Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease with multi-organ involvement, including the skin, heart, kidneys, joints, and central nervous system. Symptoms of SLE may vary from fever, fatigue, myalgia, and rash to the development of anaemia, thrombocytopenia, arthritis, nephritis, serositis, and neurological problems at a later stage []. Pulmonary arterial hypertension (PAH) is a rare but severe cardiopulmonary complication of SLE, which is associated with a significant disease burden [].
PAH is characterized by the narrowing of the pulmonary arterial bed due to extensive endothelial, adventitial, and smooth muscle dysfunction. Although there may be various causes of PAH (i.e. hereditary, drug/toxin induced, human immunodeficiency virus, portal hypertension, and congenital heart disease), it is commonly associated with connective tissue diseases (CTDs) including systemic sclerosis (SSc) and SLE []. The prevalence of PAH in SLE patients has been reported to vary widely, i.e. 0.5–43%; however, some recent studies have shown it to be in the range of 0.5–17.5% [, ]. Studies from Europe and the USA have shown SSc to be the most commonly associated CTD with PAH [], whereas Asian studies showed that PAH is more often a complication of SLE rather than SSc []. The time to diagnosis for PAH ranges between 4 and 5 years from SLE diagnosis, and it occurs predominantly in females [, , , ].
Late diagnosis and delayed treatment of SLE–PAH are associated with significant mortality and morbidity. PAH is one of the major causes of death in patients with SLE []. The 5-year survival among Asian patients (70%) is lower compared with European patients with SLE–PAH (84%) [, ]. Thus, the timely treatment of SLE–PAH patients is imperative.
The treatment goal in SLE–PAH is to improve lupus symptoms and reduce the pulmonary artery pressure. Several drug classes such as glucocorticoids, immunosuppressants, vasodilators, and endothelin receptor antagonists (ERAs) have been used for the treatment of SLE–PAH []. The recently published updated guidelines of the European Society of Cardiology (ESC)/European Respiratory Society (ERS) recommend that the treatment of CTD-associated PAH should be as per the specific aetiologies [, ]. For non-vasoreactive PAH patients without cardiopulmonary comorbidities presenting with low or intermediate risk, initial combination therapy with an ERA and a phosphodiesterase-5 (PDE5) inhibitor is recommended. In high-risk patients, a triple combination by adding prostacyclin is recommended. The addition of immunosuppressive therapy with glucocorticoids and cyclophosphamide is also commonly used to manage SLE–PAH patients in clinical practice [].
SLE is one of the most common CTDs in patients developing CTD-associated PAH []. Despite this, few real-world information on the patient demographics and disease characteristics of SLE–PAH is available, along with the treatment strategies adopted to manage these patients in Japan. We aimed to understand the prevalence, incidence, patient demographics and characteristics, and treatment patterns to manage SLE–PAH patients in Japan in real-world clinical practice using a well-organized Japanese database.
Materials and methods
Study design
This was a retrospective, observational study that utilized anonymized patient data from Japan’s Medical Data Vision (MDV) database. The objectives of this study were (1) to describe the demographics and clinical characteristics of SLE–PAH patients in Japan (including prevalence and cumulative incidence of PAH among SLE patients) and (2) to describe the current treatment patterns by line of therapy following PAH diagnosis among SLE patients. The data collection period for the study was from April 2008 to September 2020.
Data source
The MDV database is Japan’s largest hospital-based claims database, with >10 years of data collected. The database includes ∼40 million patients as of November 2022, of whom ∼35% were ≥65 years of age. This database covers 460 hospitals and approximately one-quarter of acute hospitalizations in Japan []. The MDV database supports the Diagnosis Procedure Combination (DPC) hospitals for the collection and reporting of administrative data and provides anonymized pharmacy claims and hospital-based and insurance-related data.
Study population
Patient data in MDV are documented as per the International Classification of Disease (ICD) coding system. It contains ICD-10 codes for SLE (M32.1 and M32.9) and PAH (I27.0). The ICD-10 code for PAH (I27.0) also covers other pulmonary diseases in addition to PAH; thus, disease code 8844804 that is used for the diagnosis of PAH in Japan was used to identify PAH patients instead. The MDV dataset was selected due to its large sample size, i.e. ∼70,000 patients with ICD-10 codes of M32.1 and M32.9.
Adults (≥18 years of age) who had at least two claims with ICD-10 (SLE: M32.1 and M32.9) were selected. For the first objective, patients were excluded further if patients did not have at least 6 months of continuous enrolment in the MDV database before their first claim of SLE, also referred to as washout period. SLE patients that subsequently developed PAH were identified using at least two claims. A history of at least one record of right heart catheterization or echocardiography prior to the PAH diagnosis code was also required. For the second objective, all adult patients with SLE–PAH were included without a washout period for first SLE diagnosis because PAH diagnosis after SLE is the point of interest and the point of first SLE diagnosis was not necessary for the analysis. The patients were stratified by SLE severity as mild (none of the criteria for moderate and severe met), moderate [had ≥1 filled prescription for an oral glucocorticoid of ≥7.5 to <60 mg/day or for an immunosuppressive agent (other than cyclophosphamide), intravenous immunoglobulin, belimumab, or calcineurin inhibitors], and severe (had ≥1 filled prescriptions for cyclophosphamide, rituximab, or oral glucocorticoids with a prednisone equivalent dose of ≥60 mg/day or intravenous glucocorticoids) []. Detailed severity algorithm is mentioned in Supplementary Table S2. All patients were followed up until death or the end of follow-up.
According to the Ethical Guidelines for Epidemiological Research issued by the Japanese Ministry of Health, Labour and Welfare, informed consent and ethics approval were not required for this study that used anonymized patient data from the MDV database [].
Outcomes
Patient demographics and clinical characteristics of newly diagnosed SLE patients were collected including gender, age at diagnosis of SLE, disease severity of SLE, and comorbidities (including diabetes, hypertension, dyslipidaemia, and chronic kidney disease) at the time of SLE diagnosis. For SLE–PAH patients, age at diagnosis of PAH and duration from diagnosis of SLE to PAH were included.
