Introduction
Stroke is the second leading cause of death and the third leading cause of disability worldwide. During the past three decades, global stroke incidence increased by 70%, its mortality increased by 43%, and its disability-adjusted life years lost increased by 32%, with a greater increase in financial burden in all countries [, ]. According to the American Heart Association and the American Stroke Association, the total annual direct medical costs attributable to stroke are projected to increase to $184.1 billion by the year 2030 []. Therefore, exploration of the pathophysiological mechanisms of stroke and development of appropriate therapeutic strategies is a key imperative.
Dysfunction of energy metabolism, toxicity of excitatory amino acids, cytoplasmic calcium influx, apoptosis, and oxidative stress have all been suggested as the underlying mechanisms of poststroke neuronal injury, including ischemic stroke and intracerebral hemorrhage (ICH) [, ]. Immunity and inflammatory reactions play a vital role in the pathophysiology of the aforementioned biological processes and are viable therapeutic targets for this disease [-] (shown in Table 1). Several immune cells, including microglia, neutrophils, natural killer (NK) cells, T lymphocytes, B lymphocytes, monocytes, and dendritic cells (DCs), have been implicated in the causation of ischemic stroke [-]. Immune cells may also trigger or promote cytotoxic edema and neuronal death through the damaged blood-brain barrier (BBB) in ICH [-]. Poststroke brain injury can be classified into primary and secondary stages (shown in Fig. 1). In ischemic stroke, interruption of the blood flow can directly result in neuronal death, involving the ischemic core and the surrounding hypoperfusion lesion, termed penumbra, which is regarded as the primary injury. Subsequently, with the regulation of proinflammatory factors, activated cerebral vascular endothelial cells (EC) may upregulate the level of intercellular adhesion molecules (ICAM), leading to adhesion of peripheral leukocytes, which exacerbates microcirculation dysfunction, causing cerebral edema, and infarction (secondary injury). Coagulation system, a potential therapeutic target, plays a key role in both primary and secondary injury after ischemic stroke. Franks et al. [] illustrated that via P-selectin/P-selectin glycoprotein ligand-1, activated platelets could release an array of prothrombotic and proinflammatory mediators, such as soluble CD40 ligand, which further bound to CD40 on ECs and induced the expression of adhesion molecules involved in leukocyte trafficking. Monocytes adherence to P-selectin bearing platelets synthesized monocyte chemotactic protein (MCP)-1 and interleukin (IL)-8, promoting the migration of leukocytes into subendothelial layers. Besides, activated platelets induced monocyte production of cyclooxygenase-2 (COX-2), an enzyme responsible for the synthesis of proinflammatory eicosanoids and the regulation of chronic inflammations [, ]. For ICH, the primary injury is caused by the space-occupying effects of hematoma, contributing to calcium overload in the vicinity of the lesion and release of excitatory toxic neurotransmitters, which further results in cytotoxic edema and neuronal death. The secondary injury is due to the damaged neurocytes and the substances migrated from peripheral blood into brain, which exacerbate the deterioration of the BBB and cause angioedema. However, the specific mechanisms by which the infiltrated cells participate in stroke and the types of inflammatory factors that are secreted still need to be fully clarified.
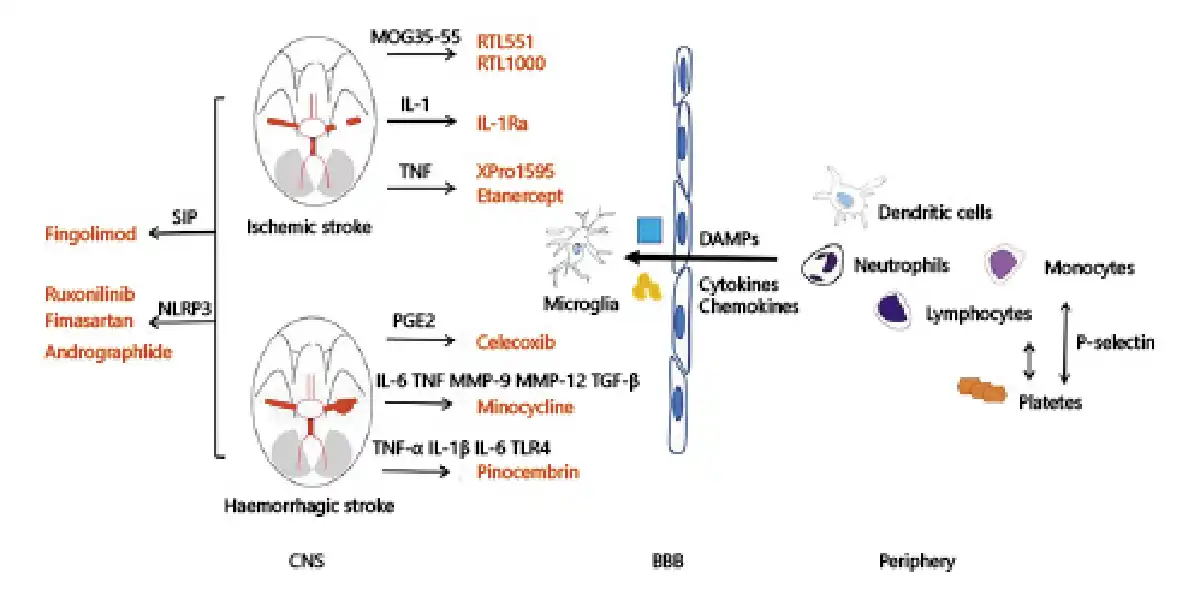
Fig. 1
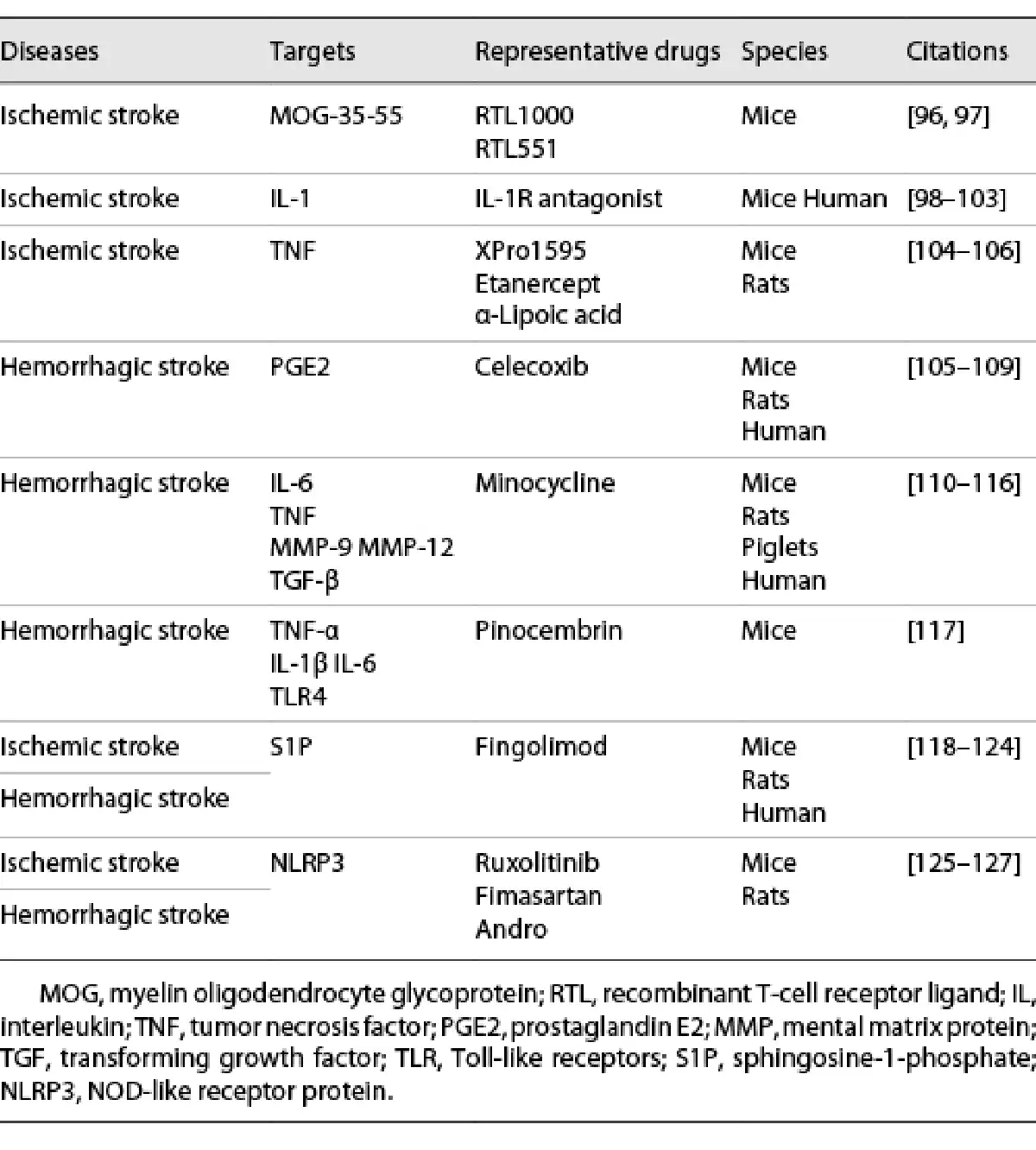
Both primary and secondary injury play a role in the pathophysiology of stroke. Interruption of blood flow or formation of hematoma leading to neuronal death and release of proinflammatory factors (i.e., DAMPs, cytokines, chemokines) is referred to as the primary injury. Primary injury triggers the migration of immune cells from periphery into the brain, causing secondary injury. The immune and inflammatory pathways are potential therapeutic targets for stroke. RTL551 and RTL1000 are recombinant T-cell receptor ligands that could inhibit the infiltration of macrophages/activated microglial cells and DCs into the infarct lesion by linking to MOG-35-55 peptide. IL-1Ra could decrease ischemic brain injury by increasing the axonal growth and neuronal plasticity. TNF antagonist, such as XPro1595, etanercept, α-lipoic acid, are found to reduce brain infarct by downregulating cerebral microglial activation. Celecoxib, a selective COX-2 inhibitor, may decrease brain edema after ICH via reducing the production of PGE2. By targeting inflammatory mediators, including TNF, IL-6, MMP-9, MMP-12, and TGF-β, minocycline can decrease the level of perihematomal iron overload, iron handling proteins. Pinocembrin is shown to downregulate the level of proinflammatory cytokines such as TNF-α, IL-1β, IL-6, and the expression of TLR4 after ICH. Fingolimod, an S1P-selective agonist, can improve the neurological dysfunction and reduce brain injury after stroke. By targeting NLRP3, ruxolitinib, fimasartan, and Andro are shown to improve neurological scores and alleviate secondary brain injury in both ischemic and hemorrhagic stroke. CNS, central nervous system; BBB, blood-brain barrier; DAMPs, damage-associated molecular patterns; COX-2, cyclooxygenase-2; MOG, myelin oligodendrocyte glycoprotein; RTL, recombinant T-cell receptor ligand; IL, interleukin; TNF, tumor necrosis factor; PGE2, prostaglandin E2; MMP, mental matrix protein; TGF, transforming growth factor; TLR, Toll-like receptors; S1P, sphingosine-1-phosphate; NLRP3, NOD-like receptor protein.
