pfeffer M.A., Assmann S.F., et al (2014). Spironolactone for heart failure with preserved ejection fraction. N. Engl. J. Med.370, 1383–1392.
- Poornima I.G., Parikh P., Shannon R.P. (2006). Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ. Res.98, 596–605.
- Pries A.R., Badimon L., Bugiardini R., et al (2015). Coronary vascular regulation, remodelling, and collateralization: mechanisms and clinical implications on behalf of the working group on coronary pathophysiology and microcirculation. Eur. Heart J.36, 3134–3146.
- Pyun J.H., Kim H.J., Jeong M.H., et al (2018). Cardiac specific PRMT1 ablation causes heart failure through CaMKII dysregulation. Nat. Commun.9, 5107.
- Radholm K., Figtree G., Perkovic V., et al (2018). Canagliflozin and heart failure in type 2 diabetes mellitus: results from the CANVAS program. Circulation138, 458–468.
- Rangaswami J., Bhalla V., Blair J.E.A., et al (2019). Cardiorenal syndrome: classification, pathophysiology, diagnosis, and treatment strategies: a scientific statement from the American Heart Association. Circulation139, e840–e878.
- Ren D.Y., Zhang Y. (2018). Cardiovascular benefit of SGLT2 inhibitors in the therapeutics of diabetes mellitus: a close look beyond the horizon. Curr. Drug Targets19, 1051–1057.
- Ren J., Anversa P. (2015). The insulin-like growth factor I system: physiological and pathophysiological implication in cardiovascular diseases associated with metabolic syndrome. Biochem. Pharmacol.93, 409–417.
- Ren J., Ceylan-Isik A.F. (2004). Diabetic cardiomyopathy: do women differ from men? Endocrine25, 73–84.
- Ren J., Pulakat L., Whaley-Connell A., et al (2010). Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J. Mol. Med.88, 993–1001.
- Ren J., Wu N.N., Wang S., et al (2021). Obesity cardiomyopathy: evidence, mechanisms, and therapeutic implications. Physiol. Rev.101, 1745–1807.
- Ruiz S., Pergola P.E., Zager R.A., et al (2013). Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int.83, 1029–1041.
- Sakashita M., Tanaka T., Inagi R. (2021). Metabolic changes and oxidative stress in diabetic kidney disease. Antioxidants10, 1143.
- Sandesara P.B., O'Neal W.T., Kelli H.M., et al (2018). The prognostic significance of diabetes and microvascular complications in patients with heart failure with preserved ejection fraction. Diabetes Care.41, 150–155.
- Santos-Gallego C.G., Vargas-Delgado A.P., Requena-Ibanez J.A., et al (2021). Randomized trial of empagliflozin in nondiabetic patients with heart failure and reduced ejection fraction. J. Am. Coll. Cardiol.77, 243–255.
- Scirica B.M., Bhatt D.L., Braunwald E., et al (2013). Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N. Engl. J. Med.369, 1317–1326.
- Seferovic P.M., Paulus W.J. (2015). Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes. Eur. Heart J.36, 1718–1727, 1727a–1727c.
- Seferovic P.M., Petrie M.C., Filippatos G.S., et al (2018). Type 2 diabetes mellitus and heart failure: a position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail.20, 853–872.
- Seferovic P.M., Polovina M., Bauersachs J., et al (2019). Heart failure in cardiomyopathies: a position paper from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail.21, 553–576.
- Seo K., Rainer P.P., Shalkey Hahn V., et al (2014). Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc. Natl Acad. Sci.111, 1551–1556.
- Sickinghe A.A., Korporaal S.J.A., den Ruijter H.M., et al (2019). Estrogen contributions to microvascular dysfunction evolving to heart failure with preserved ejection fraction. Front. Endocrinol.10, 442.
- Solomon S.D., McMurray J.J.V., Anand I.S., et al (2019). Angiotensin–neprilysin inhibition in heart failure with preserved ejection fraction. N. Engl. J. Med.381, 1609–1620.
- Soro-Paavonen A., Zhang W.Z., Venardos K., et al (2010). Advanced glycation end-products induce vascular dysfunction via resistance to nitric oxide and suppression of endothelial nitric oxide synthase. J. Hypertens.28, 780–788.
- Stahrenberg R., Edelmann F., Mende M., et al (2010). Association of glucose metabolism with diastolic function along the diabetic continuum. Diabetologia53, 1331–1340.
- Sztechman D., Czarzasta K., Cudnoch-Jedrzejewska A., et al (2018). Aldosterone and mineralocorticoid receptors in regulation of the cardiovascular system and pathological remodelling of the heart and arteries. J. Physiol. Pharmacol.69, doi: 10.26402/jpp.2018.6.01.