For treatment patterns, information was collected for SLE-specific treatments and vasodilators. SLE-specific treatments included antimalarial (hydroxychloroquine), glucocorticoids oral/parenteral (prednisolone and methylprednisolone), immunosuppressants (azathioprine, ciclosporin, cyclophosphamide, methotrexate, mycophenolate mofetil, and tacrolimus), and monoclonal antibodies (belimumab), whereas vasodilators comprised ERAs (ambrisentan, bosentan, and macitentan), nitric oxide (NO; sildenafil, tadalafil, and riociguat), and prostacyclins (beraprost, epoprostenol, iloprost, treprostinil, and selexipag).
Statistical analysis
Baseline demographics and treatment patterns were summarised descriptively. Continuous variables were expressed as the mean, standard deviation, and median. Categorical variables were expressed as a number and percentage. The characteristics of SLE patients who developed PAH were compared with those who did not develop PAH using independent Student’s t-test or Mann–Whitney’s U-test for continuous variables and Pearson’s chi-squared test or Fisher’s exact test for categorical variables. The difference between the two groups was considered statistically significant at a P-value of <.05.
The prevalence of PAH among SLE patients used the study period prevalence proportion (%), calculated by the numerator of all SLE–PAH patients divided by the denominator of all SLE patients calculated as the annual average number of corresponding patients in the database time period. A sensitivity analysis for the prevalence of PAH was conducted using the mid-interval year (2014) of the database time period divided by the denominator of all SLE patients in the mid-interval year (2014) of the database time period. The Kaplan–Meier method (K–M curve) was used to estimate the cumulative incidence of PAH among the patients with SLE. Patients were censored at the end of follow-up or death.
Treatment patterns among SLE–PAH patients were described as the use of SLE-specific treatments and vasodilators as monotherapy and in combinations. Vasodilators were also summarized by lines of treatment (first, second, and third lines). The patient was considered to change the ‘line of treatment’ each time the patient purchased a different ‘drug group’ for the first time. For example, if the patient was purchasing a drug from the NO drug group, the first time the patient purchased a drug from the ERA drug group, the patient was considered to have changed the line of treatment. The duration of transition from the main drug class in the first line to treatment to the predominant drug class in the second line to treatment was also analysed.
Results
Demographics and baseline characteristics
Patient selection cohort is presented in Figure 1. A total of 29,077 patients with SLE met the criteria for the first objective and were included in the analysis. Of these, 114 patients with SLE developed PAH. The characteristics of SLE–PAH patients versus those who did not develop PAH (SLE without PAH) are shown in Table 1. In comparison to patients with SLE alone, patients with SLE–PAH were predominantly females (88% vs. 72%; P < .001), were younger at the time of SLE diagnosis (mean age: 53 vs. 56 years; P = .070), had longer follow-up (mean duration: 3.8 vs. 2.2 years; P < .001), had more comorbidity of diabetes (59% vs. 49%; P = .052), and had more severe SLE (61% vs. 25%; P < .001). There was no significant difference in comorbidities of hypertension, dyslipidaemia, and kidney disease between the two groups of patients (P > .7) (Table 1).
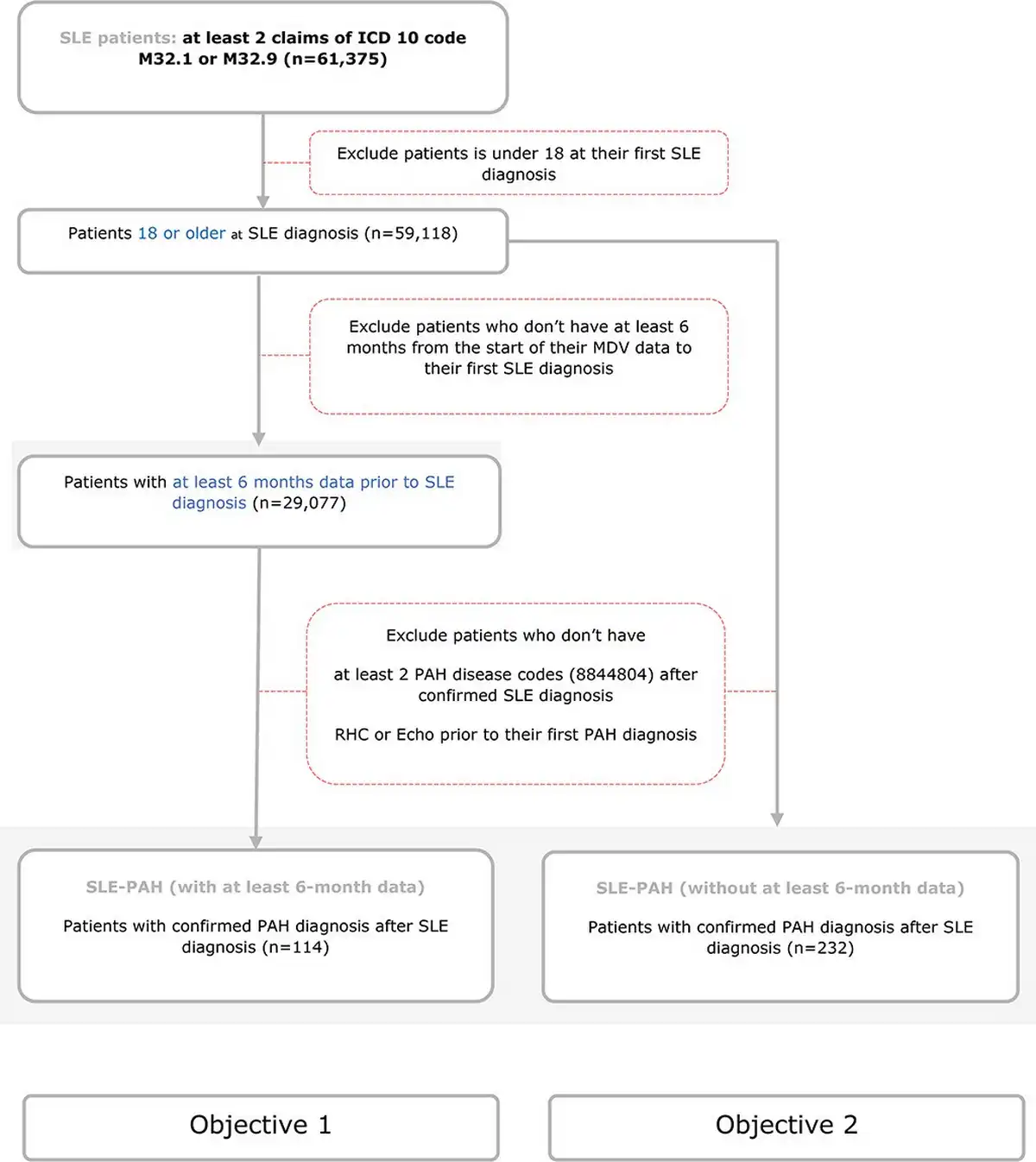
Figure 1
Patient selection cohort.