Based on the above pathophysiological mechanisms, several interventions targeting systemic inflammation have been shown to relieve neurological injury (shown in Table 2). Combining pharmacological interventions with endovascular management, both related to the regulation of immunity and inflammation, can improve neurological outcomes in patients []. Rehabilitation treatment is suggested as one of the holy grails in stroke therapy []. Additionally, neural stem-cell transplantation, a new type of remedy, has been explored in basic researches and is poised to be promoted to clinic [, ].
In this review article, we summarize the pathogenetic mechanisms that participate in ischemic stroke and ICH from the perspective of immunity and inflammation. Furthermore, we illustrate potential novel therapies targeting the aforementioned mechanisms to improve the prognosis of the disease. Lastly, we underline the future research directions for stroke therapies.
Immunity and Inflammation in Ischemic Stroke
A plethora of studies have demonstrated the critical role of immune and inflammatory factors in the progression of ischemic stroke. The immune mechanisms involved in ischemic stroke in vertebrates involve two major components: natural immunity and adaptive immunity. Although the type of immune cells varies with immune responses, there is a considerable overlap in the reaction processes. A broad spectrum of immune cells participate in the causation of ischemic brain injury, for example, microglia, neutrophils, lymphoid cells, monocytes, and DCs [-, -].
Microglia
As brain resident cells, microglia are the first responders to ischemia and are engaged in intimate cross-talk with other intrinsic brain cells and infiltrating leukocytes. In the acute stage of ischemia, microglia are attracted to blood vessels and extensively interact with ECs and pericytes, regulating the capillary diameter and cerebral blood flow via altering pericyte or astrocyte coverage, which may contribute to the impaired integrity of the blood vessels [, ]. Activated microglia were also shown to upregulate the level of reactive oxygen species (ROS) and the secretion of inflammatory cytokines such as interferon (IFN)-γ, IL-1β, and tumor necrosis factor (TNF)-α to increase the extent of cytotoxic responses []. The redox signal mediated by NADPH oxide 2 was shown to enhance the sensitivity of microglia based on proinflammatory stimulation, thereby increasing the secretion of neurotoxic cytokines []. Under the regulatory effect of NADPH oxide 2, microglia can prominently secrete matrix metalloproteinases (MMPs), especially MMP-3 and MMP-9, and further degrade the cytoskeleton protein to exaggerate the deterioration of BBB. In a study, inhibitors of MMPs were found to decrease the cerebral infarct volume, alleviate peri-lesional edema, and ameliorate ICH caused by recombinant tissue plasminogen activators []. On the other hand, stimulated microglia have been shown to play a protective role against ischemic injury to a certain extent. In the chronic recovery phase of stroke, microglia contribute to infarct resolution and tissue repair as they have the ability to engulf dead cells, debris, and newborn neurons that fail to integrate into the neural circuit [, ]. At 24 h after ischemic injury, branched and phagocytic microglia form a barrier around the lesion to hinder exaggeration of ischemia. At the same time, globular microglia via expressing Ym1 and CD206 at the core region of the lesion was found to downregulate the inflammatory response and alleviate neurological deficits []. The potential mechanisms of the protective effect of microglia in ischemic stroke are (1) Increase in the levels of various neurotrophic factors, including brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor []; (2) phagocytosis and clearance of intracerebral neutrophils []; (3) elimination of necrotic cells. To achieve therapeutic goals in a clinic, future studies should focus on elucidating the process of subtype-transdifferentiation rather than simple activation or depletion of microglia.
Studies have shown that M1 is the main subtype of microglia in the initial stage of ischemia, and reaches a peak at 3–5 days after ischemic damage. Subsequently, M2 becomes the prominent subtype of microglia and lasts nearly 14 days. The above phenotypic switch from M1 to M2 is more prominent around the ischemic lesion. Moreover, studies have indicated that M1 may exaggerate neuronal death triggered by oxygen-glucose deprivation in cell culture medium, while M2 was shown to inhibit the above progression []. The modulators of microglia polarization can be classified into four categories: (1) transcription factors: nuclear transcription factor (NF)-κB and STAT family members are closely related to M1 polarization, whereas nuclear factor erythroid 2-related factor 2 and PPARγ are associated with M2 polarization [-]; (2) receptors: Toll-like receptor (TLR) 4 and sphingosine 1 phosphate receptors are involved in shifting microglia toward M1 phenotype poststroke [, ]; (3) ion channels: the voltage-gated potassium channel Kv1.3 may be one of the main mediators of M1 microglia polarization via modulating Ca2+ signaling and induced neuroinflammation []; (4) gene modulators: H19 and miRNA-155 have been demonstrated to promote M1 polarization, whereas miRNA-124 and chemokine-like protein TAFA-3 (FAM19A3) are associated with M2 polarization [].
Neutrophils
Neutrophils, the type of rapid reaction cells, infiltrate into the lesion at 30 min after ischemic stroke. Stimulated by various cytokines and damage-associated molecular patterns, neutrophils migrate from bone marrow, spleen, and peripheral blood into the damaged region, and play a key role in triggering the inflammatory cascade []. Whether at the time of ischemia or after reperfusion, neutrophils are considered critical sources of ROS, which could damage neurovascular units and BBB by regulating ECs, smooth muscle cells, perivascular cells, and contribute to the exaggeration of cerebral edema and hemorrhagic transformation. Proteases released by neutrophils such as MMPs and elastase may degrade the basement membrane and extracellular matrix, aggravating the impairment of BBB []. However, some authors have also suggested a beneficial effect of neutrophils in the postischemic repair process. The core pathophysiological mechanism of neutrophil-driven repair is angiogenesis. At the site of arterial injury, activated neutrophils deposit cathelicidin antimicrobial peptide, and further promote the release of vascular endothelial growth factor and epidermal growth factor from EC progenitors in a paracrine manner, which stimulates the recovery of ECs [, ].
Lymphoid Cells
Lymphoid cells mainly include T, B, and NK cells. These play an important role in ischemic stroke. T lymphoid cells, which play a central role in ischemic stroke, can upregulate the production of cytotoxic substances and the secretion of oxidative stress-related ingredients such as ROS. Helper T cells (Th) 1 in the vicinity of the infarct area have been shown to stimulate the release of proinflammatory mediators, including IFN-γ, TNF-α, augmenting intercellular adhesion and enhancing vascular permeability. Activation of Th1 cells may cause persistent damage to cortical neurons. Conversely, Th1-defective mice showed significantly reduced volume of infarct area and decreased the number of necrotic neurons. These findings indicated that the pathway by which Th1 promotes the transformation of macrophages into the active state may regulate the inflammation reactions []. Another subset of T cells (Tc) that aggravates the progression of ischemia is cytotoxic Tc via killing the pathogens directly and by releasing toxins such as perforins and granzymes. Tc were shown to induce poststroke neuronal apoptosis through the Fas-FasL signaling pathway []. Further studies are required to obtain direct evidence of whether Tc can alleviate ischemic injury. While Tc contribute the least to the early dysfunction after stroke, this does not deny the critical effects of Tc on the chronic recovery of ischemic stroke. Shi et al. [] found that regulatory T (Treg) cell-derived osteopontin could enhance the repair activity of microglia via integrin receptors, thereby facilitating oligodendrocyte regeneration and remyelination during the chronic stages of stroke. Increasing Treg cells with IL-2:IL-2 antibody complex improved long-term stroke recovery [].
