Introduction
A little more than 20 years ago, Clifford Saper and Christopher Breder summarized in an authoritative review in The New England Journal of Medicine what was known at that time about “The Neurological Basis of Fever” (). While the critical role of peripherally released cytokines for the febrile response was recognized, it was not clear how these substances could signal to the brain since they could not pass the blood-brain barrier. And although it also was known that prostaglandins were involved in the elaboration of fever, it was not clear where and by which cells the fever-inducing prostaglandins were produced. Furthermore, although it was known that the elevated body temperature was generated by increased energy production and diminished energy loss (by peripheral vasoconstriction), little was known about central neural circuits involved. In this review, we will address our current knowledge of these issues and also point out outstanding questions that deserve further investigation.
Fever is a hallmark of infectious and inflammatory diseases. It is generated by the concerted action of various autonomic responses, such as peripheral vasoconstriction and decreased sweating, reducing heat loss, and shivering, and possibly also non-shivering, thermogenesis. Fever is considered beneficial because an elevated body temperature enhances the activity of the immune cells while at the same time it impairs the replication of many microorganisms (; ), although controlled clinical studies of the benefit of fever are lacking (). The elevation of the body temperature on immune challenge is a stereotypic response seen in all vertebrates, including poikilotherms, which have been shown to prefer a warmer environment when they have an infection ().
It was demonstrated already at the end of the 19th century that fever required the involvement of the brain (see ). The American pathologist/bacteriologist William H. Welch showed that animals with cervical spinal cord transection did not respond with fever when given an intravenous (i.v.) injection of a pyrogen. It was also understood at that time that the inflammatory process resulted in the release of substances that produced the fever. However, it remained for long unclear how these substances, later named endogenous pyrogens and subsequently identified as cytokines (), could influence the brain, since the brain was protected by the blood-brain barrier, described early in the 20th century (). Nevertheless, injection of endogenous pyrogens into the carotid artery was demonstrated to result in a rapid and strong febrile response, suggesting a direct action on the thermoregulatory center in the brain (), and this idea was further supported by the finding that when injected directly into the brain, endogenous pyrogens elicited fever when administered into the anterior hypothalamus/preoptic region, but not when injected into other brain areas (). Based on subsequent observations that prostaglandins of the E-series, when injected into the cerebral ventricles, elicited fever (), it was further suggested that the endogenous pyrogens acted by releasing prostaglandins (), an idea that was reinforced by the demonstration that antipyretic drugs like aspirin exerted their mode of action by prostaglandin inhibition ().
Routes for Immune-to-Brain Signaling in Fever
Over the years, several different hypotheses emerged on how peripheral immune signals could traverse, or circumvent, the blood-brain barrier to influence the brain and elicit fever. These hypotheses include direct pyrogen action on the organum vasculosum of the lamina terminalis, activation of cells in the blood-brain barrier, transfer of blood-borne prostaglandin E2 (PGE2) into the brain, and activation of peripheral nerves by immune signals (Fig. 1). We will critically examine each of these.
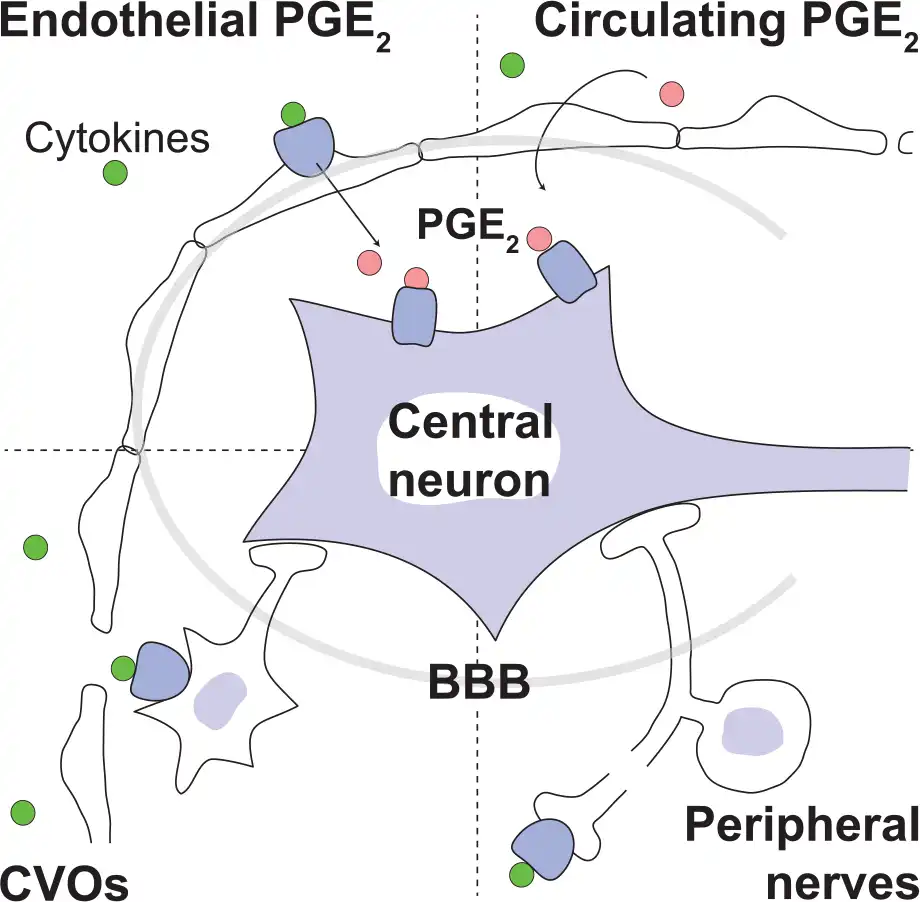
Figure 1
Different suggested routes by which peripherally released inflammatory signals can bypass the blood-brain barrier (BBB) and activate the central nervous system: Peripherally released proinflammatory cytokines (green circles) (i) bind to receptors on cells of brain blood vessels to induce synthesis of prostaglandin E2 (PGE2; pink circles), which then is transported into the brain parenchyma (upper left); (ii) activate neurons of circumventricular organs (CVOs), which contain fenestrated capillaries (lower left); or (iii) activate peripheral nerves (lower right). (iv) Peripheral inflammation may also release circulating PGE2 that enters the brain (upper right).