This figure demonstrates the criteria for patient selection for both cohorts. Cohort 1 included patients who had at least 6 months of continuous enrolment in the MDV database before their first claim of SLE. Cohort 2 included all patients with a diagnosis of SLE–PAH.

Prevalence, incidence, and time to PAH diagnosis
The prevalence of PAH among SLE patients was reported to be 0.392% during the study period between April 2008 and September 2020 in the MDV database. In the sensitivity analysis, the prevalence was found to be 0.241% in mid-interval year (2014).
The cumulative incidence of PAH among SLE patients was estimated to be 0.53% at 3 years and 0.77% at 5 years (Figure 2). Nearly half of the SLE–PAH patients (49%) developed PAH within 1 year of their SLE diagnosis (Figure 3).
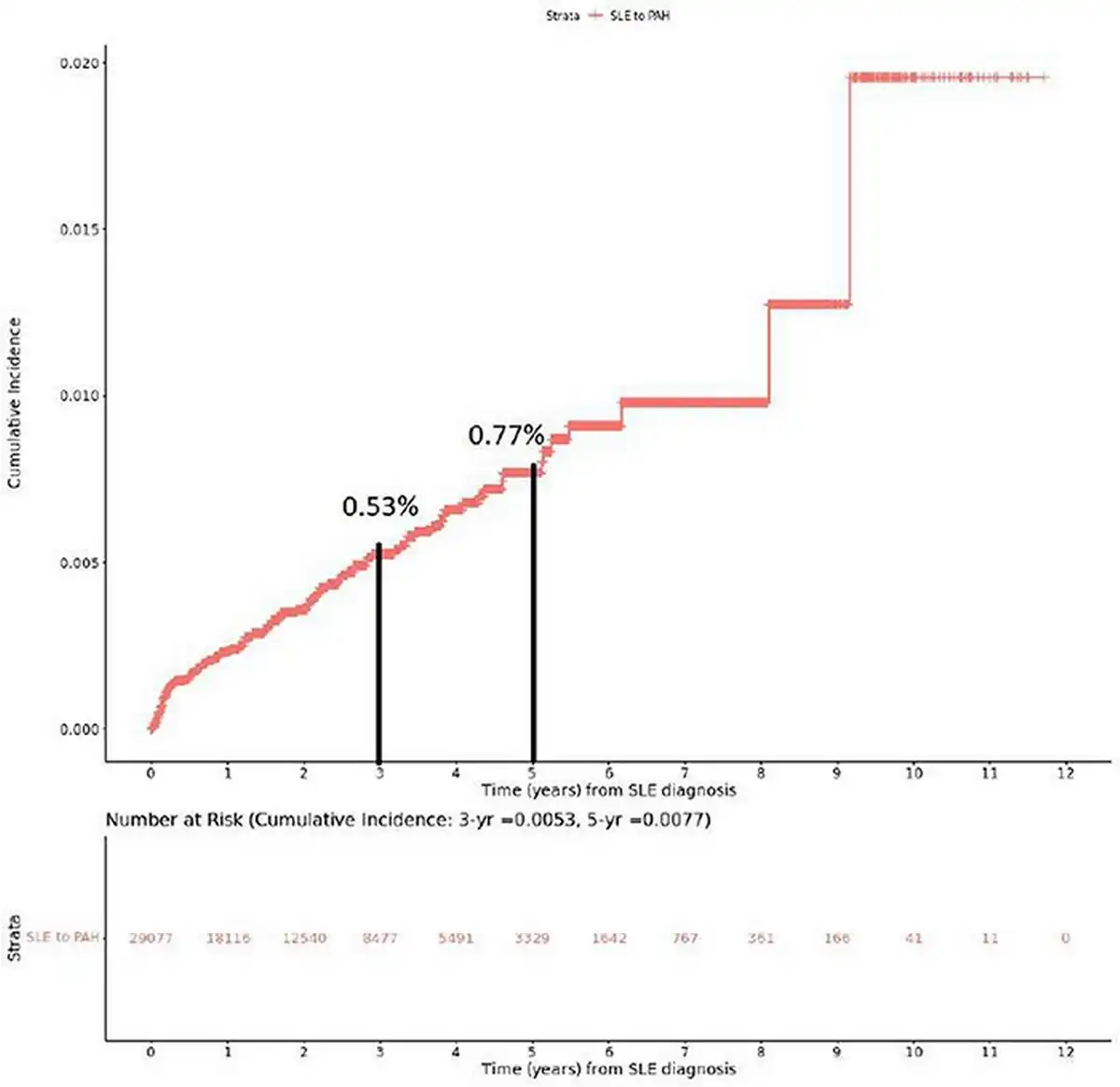
Figure 2
Cumulative incidence of PAH among SLE patients.
This figure demonstrates the cumulative incidence of PAH in patients who had at least 6 months of continuous enrolment in the MDV database before their first claim of SLE. Incidence rates at 3- and 5-year time points are also demonstrated.