B lymphoid cells, the main undertaker of fluid immune responses, is another type of crucial lymphocytes infiltrating into central nervous system (CNS) after ischemia. Recent studies found a decrease in B cells in the peripheral circulation of patients with acute cerebral ischemia, leading to the suppression of fluid immunity []. However, it is not clear whether B cells can immigrate to the CNS from the peripheral circulation like Tc, and the associated mechanisms are not clear.
NK cells are the principal players in natural immunity. These have the ability to spontaneously initiate cytotoxic response and can regulate the activity of B cells, Tc, and bone marrow stem cells. With the significant decrease in peripheral blood, NK cells may mediate low-level innate immunity and increase the risk of infection after ischemia. Therefore, researchers speculated that in the early stage of ischemia and hypoxia, certain inducible factors may promote migration of NK cells into CNS, and kill the targeted cells directly without secondary response or memory effect and regulate the function of other immune cells by secreting cytokines such as IFN-γ [-].
Monocytes
Recruitment of monocytes at the site of lesion is initiated in the early stage after ischemic stroke. Researchers have consistently shown that as early as 24-h post-MCAO, monocytes originating from bone marrow begin to immigrate into the ischemic core and the peri-infarct area, which might transit to microglia by expressing Iba-1 []. In the study by Chu et al. [] Ly6Chi monocytes showed a 3-fold increase in the blood and 10-fold increase in the CNS after MCAO in C57BL/6J mice and exerted a protective effect via limiting brain injury and neurological deficit. These effects were associated with promoting M2 microglia/macrophage polarization. In addition, CCR2-CCL2 axis is reported to be essential for the immigration of monocytes to the brain []. After recruitment, Ly6Chi monocytes begin to increase the expression of macrophage marker F4/80 []. Grosse et al. [] reported that the proportion of classical monocytes (CD14++CD16−; Mon1) was higher in patients with moderate/severe stroke, while the proportion of nonclassical monocytes (CD14+CD16++; Mon3) was higher in patients with mild stroke. They suggested further investigation to elucidate the mechanism of monocyte subsets in human ischemic injury.
Dendritic Cells
As classic antigen presenting cells, DCs form the links and bridges between natural and adaptive immunity. After ischemic injury, granulocyte-macrophage colony stimulating factor may induce the transformation of microglia into DCs with membrane expression of major histocompatibility complexes II [, ]. However, the specific mechanisms by which DCs take part in ischemic stroke are not clear.
Immune and Inflammatory Processes in ICH
The mechanisms of immunity and inflammation in ICH are not well characterized. Primary and secondary injury constitute two major aspects after hemorrhagic stroke and the immune response is suggested to have a significant effect on the secondary stage. The potential immune and inflammatory pathophysiological processes involved in secondary injury include the following aspects: (1) blood clots and thrombin can activate the complements and proteases, and further activate enzyme receptors resulting in cell lysis. Erythrocytes are believed to exacerbate the expression of heme oxygenase-1 in microglia and the hemoglobin can be decomposed into Fe, CO, and bilirubin. Fe produces free radicals through redox reactions which may contribute to the destruction of BBB and activation of inflammatory response. CO restricts the cellular respiration and oxygen release leading to the explosion of oxygen free radicals in mitochondria. Based on this theory, deferoxamine, a potent iron chelating agent, has been shown to alleviate neurological deficit and the degree of cerebral edema [] (2) Inflammation related to secondary injury could deteriorate the integrity of BBB mediated via MMP and other cytokines, and then increase the permeability of capillaries and exacerbate edema after hemorrhagic injury [, ].
Microglia
The most rapidly activated cells after ICH are microglia, with membrane expression of receptors such as TLR []. Upon stimulation, microglia can produce proinflammatory cytokines, including IL-1β, IL-6, TNF-α, ROS, and NF-κB; this further exacerbates cerebral water content and aggravates neurological deficit. Yang et al. [] demonstrated that inhibition of microglial inflammation provided a novel treatment for ICH-induced brain injury via the intervention of ICH-challenged BV2 microglial cells in vitro and male C57BL/6 mice in vivo. Preclinical studies have suggested that M2 microglia/macrophage play a role in brain repair and recovery via engulfing the hematoma and cells’ debris, removing harmful substances, and providing space for tissue regeneration. With increase in the number of M2 microglia, the lesion of the hematoma is eliminated in 7–21 days after ICH []. M2 microglia express a series of growth factors and trophic factors, including insulin-like growth factors-1, brain-derived neurotrophic factor, glial cell line-derived neurotrophic factor, neurotrophin 3, NT-4/5, which could facilitate neurogenesis and neural circuit reframing [, ].
Very little is known about the transition of microglial polarization. Lan et al. [] suggested that transcription factors, prostaglandin E2 receptors, and sphingosine 1 phosphate receptors may mediate the polarization of microglia. By interacting with various cells, microglia act as guardians of the brain and play a key role after ICH. In a mouse model of blood-induced ICH, Treg cells were found to promote M2-like polarization of microglia and macrophages by activating IL-10-glycogen synthase kinase-3β signaling pathway []. Besides, the signaling pathway involving soluble CX3CL1 and CX3C chemokine receptor 1 (CX3CR1) may mediate the interactions between neurons and microglia, associated with microglial activation in the hemorrhagic injury []. However, monocyte-specific CX3CR1 deficiency showed no effect on functional improvement after ICH.
The following are the potential mechanisms by which microglia participate in ICH: (1) damage-associated molecular patterns: hemoglobin can induce the release of high-mobility group protein B1 and activate TLR4 of microglia to exacerbate post-ICH inflammation. Studies have shown that inhibiting high-mobility group protein B1 and activating the microglia around the lesion may ameliorate edema and neuronal apoptosis []. (2) TLR: hemoglobin may promote the secretion of NF-κB by activating the TLR of microglia, then upregulate the inflammatory cytokines to exacerbate hemorrhagic injury []. (3) Inflammatory corpuscle: as the classic pattern recognition receptor of natural immune responses, inflammatory corpuscle is proven to be an active regulatory factor of caspase-1, such as NOD-like receptor protein (NLRP) 3. By increasing the secretion of inflammatory factors such as IL-1β, IL-18, the inflammatory corpuscle expressed by microglia can aggravate the immune and inflammatory damage after ICH [, ].
Neutrophils
Neutrophils infiltrate into the lesion after ICH. In the ICH animal model prepared by collagenase, neutrophils were detected in the vicinity of the lesion at 4 h after ICH; the extent of neutrophilic infiltrate peaked at 3–5 days after hemorrhagic event []. Moreover, apoptosis of neutrophils can activate the microglia and further lead to neuroinflammatory injury. Neutrophils are divided into two major subtypes: N1, the proinflammatory subtype, can exacerbate neurological deficit and the level of encephaledema; N2, the anti-inflammatory subtype, can attenuate the inflammatory reaction to preserve the activity of neurocytes. In addition, polymorphonuclear neutrophils (PMN) may exaggerate the deterioration of BBB, resulting in cerebral edema, recurrence of hemorrhage, and neurological deficits [].
Lymphoid Cells
T lymphoid cells play vital roles in the pathogenesis of ICH. A study illustrated that CD4+Tc migrate into hemorrhagic hemisphere at 1 day, peak at 4 days, and gradually decline up to 14 days after ICH. Treg cells likely showed a similar pattern of variation as CD4+Tc, but peaked at 7 days after hemorrhagic injury. Treg cells can improve ICH-induced inflammatory damage via regulating microglia/macrophage polarization toward the M2 phenotype, which was related to the activation of IL-10/GSK3/PTEN axis [].
The effects of B lymphoid cells and NK cells after intracerebral injury may be limited owing to their lesser permeability across the BBB [, ]. A recent research suggested that brain-bound NK cells may contribute to perihematomal edema and neurological deficit via cytotoxicity toward brain ECs and recruitment of neutrophils at the early stages of ICH [].
Monocytes
Studies have shown that monocytes infiltrate into the brain at 12 h after ICH, and reach peak level on the 5th day []. MCP-1 and CC-chemokine receptor (CCr) 2 play an essential role in inducing immigration of monocytes into the CNS; thus, these are potential therapeutic targets for alleviation of inflammatory response after hemorrhagic stroke. Comparing the CCr2−/− hematopoietic cells with the control, the former exhibited a prominent increase in restoration of neurological function by decreasing the number of downstream monocytes. In addition to the aforementioned experimental studies, clinical prospective studies have also shown a close relation of the level of CCr with the poor prognosis of neurological function at the first week after ICH []. In a clinical study of 115 ICH patients, levels of MCP-1 and chemokine ligand (CXCL)-10 showed a negative correlation with modified Ranking Scale (mRS) score, and were independent predictors of prognosis of ICH []. Walsh et al. [] found that the median number of M2 monocyte microparticles was significantly higher in ICH group compared with controls and they suggested that the results represented differences in the chronic inflammatory status in patients susceptible to ICH, such as cellular activation and apoptosis. In addition, the inflammatory monocytes may be responsible for the M1-type macrophage response, characterized by phagocytosis, proteolysis, inflammation, and neurological deficit in the first 3 days after ICH [].