- Tadic M., Sala C., Saeed S., et al (2021). New antidiabetic therapy and HFpEF: light at the end of tunnel? Heart Fail. Rev.doi: 10.1007/s10741-021-10106-9.
- Tan Y., Zhang Z., Zheng C., et al (2020). Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat. Rev. Cardiol.17, 585–607.
- Taqueti V.R., Solomon S.D., Shah A.M., et al (2018). Coronary microvascular dysfunction and future risk of heart failure with preserved ejection fraction. Eur. Heart J.39, 840–849.
- Tayanloo-Beik A., Roudsari P.P., Rezaei-Tavirani M., et al (2021). Diabetes and heart failure: multi-omics approaches. Front. Physiol.12, 705424.
- Tomasoni D., Adamo M., Lombardi C.M., et al (2019). Highlights in heart failure. ESC Heart Fail.6, 1105–1127.
- Tomasoni D., Fonarow G.C., Adamo M., et al (2022). Sodium–glucose co-transporter 2 inhibitors as an early, first line therapy in patients with heart failure and reduced ejection fraction. Eur. J. Heart Fail.24, 431–441.
- Triposkiadis F., Xanthopoulos A., Bargiota A., et al (2021). Diabetes mellitus and heart failure. J. Clin. Med.10, 3682.
- Tschope C., Elsanhoury A., Nelki V., et al (2021). Heart failure with preserved ejection fraction as a model disease for the cardio–pulmonary–renal syndrome: importance of visceral fat expansion as central pathomechanism. Internist62, 1141–1152.
- Tucker W.J., Nelson M.D., Beaudry R.I., et al (2016). Impact of exercise training on peak oxygen uptake and its determinants in heart failure with preserved ejection fraction. Card. Fail. Rev.2, 95–101.
- Uthman L., Baartscheer A., Bleijlevens B., et al (2018). Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na+/H+ exchanger, lowering of cytosolic Na+ and vasodilation. Diabetologia61, 722–726.
- van Heerebeek L., Hamdani N., Handoko M.L., et al (2008). Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation117, 43–51.
- van Woerden G., Gorter T.M., Westenbrink B.D., et al (2018). Epicardial fat in heart failure patients with mid-range and preserved ejection fraction. Eur. J. Heart Fail.20, 1559–1566.
- Wang D., Yin Y., Wang S., et al (2021). FGF1ΔHBS prevents diabetic cardiomyopathy by maintaining mitochondrial homeostasis and reducing oxidative stress via AMPK/Nur77 suppression. Signal Transduct. Target. Ther.6, 133.
- Wiviott S.D., Raz I., Bonaca M.P., et al (2019). Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med.380, 347–357.
- Wold L.E., Ceylan-Isik A.F., Ren J. (2005). Oxidative stress and stress signaling: menace of diabetic cardiomyopathy. Acta Pharmacol. Sin.26, 908–917.
- Xu G., Liu B., Sun Y., et al (2018). Prevalence of diagnosed type 1 and type 2 diabetes among US adults in 2016 and 2017: population based study. BMJ362, k1497.
- Xu H., Yu W., Sun S., et al (2021). TAX1BP1 protects against myocardial infarction-associated cardiac anomalies through inhibition of inflammasomes in a RNA34/MAVS/NLRP3-dependent manner. Sci. Bull.66, 1669–1683.
- Xu H.X., Cui S.M., Zhang Y.M., et al (2020). Mitochondrial Ca2+ regulation in the etiology of heart failure: physiological and pathophysiological implications. Acta Pharmacol. Sin.41, 1301–1309.
- Yamazaki T., Mimura I., Tanaka T., et al (2021). Treatment of diabetic kidney disease: current and future. Diabetes Metab. J.45, 11–26.
- Yancy C.W., Jessup M., Bozkurt B., et al (2017). 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation136, e137–e161.
- Yang L., Zhao D., Ren J., et al (2015). Endoplasmic reticulum stress and protein quality control in diabetic cardiomyopathy. Biochim. Biophys. Acta1852, 209–218.
- Zhang W., Liang J., Han P. (2021). Cardiac cell type-specific responses to injury and contributions to heart regeneration. Cell Regen.10, 4.
- Zhang Y., Sowers J.R., Ren J. (2018). Targeting autophagy in obesity: from pathophysiology to management. Nat. Rev. Endocrinol.14, 356–376.
- Zhao X., Ren Y., Li H., et al (2019). The effect of diuretics on patients with heart failure: a network meta-analysis: diuretics effect on heart failure patients. J. Pharm. Pharm. Sci.22, 270–280.
- Zhou H., Wang S., Zhu P., et al (2018). Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox. Biol.15, 335–346.
- Zinman B., Wanner C., Lachin J.M., et al (2015). Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med.373, 2117–2128.