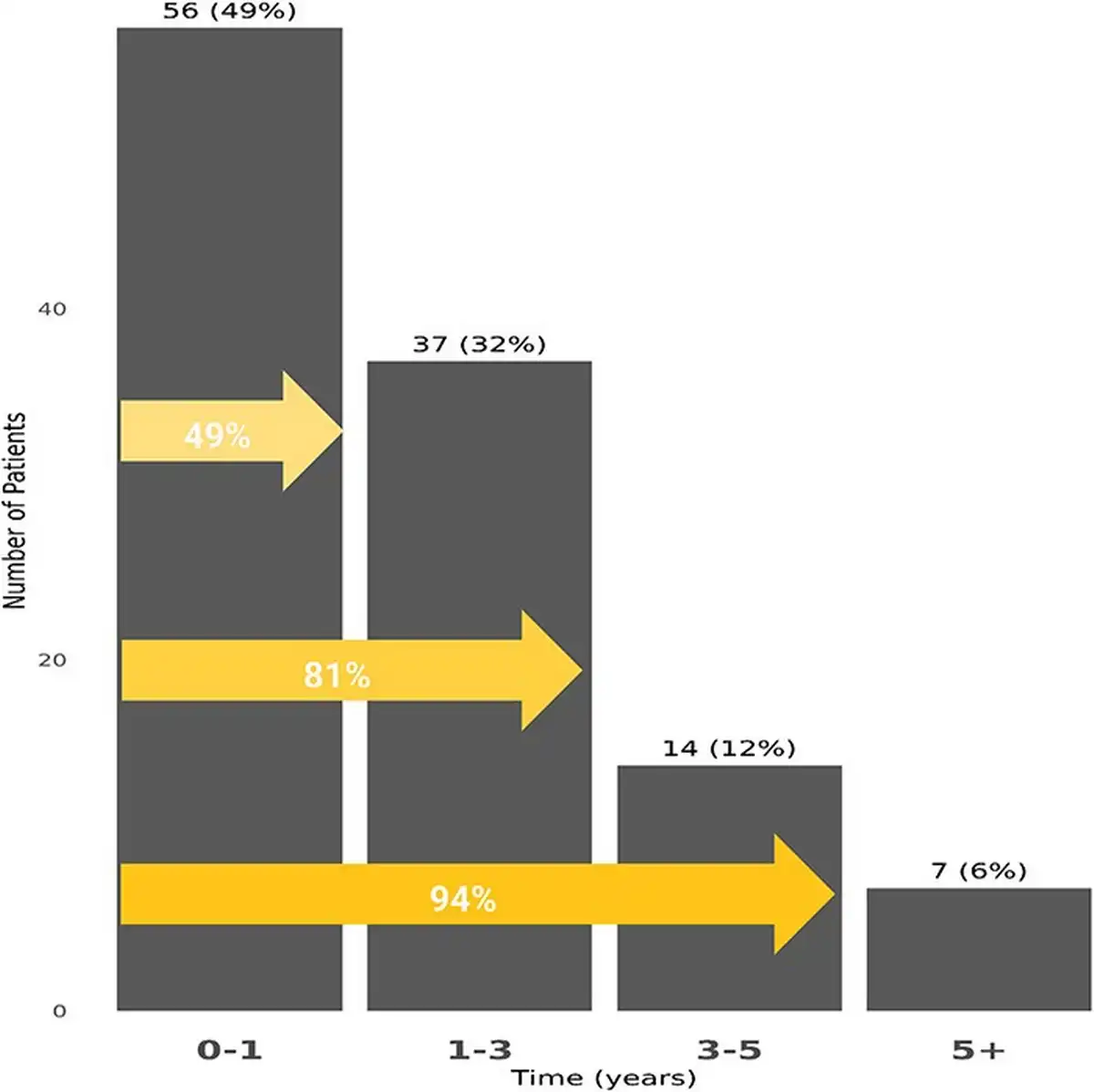
Figure 3
The K–M curve for cumulative incidence and time of PAH among SLE patients.
This figure shows the K–M curve for cumulative incidence and time of PAH. The cumulative incidence and time of PAH were evaluated for patients at least 6 months of continuous enrolment in the MDV database before their first claim of SLE. Patients were censored at the end of follow-up or death.
Treatment patterns in SLE–PAH
A total of 232 SLE–PAH patients who also included patients without at least 6 months of continuous enrolment in MDV for first SLE diagnosis were identified for the objective related to treatment patterns from the time of PAH diagnosis. The demographics and characteristics of these patients are presented in Supplementary Table S1.
SLE and PAH drugs
In the overall categories of SLE and PAH drugs, glucocorticoids (58%) were predominantly used in the first line of therapy among SLE–PAH patients and their use decreased rapidly in second- (27%) and third-line (8%) therapies. Of note, PAH drugs were used in approximately a quarter of patients in the first line (27%), but their use increased sharply in the second (38%) and third lines (44%) of therapies. The same trend was observed regarding the use of immunomodulators (Table 2). The information on transition from glucocorticoids in the first line to PAH drugs in the second line was available for 66 patients, which showed that the median time for transition from first-line glucocorticoids to second-line PAH drugs among these patients was 1.6 months [mean (Q1, Q3): 13.3 (0.0, 20.9) months].