Representative Drugs of Anti-Inflammatory Therapies in Ischemic Stroke
Owing to the complex interplay between immune system and inflammation in ischemia, use of methods to intervene the aforementioned processes may improve the clinical outcomes of patients with stroke. Thrombolytic therapy with tissue-type plasminogen activator (t-PA) within a specific time window (4.5 h after onset), as the classic pharmacological treatment for acute ischemic stroke, has been reported to weaken the immune response and contribute to a higher risk of infection and impairment of neurological recovery. t-PA via promoting plasmin generation was shown to further reduce lymphocyte and monocyte counts in blood, decrease DC subsets in the spleen and cervical lymph nodes, and enhance the plasma level of IL-10, TNF-α, contributing to immunosuppression and systemic inflammation poststroke [, ]. During the subacute and chronic recovery phase of ischemia (>2–3 days), neurological rehabilitation is considered the major strategy for stroke. Researchers have suggested that poststroke exercise initiated after 3 days of reperfusion was related to decreased expression of the pro-inflammatory mediators, including ICAM-1, vascular cell adhesion molecule (VCAM)-1, TNF-α, IL-1β, and cell stress markers, including Hsp70, hypoxia-inducible factor (HIF)-1α. However, very early initiation of poststroke exercise was found to increase the expression of these molecules. The results provide evidence for the optimal timing of exercise initiation in poststroke rehabilitation []. Therefore, in order to achieve maximal treatment success and safety, a more personalized therapeutic approach is required for patients with ischemic stroke. In addition, to decrease the risk of recurrence of ischemic stroke, clinicians advocate the use of anticoagulant and/or antiplatelet drugs from presentation until 3 or 6 months after revascularization, the mechanism of which is partly associated with anti-inflammation [].
Recombinant T-Cell Receptor Ligand
RTL551 (I-Ab molecule linked to MOG-35-55 peptide) was shown to decrease cortical and total infarct volume by approximately 50% and inhibit the infiltration of immune cells, including macrophages/activated microglial cells and DCs []. Administration of RTL551 4 h after middle cerebral artery occlusion (MCAO) in C57BL/6 mice decreased the infarct size and reduced inflammatory cell infiltration into the CNS, which was related to reduced expressions of T-cell activation (CD44) and CNS homing receptor (CCR5 and CCR7) markers. Similarly, RTL1000 (HLA-DR2 moiety linked to human MOG-35-55 peptide) was also shown to reduce stroke lesion volume in HLA-DR2 transgenic mice []. To promote the clinical translation, future studies are underway to determine the optimal duration for which RTL can be administered after ischemia to exert therapeutic benefit.
IL-1 Receptor Antagonist
IL-1Ra is classified into two structural variants, i.e., secreted IL-1Ra and intracellular IL-1Ra (icIL-1Ra). It effectively regulates the activity of IL-1 in the acute phase of ischemic injury. Administration of IL-1Ra was shown to decrease ischemic brain injury in animal models of both transient(t) and permanent(p) MCAO. The specific mechanisms might be related to the neuroprotective effects on microglia at the ischemic lesion [-]. In a study, injecting IL-1Ra-producing bone marrow cells into mice was found to modulate JNK/SAPK signaling pathway resulting in outgrowth of axons and improved neuronal plasticity in the acute phase after ischemic stroke. However, the efficacy and safety of IL-1Ra are yet to be established in clinical studies; in addition, achieving a therapeutic concentration in brain is another challenge as it shows poor permeability across the BBB [, ].
XPro1595, Etanercept, α-Lipoic Acid
In preclinical experiments, topical administration of TNF antagonists, such as XPro1595 (a dominant-negative inhibitor of soluble TNF), to C57BL/6 mice reduced the infarct injury at both 1 and 3 days after ischemia; the effect may be attributable to microglial activation toward a phagocytic phenotype. Compared with the transmembrane TNF, the study showed that soluble TNF may play the main role in relieving ischemic injury by regulating systemic and local inflammation in the CNS []. In another study, both XPro1595 and etanercept (inhibitor of both soluble TNF and transmembrane TNF) improved neurological deficit, but without decreasing the infarct volume, via altering microglial responses, modifying liver acute phase response (APR), and changing spleen Tc and microvesicle numbers. Systemically administered XPro1595 was as efficient as etanercept in alleviating infarct damage, improving clinical outcome, and reducing inflammation after focal cerebral ischemia []. The therapeutic effects of etanercept and α-lipoic acid were also tested in rats subjected to MCAO. Etanercept and α-lipoic acid, administered either alone or in combination, were found to reduce brain infarct, alleviate BBB disruption and neurological dysfunction by decreasing serum level of TNF-α and downregulating cerebral microglial activation []. However, more research about anti-TNF therapy is required to testify its effect on stroke.
Representative Drugs of Anti-Inflammatory Therapies in ICH
Celecoxib
Based on the aforementioned role of pro- and anti-inflammatory cytokines, several therapies targeting these molecules have been investigated on a large scale. The most common anti-inflammatory therapy for ICH is selective COX-2 inhibitor []. Injection of celecoxib (a selective COX-2 inhibitor) at concentrations of 10 or 20 mg/kg to adult rats at 20 min, 6 h, and 24 h after hemorrhagic injury, respectively, showed potent effects on neurological function recovery []. Compared with the ICH-only group, the group of ICH treated with 10 or 20 mg/kg celecoxib showed a significant decrease of inflammatory cells such as myeloperoxidase-positive cells and OX42-positive cells in the periphery of hematoma. These findings support the theory that COX-2 inhibition may attenuate immune and inflammatory reactions by reducing the production of prostaglandin E2 and decreasing brain edema after ICH []. The small size of the study population constrains us from determining the most effective dose of celecoxib for ICH, providing the direction for future investigations.
Minocycline
In a study, minocycline improved the neurobehavioral results and exerted neuroprotective effects on ICH by targeting inflammatory mediators, including TNF, IL-6, MMP-9, and MMP-12 []. A wide body of evidence from experimental studies on rodents suggest that minocycline can decrease the level of perihematomal iron overload, iron handling proteins, and further ameliorate the disruption of BBB, brain edema, neuronal apoptosis, and autophagy; this phenomenon may be related with the DKK1-Wnt signaling pathway [-]. Similarly, in piglets, minocycline attenuated ICH-induced white matter damage by activating TGF-β [, ]. The significant effects of minocycline in animal models were also mirrored in patients. A clinical trial demonstrated the safety of high-dose (10 mg/kg) intravenous minocycline in patients with ICH owing to its effect in decreasing the level of MMP-9 in serum []. However, larger clinical trials evaluating the efficacy of minocycline in ICH patients with poor prognoses are still needed.
Pinocembrin
Pinocembrin (5,7-dihydroxyflavanone) was shown to be a promising strategy to inhibit post-ICH inflammatory reactions in vivo. It was shown to reduce the lesion volume in a dose-dependent manner and further ameliorate the neurologic deficits. Pinocembrin decreased the level of proinflammatory cytokines related to M1-like microglia such as TNF-α, IL-1β, IL-6, and the expression of TLR4, including its downstream target proteins TRIF and MyD88 in C57BL/6 mice with ICH. In vitro, pinocembrin downregulated the expression of NF-κB in lipopolysaccharide-stimulated BV-2 cells or primary microglia []. Although pinocembrin was demonstrated to have an anti-inflammatory effect in the CNS, it is unknown whether it targets microglia. Additional studies of the effects of pinocembrin on microglial polarization are required prior to exploration of its clinical use.
Representative Drugs of Anti-Inflammatory Strategies in Ischemic and Hemorrhagic Stroke
Fingolimod
Fingolimod, a S1P-selective agonist, has shown therapeutic effects on ischemic and hemorrhagic stroke in both animal models and small-scale clinical trials []. Intravenous administration of fingolimod (0.5 mg/kg) to C57BL/6 mice attenuated the neurological deficit and reduced lesion size after ischemic stroke. Combination of fingolimod and t-PA improved the outcomes of thrombolytic treatment and decreased the risk of hemorrhagic transformation associated with delayed administration of t-PA []. In diabetic mice after MCAO, fingolimod decreased the level of infiltrated inflammatory cells and TNF-α, and increased the ratio of Bcl-2/Bax. However, fingolimod remarkably exacerbated brain edema and the expression of tight junction proteins ZO-1 and occludin, the detrimental effects of which outweighed the beneficial effects leading to the lack of improvement in neurological outcomes at 24 h after tMCAO []. In a clinical study, a combination of fingolimod with alteplase ameliorated endpoint outcomes, and attenuated reperfusion injury in patients with acute ischemic stroke was treated in the first 4.5 h after symptom onset []. Furthermore, co-administration of fingolimod with alteplase in acute ischemic stroke patients in the 4.5–6 h after symptom onset promoted both anterograde reperfusion and retrograde collateral flow resulting in favorable neurological outcomes []. The small sample size, absence of blinded treatment administration, the lack of an independent data safety and monitoring entity, and suboptimal randomization method may have introduced an element of bias. However, the findings indicated that fingolimod extends the time window for alteplase in acute ischemic stroke.
CD-1 mice treated with fingolimod showed improved neurological function and decreased brain edema at 24 and 72 h following experimental ICH. It reduced the number of lymphocytes in blood and CNS and the expressions of ICAM-1, IFN-γ, IL-17 at 72 h after hemorrhagic stroke. Intraperitoneal injection of fingolimod in rats decreased the spatial and motor learning deficit, along with reduced brain atrophy and neuron loss in the basal ganglia []. The beneficial impacts of fingolimod were also mirrored in patients with ICH. Administration of oral fingolimod within 72 h of primary supratentorial ICH decreased perihematomal edema and neurological dysfunction. Investigations revealed that fingolimod reduced the circulating immune cells such as CD4+T, CD8+T, CD19+B, NK, NKTc, and plasma ICAM. The level of anti-inflammatory cytokine IL-10 was increased in the fingolimod group. In addition, it helped preserve vascular integrity by reducing the level of plasma MMP9 and rT1% []. Whether the administration of fingolimod beyond 3 days can prolong the decrease of edema deserves further investigations, for its effect on an immune-deficient state and compromise the neural repair mechanism.