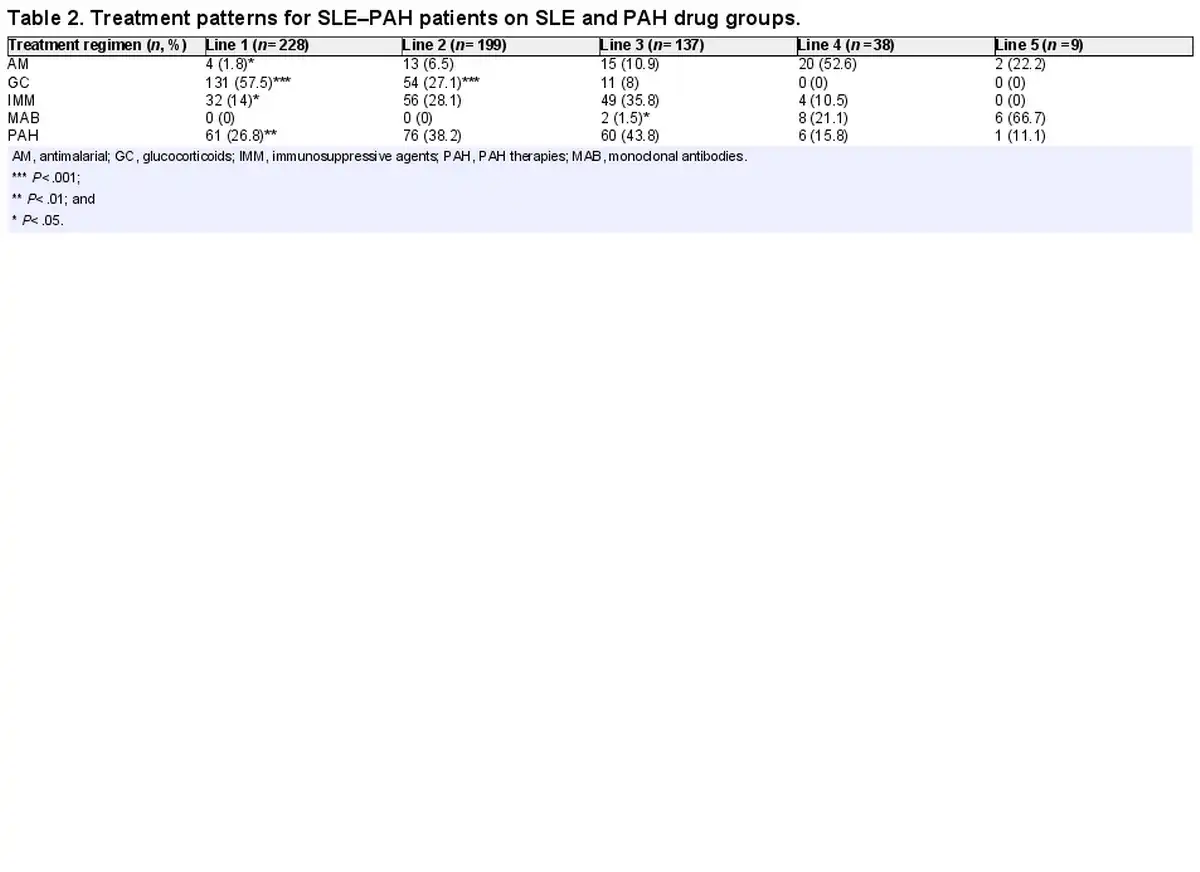
Within combination therapies that were prescribed for SLE–PAH patients, a combination of glucocorticoids and immunosuppressants was the most frequently used combination in the first line (19%), followed by the combinations of PAH, glucocorticoids, and immunomodulators (17%), and PAH and glucocorticoids (16%). In the second-line treatment, a combination of PAH, glucocorticoids, and immunomodulators (25%) was the most commonly prescribed combination, followed by the combination of PAH and glucocorticoids (22%). In the third line of therapy, 17% of all patients were prescribed a combination of PAH and glucocorticoids (Table 3).
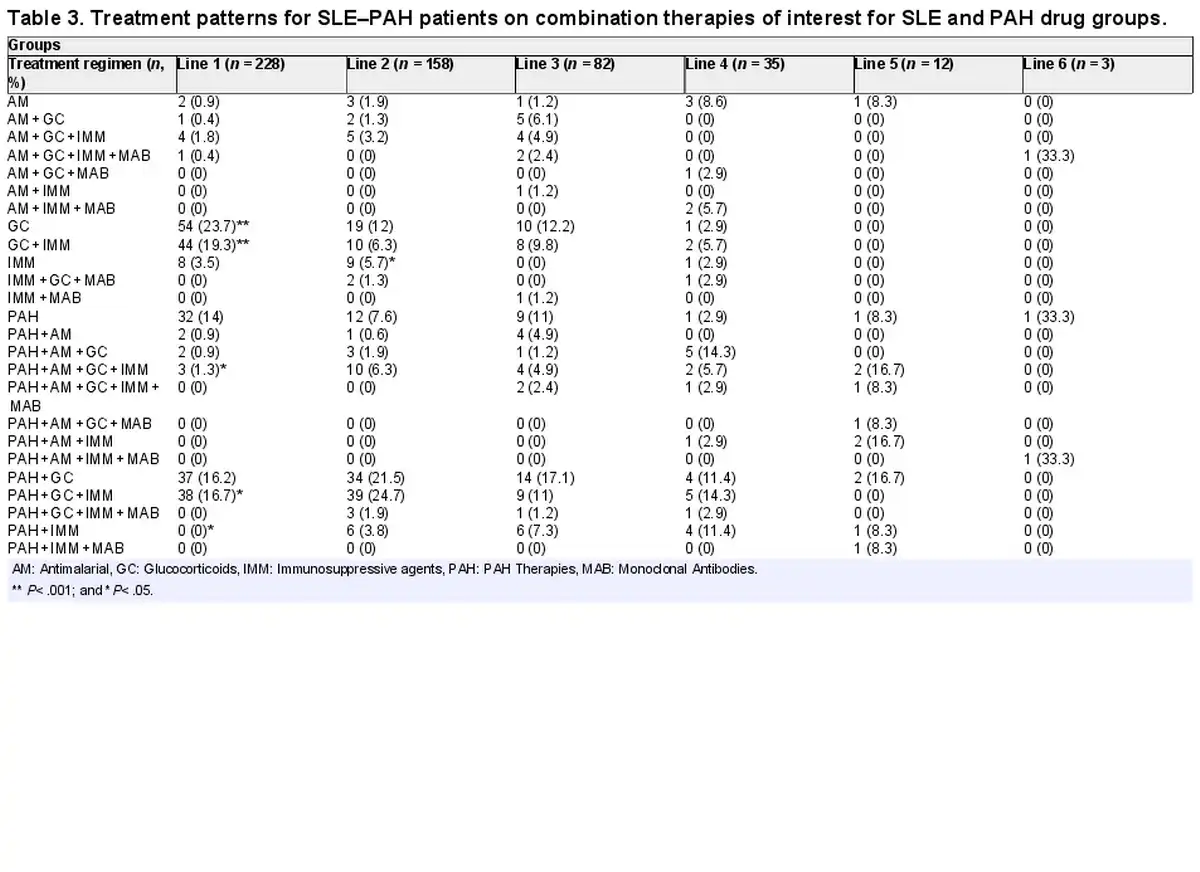
Vasodilators
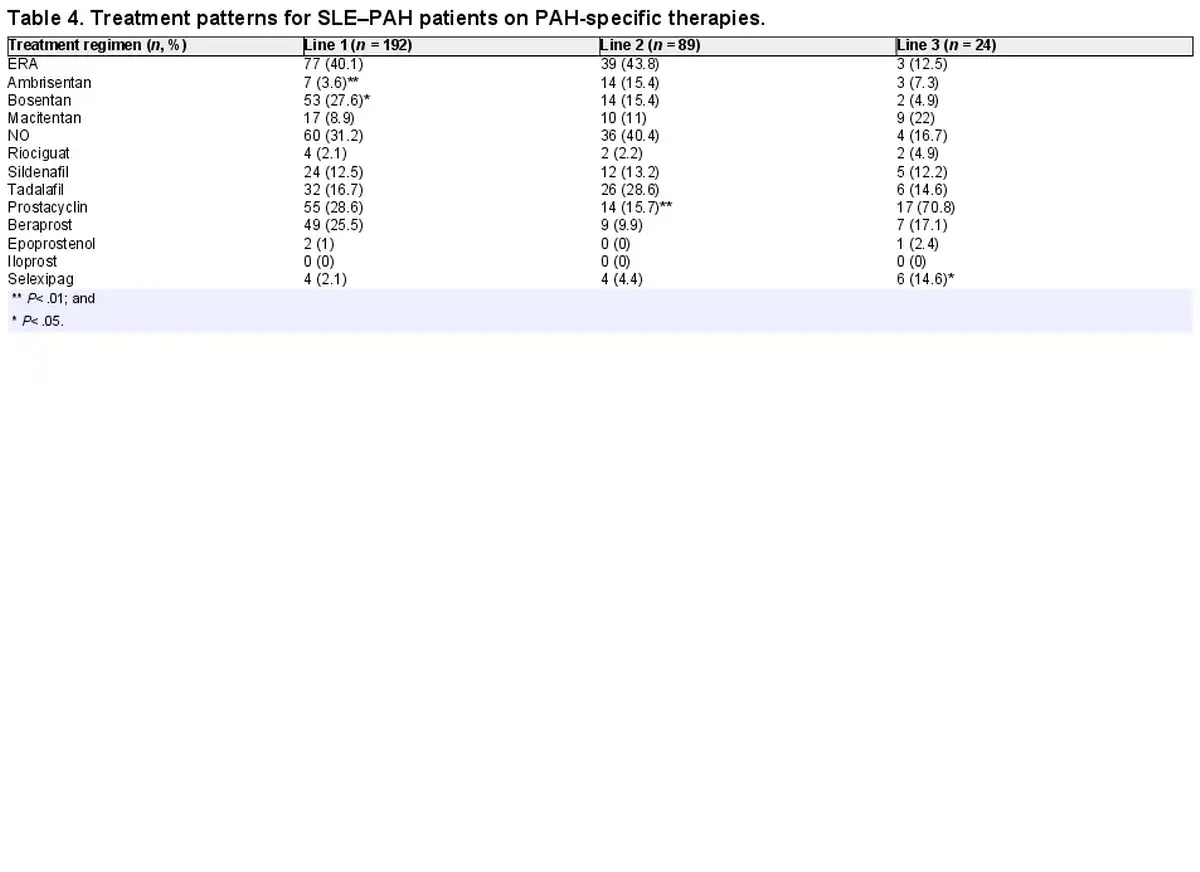
Within vasodilators, in the first- and second-line monotherapies, ERA and NO were the most frequently prescribed drugs (40% and 31%, respectively) in >70% of patients. Of note, prostacyclins were the treatment of choice in the third-line therapy (71%; Table 4). A preference for monotherapies was observed for the first-line therapies, whereas combination therapies were preferred in subsequent lines of treatment. Drug-specific analysis of PAH-specific drugs showed that bosentan (28%) and beraprost (26%) were the first-line drugs of choice, whereas tadalafil (27%) was the most prescribed drug in the second-line treatment. Macitentan and selexipag was the drug of choice in the third and fourth lines of therapy, respectively (Table 4).
Discussion
This retrospective, observational study was conducted to identify patient characteristics, prevalence, and clinical practice patterns among patients with SLE–PAH in a real-world setting in Japan using the MDV database. The overall findings indicated that of the 29,077 patients identified with newly diagnosed SLE, 114 developed PAH, with a period prevalence of 0.392% in Japan, and about half of these patients developed PAH within 1 year of SLE diagnosis. Patients with SLE–PAH were aged 53 years on average at SLE diagnosis, with a predominance of females, and a majority had severe grade SLE. The clinical practice trend revealed that glucocorticoids were the dominant treatment used in the first line among SLE–PAH patients, followed by vasodilators. Within vasodilators, ERA dominated the prescription in the first and second lines, followed by NO. More than half of the patients were prescribed bosentan and beraprost in the first line. We found that patients with SLE–PAH were predominantly females, which is consistent with worldwide data.
The prevalence in this study was 0.392%, while cumulative 3-year incidence was 0.53% and 5-year incidence was 0.77%. Almost half of all the patients with SLE–PAH developed PAH within 1 year of SLE diagnosis. As per the Japanese clinical practice guidelines for PAH associated with CTDs, PAH is often diagnosed at the onset of SLE or at the time of increased SLE disease activity. Hence, the guidelines recommend screening PAH at the onset of SLE, as well as during SLE flare-ups []. The reported prevalence of PAH in SLE patients ranges from 0.5% to 14% [, ]. In a Taiwanese study, 5-year incidence was reported to be 1.8% []. The lower incidence and prevalence observed in the present study can be explained due to the short median follow-up period, which was only 1.58 years, indicating a higher censorship. Further explanation could be that the MDV database is restricted only to the DPC hospitals; thus, any movement of the patients outside the DPC hospital may have affected the prevalence/incidence. The true incidence of SLE–PAH can be higher; however, earlier PAH diagnosis may not be feasible because of the extensive diagnostic testing required to identify such patients [, ].
The general treatment goal for SLE–PAH per the 2017 Japanese and 2022 ESC/ERS guidelines is to maintain the functional class and to move the patient to a lower risk level [, ]. Treatment patterns in the present study demonstrated a strong preference for glucocorticoid monotherapy as a first line of treatment, which was prescribed in more than half of all the SLE–PAH patients. In subsequent lines of therapies, vasodilators and immunosuppressants became more prominent. This is in line with the 2017 Japanese guidelines for the treatment of PAH, which suggest focusing initial therapy on mitigation of the underlying disease (SLE). As PAH is an organ-specific manifestation of SLE, it is highly likely that immunological mechanism is involved in its pathophysiology []. Immunosuppressants and glucocorticoids or their combination is recommended depending on the severity of the disease. The 2022 ESC/ERS guidelines also suggest immunosuppressive therapy as a combination of glucocorticoids and cyclophosphamide []. Furthermore, a very limited use of hydroxychloroquine was observed in this study. This reduced use, compared with clinical experience, may be attributable to the fact that the drug only became available in 2015 in Japan [].
Within vasodilators, the majority of the patients with SLE–PAH received ERA drugs, followed by NO drugs and prostacyclin analogues. The same pattern was observed in the second-line treatment. While in the third line, prostacyclin analogues were most prescribed. Bosentan and beraprost were most prescribed for the first line, while tadalafil had a large share in the second line. In the third and fourth lines of treatment, macitentan and selexipag were dominant, respectively. The preference for these therapies may be linked to the fact that selexipag was launched in the Japanese market in late 2016 []. By the third line, a majority of patients were switched to macitentan (n = 9), compared with bosentan, where only two patients were receiving bosentan. The use of bosentan may have been influenced by multiple factors such as the presence of Raynaud’s phenomenon or digital ulcers complicated by SSc; however, further analysis into the aspects affecting treatment choice can be a future research question and was out of scope for this analysis.
The high proportion of patients prescribed on first-line bosentan and the treatment shift from bosentan to macitentan appear due to the timing of macitentan introduction to Japan, which was launched in mid-2015 (Table 4). The MDV database covers a few non-specialist PAH facilities. According to a study conducted by Tamura et al., post-2016, 83.5% of the patients with CTD–PAH were prescribed macitentan []. As shown in the SERAPHIN study, macitentan was associated with reduced mortality and morbidity in PAH patients, regardless of previous treatment status []. Switching from bosentan to macitentan is also shown to reduce the risk of hepatotoxicity, which is a major limitation of bosentan therapy [, ]. Another study demonstrated that macitentan was associated with an 18% lower risk of mortality than ambrisentan and 39% lower than bosentan []. In the recent 2022 ESC/ERS guidelines, an initial combination of ERA and PDE5 inhibitors is also recommended in patients with no cardiovascular comorbidities and at low to intermediate risk. For the patients in the high-risk group, initial triple-combination therapy adding an intravenous or subcutaneous prostacyclin analogue is recommended [].
Treatment shift from beraprost in the first line to selexipag in the fourth line also corresponds with selexipag availability in Japan, which was introduced at the end of 2016. In clinical trials, selexipag was shown to be more efficacious than other prostanoid therapeutics, such as beraprost and treprostinil, while showing comparable safety. Selexipag is the only orally administered therapy that targets the prostacyclin pathway, whereas beraprost is a PGI2 analogue, which leads to unwanted off-target effects. Beraprost also has a short-term effect, which is lost by 1 year of treatment. Consequently, the drug is not available in Europe or the USA []. The 2022 ESC/ERS guidelines do not provide any recommendations for the use of beraprost in clinical settings. However, the 2022 guidelines grant the highest level of recommendation for selexipag [].
There are certain limitations to this study. First, the claims data have inherent limitations including potential miscoding to the condition and/or missing data. The data analysed in the present study were longitudinal patient data which are available only within the same DPC hospital. In case of movement of patients outside the same hospital, their data could not be tracked, and those patients were considered ‘lost to follow-up’ and the median follow-up (1.58 years) is relatively short. Another limitation is that there were no clinical details for disease severity. An algorithm using medication usage and the presence of comorbidities was utilized to categorize the SLE severity at diagnosis. Furthermore, only the data available after the patients were registered in the MDV database were utilized for the analyses; hence, treatment patterns may change if the historical patient data are taken into consideration (e.g. percentage of patients receiving glucocorticoids as first-line treatment would change if historical data were considered). Given the limited number of patients identified with PAH, the interpretation of findings should be done cautiously.
Conclusion
PAH development was observed predominantly in females, younger and more severe SLE patients. The initial treatment goal among SLE–PAH patients appears to be mitigation of SLE, which is in line with the Japanese guidelines for the treatment of PAH, whereas vasodilators are preferred more in the subsequent lines of therapies in Japan’s real-world clinical practice. For optimised treatment outcomes, treatment patterns must be aligned with the 2022 ESC/ERS guidelines. Given that only a small number of patients with SLE–PAH were identified in this study, future research with a large sample size is recommended.
References
- [1]. Kamen DL, Strange C. Pulmonary manifestations of systemic lupus erythematosus. Clin Chest Med 2010;31:479–88.
- [2]. Leuchten N, Milke B, Winkler-Rohlfing B et al. Early symptoms of systemic lupus erythematosus (SLE) recalled by 339 SLE patients. Lupus 2018;27:1431–6.