Ruxolitinib, Fimasartan, Andrographolide
Targeting NLRP3 is a viable strategy for ameliorating neurological injury both in ischemic stroke and ICH. In C57BL/6 mice, ruxolitinib treatment improved neurological scores, attenuated the infarct volume, and improved brain edema 3 days after ischemia attack via activating JAK2/STAT3 pathway, which might be related to the downregulation NLRP3 inflammasome expression. A limitation of this study is that the researchers mainly focused on the anti-inflammatory effect of ruxolitinib rather than its other roles, including angiogenic effect on rescuing cerebral ischemia injury [].
Pretreatment with a low-dose (0.5 mg/kg) fimasartan to Sprague-Dawley rats alleviated ICH-induced secondary brain damage by inhibiting the NLRP3 []. The effects of regular doses of fimasartan in post-ICH treatment still need to be studied further to determine its clinical and translational relevance. Besides, it may be required to validate the translational potential of ICH models in elder animals for future studies. Andrographolide (Andro), the major active constituent of Andrographis paniculata, was tested in male Sprague-Dawley mice after ICH. Researchers investigated the potential effects of Andro on ICH-induced secondary brain injury. Andro was found to decrease the level of IL-1β and lactate dehydrogenase and inhibited microglia pyroptosis triggered by ICH via suppressing the NLRP3. The results suggested that Andro could be a potential candidate for reducing neuronal cell death and degeneration. However, there are a few limitations to the study. Future studies are required to testify the treatment of Andro in female and elder ICH models. In addition, the specific mechanisms by which Andro blocks NK-κB, NLRP3 inflammasome, and other pathways are still not entirely clear [, ].
Conclusion and Future Perspective
Immunity and systemic inflammation play a key role in the occurrence and development of stroke. We summarized the available evidence of the mechanisms by which the immune cells and cytokines participate in ischemic and hemorrhagic stroke to explore potential therapies for anti-inflammatory intervention. Some therapeutic methods targeting inflammation still need to be tested. For example, anti-IL-6 treatment is a potential target to prevent focal injury, which is associated with the infiltration of leukocytes. Using the chimeric molecule sgp130Fc via binding to the complex of IL-6/soluble(sol)IL-6R might be a promising method in future stroke research [, ]. In addition, although in-depth basic researches related to anti-inflammatory treatment have been carried out, the results of these studies have not been entirely replicated in clinical studies. Promotion of anti-inflammatory therapies still needs to be further verified in both experiment models and clinical studies.
Conflict of Interest Statement
Hui Zhao, Yan Li, Ying Zhang, Wen-Yan He, and Wei-Na Jin declare that they have no conflict of interest.
Funding Sources
This study was supported in part by the National Natural Science Foundation of China (81971094, 81771274), the Advanced Innovation Center for Human Brain Protection, Capital Medical University, Beijing, China.
Author Contributions
Wei-Na Jin formulated the concept. Hui Zhao, Yan Li, and Ying Zhang searched the articles. All the authors drafted the manuscript.
References
- 1. Stroke Experts Collaboration Group. Primary stroke prevention worldwide: translating evidence into action. Lancet Public Health. 2022 Jan;7(1):e74–e85.
- 2. GBD 2019 Stroke Collabrators. Global, regional, and national burden of stroke and its risk factors, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021 Oct;20(10):795–820.
- 3. Ovbiagele B, Goldstein LB, Higashida RT, Howard VJ, Johnston SC, Khavjou OA, et al. Forecasting the future of stroke in the United States: a policy statement from the American Heart Association and American Stroke Association. Stroke. 2013 Aug;44(8):2361–75.
- 4. Moskowitz MA, Lo EH, Iadecola C. The science of stroke:mechanisms in search of treatments. Neuron. 2010 Jul;67(2):181–98.
- 5. Urra X, Cervera A, Villamor N, Planas AM, Chamorro A. Harms and benefits of lymphocyte subpopulations in patients with acute stroke. Neuroscience. 2009 Feb;158:1174–83. http://dx.doi.org/10.1016/j.neuroscience.2008.06.014.
- 6. Shi K, Tian DC, Li ZG, Ducruet AF, Lawton MT, Shi FD. Global brain inflammation in stroke. Lancet Neurol. 2019 Nov;18(11):1058–66. http://dx.doi.org/10.1016/S1474-4422(19)30078-X.
- 7. Hankey GJ. Stroke. Lancet. 2017 Feb;389(10069):641–54. http://dx.doi.org/10.1016/s0140-6736(16)30962-x.
- 8. Chen Z, Venkat P, Seyfried D, Chopp M, Yan T, Chen J. Brain-heart interaction: cardiac complications after stroke. Circ Res. 2017 Aug;121(4):451–68. http://dx.doi.org/10.1161/CIRCRESAHA.117.311170.
- 9. Li Z, Ye H, Cai X, Sun W, He B, Yang Z, et al. Bone marrow-mesenchymal stem cells modulate microglial activation in the peri-infarct area in rats during the acute phase of stroke. Brain Res Bull. 2019 Nov;153:324–33.
- 10. Tang C, Wang C, Zhang Y, Xue L, Li Y, Ju C, et al. Recognition, intervention, and monitoring of neutrophils in acute ischemic stroke. Nano Lett. 2019 Jul;19(7):4470–7.
- 11. Kong Y, Li S, Cheng X, Ren H, Zhang B, Ma H, et al. Brain ischemia significantly alters microRNA expression in human periphery blood natural killer cell. Front Immunol. 2020 May;11:759.
- 12. Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδT cells. Nat Med. 2016 May;22(5):516–23.
- 13. Novotny J, Oberdieck P, Titova A, Pelisek J, Chandraratne S, Nicol P, et al. Thrombus NET content is associated with clinical outcome in stroke and myocardial infarction. Neurology. 2020 Jun;94(22):e2346–e2360.
- 14. Pedragosa J, Miró-Mur F, Otxoa-de-Amezaga A, Justicia C, Ruíz-Jaén F, Ponsaerts P, et al. CCR2 deficiency in monocytes impairs angiogenesis and functional recovery after ischemic stroke in mice. J Cere Blood Flow Metab. 2020 Dec;40:S98–116.
- 15. Sun L, Zhang W, Zhao Y, Wang F, Liu S, Liu L, et al. Dendritic cells and T cells, partners in atherogenesis and the translating road ahead. Front Immunol. 2020 Jul;11:1456.
- 16. Drieu A, Lanquetin A, Levard D, Glavan M, Campos F, Quenault A, et al. Alcohol exposure-induced neurovascular inflammatory priming impacts ischemic stroke and is linked with brain perivascular macrophages. JCI Insight. 2020 Feb;5(4):e129226.
- 17. Huttunen J, Kurki MI, von Und Zu Fraunberg M, Koivisto T, Ronkainen A, Rinne J, et al. Epilepsy after aneurysmal subarachnoid hemorrhage: a population-based, long-term follow-up study. Neurology. 2015 Dec;84:1997.
- 18. Lattanzi S, Brigo F, Trinka E, Cagnetti C, Di Napoli M, Silvestrini M. Neutrophil-to-lymphocyte ratio in acute cerebral hemorrhage: a system review. Transl Stroke Res. 2019 Apr;10(2):137–45. http://dx.doi.org/10.1007/s12975-018-0649-4.
- 19. Ren H, Han R, Chen X, Liu X, Wan J, Wang L, et al. Potential therapeutic targets for intracerebral hemorrhage-associated inflammation: an update. J Cereb Blood Flow Metab. 2020 Sep;40(9):1752–68.
- 20. Zhu H, Wang Z, Yu J, Yang X, He F, Liu Z, et al. Role and mechanisms of cytokines in the secondary brain injury after intracerebral hemorrhage. Prog Neurobiol. 2019 Jul;178:101610.
- 21. Franks ZG, Campbell RA, Weyrich AS, Rondina MT. Platelet-leukocyte interactions link inflammatory and thromboembolic events in ischemic stroke. Ann N Y Acad Sci. 2010 Oct;1207:11–7. http://dx.doi.org/10.1111/j.1749-6632.2010.05733.x.
- 22. Schmidt R, Bültmann A, Fischel S, Gillitzer A, Cullen P, Walch A, et al. Extracellular matrix metalloproteinase inducer (CD147) is a novel receptor on platelets, activates platelets, and augments nuclear factor kappaB-dependent inflammation in monocytes. Circ Res. 2008 Feb;102:302–9. http://dx.doi.org/10.1161/CIRCRESAHA.107.157990.
- 23. Chen CJ, Ding D, Starke RM, Mehndiratta P, Crowley RW, Liu KC, et al. Endovascular vs medical management of acute ischemic stroke. Neurology. 2015 Dec;85(22):1980–90.
- 24. Xu AH, Sun YX. Research hotspots and effectiveness of repetitive transcranial magnetic stimulation in stroke rehabilitation. Neural Regen Res. 2020 Nov;15(11):2089–97. http://dx.doi.org/10.4103/1673-5374.282269.