- [3]. Asif S, Rasheed A, Mahmud T-H et al. Frequency and predictors of pulmonary hypertension in patients with systemic lupus erythematosus. Pakistan J Med Sci 2019;35:86–9.
- [4]. Chen H-A, Hsu T-C, Yang S-C et al. Incidence and survival impact of pulmonary arterial hypertension among patients with systemic lupus erythematosus: a nationwide cohort study. Arthritis Res Ther 2019;21:82.
- [5]. Zanatta E, Polito P, Famoso G et al. Pulmonary arterial hypertension in connective tissue disorders: pathophysiology and treatment. Exp Biol Med (Maywood) 2019;244:120–31.
- [6]. Arnaud L, Agard C, Haroche J et al. Hypertension artérielle pulmonaire associée au lupus systémique. Rev Méd Interne 2011;32:689–97.
- [7]. Chung L, Liu J, Parsons L et al. Characterization of connective tissue disease-associated pulmonary arterial hypertension from REVEAL. Chest 2010;138:1383–94.
- [8]. Jing Z-C, Xu X-Q, Han Z-Y et al. Registry and survival study in Chinese patients with idiopathic and familial pulmonary arterial hypertension. Chest 2007;132:373–9.
- [9]. Feng Y, Wang J, Lei Y et al. AB0818 The comparative study of systemic sclerosis and systemic lupus erythematosus-associated pulmonary arterial hypertension. Scleroderma, myositis and related syndromes. Ann Rheum Dis 2018;1539.
- [10]. Hao Y-J, Jiang X, Zhou W et al. Connective tissue disease-associated pulmonary arterial hypertension in Chinese patients. Eur Respir J 2014;44:963–72.
- [11]. Lin C-Y, C-h K, Hsu C-Y et al. Epidemiology and mortality of connective tissue disease-associated pulmonary arterial hypertension: a national cohort study in Taiwan. Semin Arthritis Rheum 2020;50:957–62.
- [12]. Hachulla E, Jais X, Cinquetti G et al. Pulmonary arterial hypertension associated with systemic lupus erythematosus. Chest 2018;153:143–51.
- [13]. Humbert M, Kovacs G, Hoeper MM et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2022;43:3618–731.
- [14]. Galiè N, Humbert M, Vachiery J-L et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 2016;37:67–119.
- [15]. Zhao J, Wang Q, Liu Y et al. Clinical characteristics and survival of pulmonary arterial hypertension associated with three major connective tissue diseases: a cohort study in China. Int J Cardiol 2017;236:432–7.
- [16]. About MDV Database|Medical Data Vision Co., Ltd. n.d. https://en.mdv.co.jp/about-mdv-database/ (19 April 2023, date last accessed).
- [17]. Garris C, Jhingran P, Bass D et al. Healthcare utilization and cost of systemic lupus erythematosus in a US managed care health plan. J Med Econ 2013;16:667–77.
- [18]. Ethical Guidelines for Epidemiological Research. [Internet] Ministry of Health, Labour, and Welfare. Ethical Guidelines for Medical and Health Research Involving Human Subjects. 2015. https://www.mhlw.go.jp/file/06-Seisakujouhou-10600000-Daijinkanboukouseikagakuka/0000080278.pdf (18 April 2023, date last accessed).
- [19]. Pulmonary arterial hypertension associated with connective tissue disease – clinical practice guidelines. Japanese Pulmonary Circulation and Pulmonary Hypertension Society. 2019. http://jpcphs.org/pdf/guideline/ketugou_guideline.pdf (18 April 2023, date last accessed).
- [20]. Pérez-Peñate GM, Rúa-Figueroa I, Juliá-Serdá G et al. Pulmonary arterial hypertension in systemic lupus erythematosus: prevalence and predictors. J Rheumatol 2016;43:323–9.
- [21]. Li M, Wang Q, Zhao J et al. Chinese SLE Treatment and Research group (CSTAR) registry: II. Prevalence and risk factors of pulmonary arterial hypertension in Chinese patients with systemic lupus erythematosus. Lupus 2014;23:1085–91.
- [22]. Fukuda K, Date H, Doi S et al. Guidelines for the treatment of pulmonary hypertension (JCS 2017/JPCPHS 2017). Circ J 2019;83:842–945.
- [23]. Cutaneous lupus erythematosus and systemic lupus erythematosus drugs Launch of Plaquenil® Tablets 200 mg. Sanofi Japan. https://www.sanofi.co.jp/dam/jcr:c8ddf93f-a6a9-4a96-a0d8-645e12668aa8/201509072.pdf (18 April 2023, date last accessed).
- [24]. Nippon Shinyaku. Launch of Uptravi® tablet for the treatment of pulmonary arterial hypertension. IR News 2016. n.d. https://www.nippon-shinyaku.co.jp/english/ir/ir_news.php?id=992 (Accessed 19 April 2023, date last accessed).
- [25]. Tamura Y, Kumamaru H, Inami T et al. Changes in the characteristics and initial treatments of pulmonary hypertension between 2008 and 2020 in Japan. JACC Asia 2022;2:273–84.
- [26]. Pulido T, Adzerikho I, Channick RN et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013;369:809–18.
- [27]. Tynan T, Hird K, Hannon T et al. Pulmonary arterial hypertension outcomes upon endothelin-1 receptor antagonist switch to macitentan. J Int Med Res 2019;47:2177–86.
- [28]. Aypar E, Alehan D, Karagöz T et al. Clinical efficacy and safety of switch from bosentan to macitentan in children and young adults with pulmonary arterial hypertension: extended study results. Cardiol Young 2020;30:681–5.
- [29]. Benza RL, Lickert CA, Xie L et al. Comparative effectiveness of endothelin receptor antagonists on mortality in patients with pulmonary arterial hypertension in a US Medicare population: a retrospective database analysis. Pulm Circ 2020;10:1–11.
- [30]. Coghlan JG, Picken C, Clapp LH. Selexipag in the management of pulmonary arterial hypertension: an update. Drug Healthc Patient Saf 2019;11:55–64.