- 25. Chen L, Qiu R, Li L, He D, Lv H, Wu X, et al. The role of exognous neural stem cells transplantation in cerebral ischemic stroke. J Biomed Nanotechnol. 2014 Nov;10(11):3219–30.
- 26. Hou B, Ma J, Guo X, Ju F, Gao J, Wang D, et al. Exogenous neural stem cells transplantation as a potential therapy for photothrombotic ischemia stroke in kunming mice model. Mol Neurobiol. 2017 Mar;54(2):1254–62.
- 27. Kronenberg G, Uhlemann R, Richter N, Klempin F, Wegner S, Staerck L, et al. Distinguishing features of microglia- and monocyte-derived macrophages after stroke. Acta Neuropathol. 2018 Apr;135(4):551–68.
- 28. Zhu B, Pan Y, Jing J, Meng X, Zhao X, Liu L, et al. Neutrophil counts, neutrophil ratio, and new stroke in minor ischemic stroke or TIA. Neurology. 2018 May;90(21):e1870–e1878.
- 29. Ye XC, Hao Q, Ma WJ, Zhao QC, Wang WW, Yin HH, et al. Dectin-1/Syk signaling triggers neuroinflammation after ischemic stroke in mice. J Neuroinflammation. 2020 Jan;17(1):17.
- 30. Bisht K, Okojie KA, Sharma K, Lentferink DH, Sun YY, Chen HR, et al. Capillary-associated microglia regulate vascular structure and function through PANX1-P2RY12 coupling in mice. Nat Commun. 2021 Sep;12(1):5289.
- 31. Jolivel V, Bicker F, Biname F, Ploen R, Keller S, Gollan R, et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. 2015 Feb;129(2):279–95.
- 32. Li C, Zhao Z, Luo Y, Ning T, Liu P, Chen Q, et al. Macrophage-disguised manganese dioxide nanoparticles for neuroprotection by reducing oxidative stress and modulating inflammatory microenvironment in acute ischemic stroke. Adv Sci. 2021 Oct;8(20):e210526.
- 33. Liu H, Wei X, Kong L, Liu X, Li C, Shi Y, et al. NOD2 is involved in the inflammatory response after cerebral ischemia-reperfusion injury and triggers NADPH oxidase 2-derived reactive oxygen species. Int J Biol Sci. 2015 Mar;11(5):525–35.
- 34. Saleem S, Wang D, Zhao T, Sullivan RD, Reed GL. Matrix metalloproteinase-9 expression is enhanced by ischemia and tissue plasminogen activator and induces hemorrhage, disability and mortality in experimental stroke. Neuroscience. 2021 Apr;460:120–9. http://dx.doi.org/10.1016/j.neuroscience.2021.01.003.
- 35. Ma Y, Yang S, He Q, Zhang D, Chang J. The role of immune cells in post-stroke angiogenesis and neuronal remodeling: the known and the unknown. Front Immunol. 2021 Dec;12:784098. http://dx.doi.org/10.3389/fimmu.2021.784098.
- 36. Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009 Feb;132:288–95. http://dx.doi.org/10.1093/brain/awn109.
- 37. Perego C, Fumagalli S, De Simoni MG. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J Neuroinflammation. 2021 Dec;8:174. http://dx.doi.org/10.1186/1742-2094-8-174.
- 38. Wei L, Fraser JL, Lu ZY, Hu X, Yu SP. Transplantation of hypoxia preconditioned bone marrow mesenchymal stem cells enhances angiogenesis and neurogenesis after cerebral ischemia in rats. Neurobiol Dis. 2012 Jun;46(3):635–45. http://dx.doi.org/10.1016/j.nbd.2012.03.002.
- 39. Otxoa-de-Amezaga A, Miró-Mur F, Pedragosa J, Gallizioli M, Justicia C, Gaja- Capdevila N, et al. Microglial cell loss after ischemic stroke favors brain neutrophil accumulation. Acta Neuropathol. 2019 Feb;137:321–41.
- 40. Li QQ, Ding DH, Wang XY, Sun YY, Wu J. Lipoxin A4 regulates microglial M1/M2 polarization after cerebral ischemia-reperfusion injury via the Notch signaling pathway. Exp Neurol. 2021 May;339:113654.
- 41. Yang S, Wang H, Yang Y, Wang R, Wang Y, Wu C, et al. Baicalein administered in the subacute phase ameliorates ischemia-reperfusion-induced brain injury by reducing neuroinflammation and neuronal damage. Biomed Pharmacother. 2019 Sep;117:109102.
- 42. Ding Y, Qian J, Li H, Shen H, Li X, Kong Y, et al. Effects of SC99 on cerebral ischemia-perfusion injury in rats: selective modulation of microglia polarization to M2 phenotype via inhibiting JAK2-STAT3 pathway. Neurosci Res. 2019 May;142:58–68.
- 43. Zhang W, Wei R, Zhang L, Tan Y, Qian C. Sirtuin 6 protects the brain from cerebral ischemia/reperfusion injury through NRF2 activation. Neuroscience. 2017 Dec;366:95–104. http://dx.doi.org/10.1016/j.neuroscience.2017.09.035.
- 44. Zhao X, Wang H, Sun G, Zhang J, Edwards NJ, Aronowski J. Neuronal interleukin-4 as a modulator of microglial pathways and ischemic brain damage. J Neurosci. 2015 Aug;35(32):11281–91. http://dx.doi.org/10.1523/JNEUROSCI.1685-15.2015.
- 45. Palma-Tortosa S, Hurtado O, Pradillo JM, Ferreras-Martin R, Garcia-Yebenes I, Garcia-Culebras A, et al. Toll-like receptor 4 regulates subventrivular zone proliferation and neuroblast migration after experimental stroke. Brain Behav Immun. 2019 Aug;80:573–82.
- 46. Qin C, Fan WH, Liu Q, Shang K, Murugan M, Wu LJ, et al. Fingolimod protects against ischemic white matter damage by modulating microglia towards M2 polarization via STAT3 pathway. Stroke. 2017 Dec;48(12):3336–46.
- 47. Di Lucente J, Nguyen HM, Wulff H, Jin LW, Maezawa I. The voltage-gated potassium channel Kv1.3 is required for microglial pro-inflammatory activation in vivo. Glia. 2018 Sep;66(9):1881–95. http://dx.doi.org/10.1002/glia.23457.
- 48. Jiang CT, Wu WF, Deng YH, Ge JW. Modulators of microglia activation and polarization in ischemic stroke (review). Mol Med Rep. 2020 May;21(5):2006–18. http://dx.doi.org/10.3892/mmr.2020.11003.
- 49. Herz J, Sabellek P, Lane TE, Gunzer M, Hermann DM, Doeppner TR. Role of neutrophils in exacerbation of brain injury after focal cerebral ischemia in hyperlipidemic mice. Stroke. 2015 Oct;46(10):2916–25. http://dx.doi.org/10.1161/STROKEAHA.115.010620.
- 50. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013 Mar;13(3):159–75. http://dx.doi.org/10.1038/nri3399.
- 51. Brea D, Sobrino T, Ramos-Cabrer P, Castillo J. [Reorganisation of the cerebral vasculature following ischaemia]. Rev Neurol. 2009 Dec;49:645–54. http://dx.doi.org/10.33588/rn.4912.2009564.
- 52. Hermann DM, Zechariah A. Implications of vascular endothelial growth factor for postischemic neurovascular remodeling. J Cereb Blood Flow Metab. 2009 Oct;29:1620–43. http://dx.doi.org/10.1038/jcbfm.2009.100.
- 53. Gu L, Xiong X, Zhang H, Xu B, Steinberg GK, Zhao H. Distinctive effects of T cell subsets in neuronal injury induced by cocultured splenocytes in vitro and by in vivo stroke in mice. Stroke. 2012 Jul;43(7):1941–6. http://dx.doi.org/10.1161/strokeaha.112.656611.
- 54. Li G, Wang X, Huang LH, Wang Y, Hao J, Ge X, et al. Cytotoxic function of CD8+T lymphocytes isolated from patients with acute severe cerebral infarction: an assessment of stroke-induced immune-suppression. BMC Immunol. 2013 Jan;14:1.
- 55. Shi L, Sun Z, Su W, Xu F, Xie D, Zhang Q, et al. Treg cell-derived osteopontin promotes microglia-mediated white matter repair after ischemic stroke. Immunity. 2021 Jul;54(7):1527–42.
- 56. Takahashi K, Aranami T, Endoh M, Miyake S, Yamamura T. The regulatory role of natural killer cells in multiple sclerosis. Brain. 2004 Sep;127(Pt9):1917–27. http://dx.doi.org/10.1093/brain/awh219.
- 57. Peterfalvi A, Molnar T, Banati M, Pusch G, Miko E, Bogar L, et al. Impaired function of innate T lymphocytes and NK cells in the acute phase of ischemic stroke. Cerebrovasc Dis. 2009 Sep;28(5):490–8.
- 58. Jin WN, Ducruet AF, Liu Q, Shi SXY, Waters M, Zou M, et al. Activation of JAK/STAT3 restores NK-cell function and improves immune defense after brain ischemia. FASEB J. 2018 May;32(5):2757–67.
- 59. Tanaka R, Komine-Kobayashi M, Mochizuki H, Yamada M, Furuya T, Migita M, et al. Migration of enhanced green fluorescent protein expressing bone marrow-derived microglia-macrophage into the mouse brain following permanent focal ischemia. Neuroscience. 2003 Feb;117(3):531–9.
- 60. Chu HX, Broughton BR, Kim HA, Lee S, Drummond GR, Sobey CG. Evidence that Ly6C(hi) monocytes are protective in acute ischemic stroke by promoting M2 macrophage polarization. Stroke. 2015 Jul;46(7):1929–37. http://dx.doi.org/10.1161/STROKEAHA.115.009426.
- 61. Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke. 2007 Apr;38(4):1345–53. http://dx.doi.org/10.1161/01.STR.0000259709.16654.8f.
- 62. Miro-Mur F, Perez-de-Puig I, Ferrer-Ferrer M, Urra X, Justicia C, Chamorro A, et al. Immature monocytes recruited to the ischemic mouse brain differentiate into macrophages with features of alternative activation. Brain Behav Immun. 2016 Mar;53:18–33.
- 63. Grosse GM, Schulz-Schaeffer WJ, Teebken OE, Schuppner R, Dirks M, Worthmann H, et al. Monocyte subsets and related chemokines in carotid artery stenosis and ischemic stroke. Int J Mol Sci. 2016 Mar;17(4):433.
- 64. Kato H, Kogure K, Liu XH, Araki T, Itoyama Y. Progressive expression of immunomolecules on activated microglia and invading leukocytes following focal cerebral ischemia in the rat. Brain Res. 1996 Sep;734:203–12. http://dx.doi.org/10.1016/0006-8993(96)00636-1.
- 65. Felger JC, Abe T, Kaunzner UW, Gottfried-Blackmore A, Gal-Toth J, McEwen BS, et al. Brain dendritic cells in ischemic stroke: time course, activation state, and origin. Brain Behav Immun. 2010 Jul;24(5):724–37.
- 66. Zhao J, Chen Z, Xi G, Keep RF, Hua Y. Deferoxamine attenuates acute hydrocephalus after traumatic brain injury in rats. Transl Stroke Res. 2014 Oct;5:586–94. http://dx.doi.org/10.1007/s12975-014-0353-y.
- 67. Li Z, Li M, Shi SX, Yao N, Cheng X, Guo A, et al. Brain transforms natural killer cells that exacerbate brain edema after intracerebral hemorrhage. J Exp Med. 2020 Dec;217(12):e20200213.
- 68. Lule S, Wu L, McAllister LM, Edmiston WJ, Chung JY, Levy E, et al. Genetic inhibition of receptor interacting protein kinase-1 reduces cell death and improves functional outcome after intracerebral hemorrhage in mice. Stroke. 2017 Sep;48(9):2549–56.
- 69. Yuan B, Shen H, Lin L, Su T, Huang Z, Yang Z. Scavenger receptor SRA attenuates TLR4-induced microglia activation in intracerebral hemorrhage. J Neuroimmunol. 2015 Dec;289:87–92. http://dx.doi.org/10.1016/j.jneuroim.2015.10.006.
- 70. Yang Z, Liu Y, Yuan F, Li Z, Huang S, Shen H, et al. Sinomenine inhibits microglia activation and attenuates brain injury in intracerebral hemorrhage. Mol Immunol. 2014 Aug;60(2):109–14.
- 71. Bi R, Fang Z, You M, He Q, Hu B. Microglia Phenotype and intracerebral hemorrhage: a balance of Yin and Yang. Front Cell Neurosci. 2021 Oct;15:765205. http://dx.doi.org/10.3389/fncel.2021.765205.
- 72. Xi G, Strahle J, Hua Y, Keep RF. Progress in translational research on intracerebral hemorrhage: is there an end in sight?Prog Neurobiol. 2014 Apr;115:45–63. http://dx.doi.org/10.1016/j.pneurobio.2013.09.007.
- 73. Ma Y, Wang J, Wang Y, Yang GY. The biphasic function of microglia in ischemic stroke. Prog Neurobiol. 2017 Oct;157:247–72. http://dx.doi.org/10.1016/j.pneurobio.2016.01.005.
- 74. Lan X, Han X, Li Q, Yang QW, Wang J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat Rev Neurol. 2017 Jul;13(7):420–33. http://dx.doi.org/10.1038/nrneurol.2017.69.
- 75. Zhou K, Zhong Q, Wang YC, Xiong XY, Meng ZY, Zhao T, et al. Regulatory T cells ameliorate intracerebral hemorrhage-induced inflammatory injury by modulating microglia/macrophage polarization through the IL-10/GSK3β/PTEN axis. J Cereb Blood Flow Metab. 2017 Mar;37(3):967–79.
- 76. Taylor RA, Hammond MD, Ai Y, Sansing LH. CX3CR1 signaling on monocytes is dispensable after intracerebral hemorrhage. PLoS One. 2014 Dec;9:e114472. http://dx.doi.org/10.1371/journal.pone.0114472.
- 77. Lei C, Lin S, Zhang C, Tao W, Dong W, Hao Z, et al. High-mobility group box1 protein promotes neuroinflammation after intracerebral hemorrhage in rats. Neuroscience. 2013 Jan;288:190–9.
- 78. Chen ZQ, Yu H, Li HY, Shen HT, Li X, Zhang JY, et al. Negative regulation of glial Tim-3 inhibits the secretion of inflammatory factors and modulates microglia to antiinflammatory phenotype after experimental intracerebral hemorrhage in rats. CNS Neurosci Ther. 2019 Jun;25(6):674–84.
- 79. Ren H, Kong Y, Liu Z, Zang D, Yang X, Wood K, et al. Selective NLRP3(Pyrin domain-containing protein inflammation inhibitor reduces brain injury after intracerebral hemorrhage. Stroke. 2018 Jan;49:184–92.
- 80. Feng L, Chen Y, Ding R, Fu Z, Yang S, Deng X, et al. P2X7R blockade prevents NLRP3 inflammasome activation and brain injury in a rat model of intracerebral hemorrhage: involvement of peroxynitrite. J Neuroinflammation. 2015 Oct;12:190.
- 81. Mracsko E, Javidi E, Na SY, Kahn A, Liesz A, Veltkamp R. Leukocyte invasion of the brain after experimental intracerebral hemorrhage in mice. Stroke. 2014 Jul;45:2107–14. http://dx.doi.org/10.1161/STROKEAHA.114.005801.
- 82. Yao Y, Tsirka SE. Chemokines and their receptors in intracerebral hemorrhage. Transl Stroke Res. 2012 Jul;3:70–9. http://dx.doi.org/10.1007/s12975-012-0155-z.
- 83. Carmona-Mora P, Ander BP, Jickling GC, Dykstra-Aiello C, Zhan X, Ferino E, et al. Distinct peripheral blood monocyte and neutrophil transcriptional programs following intracerebral hemorrhage and different etiologies of ischemic stroke. J Cere Blood Flow Metab. 2021 Jun;41(6):1398–416.
- 84. Hammond MD, Taylor RA, Mullen MT, Ai Y, Aguila HL, Mack M, et al. CCR2+Ly6C(hi) inflammatory monocyte recruitment exacerbates acute disability following intracerebral hemorrhage. J Neurosci. 2014 Mar;34:3901–9.
- 85. Landreneau MJ, Mullen MT, Messe SR, Brett C, Kevin S, Louise M, et al. CCL2 and CXCL10 are associated with poor outcome after intracerebral hemorrhage. Ann Clin Transl Neurol. 2018;5:962–70.
- 86. Walsh KB, Campos B, Hart K, Thakar C, Adeoye O. M2 monocyte microparticles are increased in intracerebral hemorrhage. J Stroke Cerebrovasc Dis. 2017 Oct;26(10):2369–75. http://dx.doi.org/10.1016/j.jstrokecerebrovasdis.2017.05.027.
- 87. Hammond MD, Ai Y, Sansing LH. Gr1+ Macrophages and Dendritic Cells Dominate the Inflammatory Infiltrate 12 Hours After Experimental Intracerebral Hemorrhage. Transl Stroke Res. 2012 Jul;3(1):s125–31. http://dx.doi.org/10.1007/s12975-012-0174-9.
- 88. Draxler DF, Lee F, Ho H, Keragala CB, Medcalf RL, Niego B. t-PA suppresses the immune response and aggravates neurological deficit in a murine model of ischemic stroke. Front Immunol. 2019 Mar;10:591. http://dx.doi.org/10.3389/fimmu.2019.00591.
- 89. Medcalf RL, Keragala CB. Fibrinolysis: a primordial system linked to the immune response. Int J Mol Sci. 2021 Mar;22(7):3406. http://dx.doi.org/10.3390/ijms22073406.
- 90. Li F, Pendy JT Jr, Ding JN, Peng C, Li X, Shen J, et al. Exercise rehabilitation immediately following ischemic stroke exacerbates inflammatory injury. Neurol Res. 2017 Jun;39(6):530–7.
- 91. Markus HS, Hayter E, Levi C, Feldman A, Venables G, et alCADISS trial investigators. Antiplatelet treatment compared with anticoagulation treatment for cervical artery dissection (CADISS): a randomised trial. Lancet Neurol. 2015 Apr;14(4):361–7.
- 92. Subramanian S, Zhang B, Kosaka Y, Burrows GG, Grafe MR, Vandenbark AA, et al. Recombinant T cell receptor ligand treats experimental stroke. Stroke. 2009 Jul;40(7):2539–45.
- 93. Dziennis S, Mader S, Akiyoshi K, Ren X, Ayala P, Burrows GG, et al. Therapy with recombinant T-cell receptor ligand reduces infarct size and infiltrating inflammatory cells in brain after middle cerebral artery occlusion in mice. Metab Brain Dis. 2011 Jun;26(2):123–33.
- 94. Lambertsen KL, Biber K, Finsen B. Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab. 2012 Sep;32:1677–98. http://dx.doi.org/10.1038/jcbfm.2012.88.
- 95. Boutin H, LeFeuvre RA, Horai R, Asano M, Iwakura Y, Rothwell NJ. Role of IL-1alpha and IL-1beta in ischemic brain damage. J Neurosci. 2001 Aug;21:5528–34. http://dx.doi.org/10.1523/jneurosci.21-15-05528.2001.
- 96. Spulber S, Bartfai T, Schultzberg M. IL-1/IL-1ra balance in the brain revisited: evidence from transgenic mouse models. Brain Behav Immun. 2009 Jul;23:573–9. http://dx.doi.org/10.1016/j.bbi.2009.02.015.
- 97. Smith CJ, Emsley HC, Udeh CT, Vail A, Hoadley ME, Rothwell NJ, et al. Interleukin-1 receptor antagonist reverses stroke-associated peripheral immune suppression. Cytokine. 2012 Jun;58:384–9.
- 98. Qu C, Li W, Shao Q, Dwyer T, Huang H, Yang T, et al. c-Jun N-terminal kinase 1 (JNK1) is required for coordination of netrin signaling in axon guidance. J Biol Chem. 2013 Jan;288(3):1883–95.
- 99. Gueorguieva I, Clark SR, McMahon CJ, Scarth S, Rothwell NJ, Tyrrell PJ, et al. Pharmacokinetic modelling of interleukin-1 receptor antagonist in plasma and cerebrospinal fluid of patients following subarachnoid hemorrhage. Br J Clin Pharmacol. 2008 Mar;65(3):317–25.
- 100. Yli-Karjanmaa M, Clausen BH, Degn M, Novrup HG, Ellman DG, Toft-Jensen P, et al. Topical administration of a soluble TNF inhibitor reduces infarct volume after focal cerebral ischemia in mice. Front Neurosci. 2019 Aug;13:781.
- 101. Clausen BH, Degn M, Martin NA, Couch Y, Karimi L, Ormhøj M, et al. Systemically administered anti-TNF therapy ameliorates functional outcomes after focal cerebral ischemia. J Neuroinflammation. 2014 Dec;11:203.
- 102. Wu MH, Huang CC, Chio CC, Tsai KJ, Chang CP, Lin NK, et al. Inhibition of peripheral TNF-α and downregulation of microglial activation by alpha-lipoic acid and etanercept protect rat brain against ischemic stroke. Mol Neurobiol. 2016 Sep;53(7):4961–71.
- 103. Wu T, Wu H, Wang J, Wang J. Expression and cellular localization of cyclooxygenases and prostaglandin E synthases in the hemorrhagic brain. J Neuroinflammation. 2011 Mar;8:22. http://dx.doi.org/10.1186/1742-2094-8-22.
- 104. Lee SH, Park HK, Ryu WS, Lee JS, Bae HJ, Han MK, et al. Effects of celecoxib on hematoma and edema volumes in primary intracerebral hemorrhage: a multicenter randomized controlled trial. Eur J Neurol. 2013 Aug;20(8):1161–9.
- 105. Chu K, Jeong SW, Jung KH, Han SY, Lee ST, Kim M, et al. Celecoxib induces functional recovery after intracerebral hemorrhage with reduction of brain edema and perihematomal cell death. J Cereb Blood Flow Metab. 2004 Aug;24(8):926–33.
- 106. Wasserman JK, Schlichter LC. Minocycline protects the blood-brain barrier and reduces edema following intracerebral hemorrhage in the rat. Exp Neurol. 2007 Oct;207(2):227–37. http://dx.doi.org/10.1016/j.expneurol.2007.06.025.
- 107. Lee CZ, Xue Z, Zhu Y, Yang GY, Young WL. Matrix metalloproteinase-9 inhibition attenuates vascular endothelial growth factor-induced intracerebral hemorrhage. Stroke. 2007 Sep;38(9):2563–8. http://dx.doi.org/10.1161/STROKEAHA.106.481515.
- 108. Dai S, Hua Y, Keep RF, Novakovic N, Fei Z, Xi G. Minocycline attenuates brain injury and iron overload after intracerebral hemorrhage in aged female rats. Neurobiol Dis. 2019 Jun;126:76–84. http://dx.doi.org/10.1016/j.nbd.2018.06.001.
- 109. Wu Z, Zou X, Zhu W, Mao Y, Chen L, Zhao F. Minocycline is effective in intracerebral hemorrhage by inhibition of apoptosis and autophagy. J Neurol Sci. 2016 Dec;371:88–95. http://dx.doi.org/10.1016/j.jns.2016.10.025.
- 110. Yang H, Gao XJ, Li YJ, Su JB, E TZ, Zhang X, et al. Minocycline reduces intracerebral hemorrhage-induced white matter injury in piglets. CNS Neurosci Ther. 2019 Oct;25(10):1195–206.
- 111. Yang Y, Zhang K, Yin X, Lei X, Chen X, Wang J, et al. Quantitative iron neuroimaging can be used to assess the effects of minocycline in an intracerebral hemorrhage minipig model. Transl Stroke Res. 2020 Jun;11(3):503–19.
- 112. Chang JJ, Kim-Tenser M, Emanuel BA, Jones GM, Chapple K, Alikhani A, et al. Minocycline and matrix metalloproteinase inhibition in acute intracerebral hemorrhage: a pilot study. Eur J Neurol. 2017 Nov;24(11):1384–91.
- 113. Lan X, Han X, Li Q, Li Q, Gao Y, Cheng T, et al. Pinocembrin protects hemorrhagic brain primarily by inhibiting toll-like receptor 4 and reducing M1 phenotype microglia. Brain Behav Immun. 2017 Mar;61:326–39. http://dx.doi.org/10.1016/j.bbi.2016.12.012.
- 114. Nitzsche A, Poittevin M, Benarab A, Bonnin P, Faraco G, Uchida H, et al. Endothelial S1P signaling counteracts infarct expansion in ischemic stroke. Circ Res. 2021 Feb;128(3):363–82.
- 115. Campos F, Qin T, Castillo J, Seo JH, Arai K, Lo EH, et al. Fingolimod reduces hemorrhagic transformation associated with delayed tissue plasminogen activator treatment in a mouse thromboembolic model. Stroke. 2013 Feb;44(2):505–11.
- 116. Li W, He T, Jiang L, Shi R, Song Y, Mamtilahun M, et al. Fingolimod inhibits inflammation but exacerbates brain edema in the acute phases of cerebral ischemia in diabetic mice. Front Neurosci. 2020 Aug;14:842.
- 117. Zhu Z, Fu Y, Tian D, Sun N, Han W, Chang G, et al. Combination of the immune modulator fingolimod with alteplase in acute ischemic stroke: a pilot trial. Circulation. 2015 Sep;132(12):1104–12.
- 118. Tian DC, Shi K, Zhu Z, Yao J, Yang X, Su L, et al. Fingolimod enhances the efficacy of delayed alteplase administration in acute ischemic stroke by promoting anterograde reperfusion and retrograde collateral flow. Ann Neurol. 2018 Nov;84(5):717–28.
- 119. Fu Y, Hao J, Zhang N, Ren L, Sun N, Li YJ, et al. Fingolimod for the treatment of intracerebral hemorrhage: a 2-arm proof-of-concept study. JAMA Neurol. 2014 Sep;71(9):1092–101.
- 120. Li YJ, Chang GQ, Liu Y, Gong Y, Yang C, Wood K, et al. Fingolimod alters inflammatory mediators and vascular permeability in intracerebral hemorrhage. Neurosci Bull. 2015 Dec;31(6):755–65.
- 121. Zhu H, Jian Z, Zhong Y, Ye Y, Zhang Y, Hu X, et al. Via Janus kinase inhibition ameliorates ischemic stroke injury and neuroinflammation through reducing NLRP3 inflammasome activation JAK2/STAT3 pathway inhibition. Front Immunol. 2021 Jul;12:714943.
- 122. Yang X, Sun J, Kim TJ, Kim YJ, Ko SB, Kim CK, et al. Pretreatment with low-dose fimasartan ameliorates NLRP3 inflammasome-mediated neuroinflammation and brain injury after intracerebral hemorrhage. Exp Neurol. 2018 Dec;310:22–32.
- 123. Li X, Wang T, Zhang D, Li H, Shen H, Ding X, et al. Andrographolide ameliorates intracerebral hemorrhage induced secondary brain injury by inhibiting neuroinflammation induction. Neuropharmacology. 2018 Oct;141:305–15.
- 124. Erta M, Quintana A, Hidalgo J. Interleukin-6, a major cytokine in the central nervous system. Int J Biol Sci. 2012 Oct;8:1254–66. http://dx.doi.org/10.7150/ijbs.4679.
- 125. Jostock T, Müllberg J, Ozbek S, Atreya R, Blinn G, Voltz N, et al. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor trans-signaling responses. Eur J Biochem. 2001 Jan;268:160–7.