Role of the Organum Vasculosum of the Lamina Terminalis for Fever
The organum vasculosum of the lamina terminalis (OVLT) belongs to the circumventricular organs (CVOs), which are parts of the brain that lack the normal blood-brain barrier (). The capillaries in the CVOs are fenestrated and blood-borne macromolecules can reach the cells within these structures, which hence can monitor essential information of importance for, for example, fluid balance (angiotensin II, natriuretic peptides, osmolarity), metabolic control (amylin, ghrelin, leptin), and reproduction (relaxin). By their efferent projections, neurons in the CVOs can transmit this information to control centers in the hypothalamus and brain stem that serve to maintain homeostasis ().
The CVOs were early on suggested as candidates for relaying inflammatory signals to neurons in deep brain structures. In support of this idea, it was shown that the sensory CVOs, which in addition to OVLT include the subfornical organ and the area postrema, express receptors for pathogen associated molecular patterns (PAMPs) and cytokines such as interleukin (IL)-1β, IL-6 and tumor necrosis factor alpha (TNFα) (; ; ; , ; ), although it seems as if the receptor expressing cells are endothelial cells and not neurons and preferentially located in the vicinity of the CVOs (; see also ). In addition, the sensory CVOs constitutively express microsomal prostaglandin E synthase-1 (mPGES-1) (), a terminal enzyme for PGE2 synthesis (), implying local PGE2 synthesis in these structures. It has also been demonstrated that cells in the sensory CVOs respond with a lower threshold to peripheral immune stimuli than other structures in the brain (), and that peripheral immune challenge induces cytokine expression in the sensory CVOs (; ; ). Furthermore, lesion studies indicate that the area postrema may contribute to IL-1β-induced hypothalamic-pituitary-adrenal axis activation () and to the anorexic response in various disease paradigms, including peripheral cytokine release (; ).
As for the role of sensory CVOs for fever, the OVLT has been the focus of interest, because of its location adjacent to thermoregulatory neurons in the preoptic hypothalamus. In a study in guinea pigs by Blatteis and collaborators, lesions of the anteroventral third ventricle, including the OVLT, suppressed fever induced by peripheral injection of bacterial wall lipopolysaccharide (LPS), a widely used model for peripheral inflammation (). However, subsequent studies on animals with lesions of the OVLT have provided contradictory results, with some studies showing attenuated fever, that is, supporting the findings of , and other studies showing augmented fever or no effect (for a review of the literature, see ). Because of the small size and position of the OVLT, ablation of the OVLT without damaging adjacent structures has been difficult to achieve, and OVLT lesions have been reported to elicit several acute and chronic effects, which are likely to be due to such additional damage, and which are likely to influence the febrile response. Hence, in one carefully executed study, rats with OVLT lesions were found to exhibit emaciation, hyporeactivity of osmotic stimulation, chronic hypernatremia, and hyperosmolality, and, most important, hyperthermia ().
A single study has examined the effect of ablation of the subfornical organ for immune-induced fever (). In that study, ablation of the subfornical organ attenuated fever elicited by peripheral injection of LPS, whereas ablation of the OVLT or the area postrema had no effect. This study clearly needs to be repeated in independent experiments.
After the recognition of the “side effects” seen following in particular OVLT ablation (), interest in the potential role of the CVOs for eliciting fever has faded. However, a recent report comparing the febrile response in mouse lines with different patterns of deletion in brain endothelial cells of MyD88, an adaptor protein for LPS and IL-1β signaling (), suggested that fenestrated capillaries in the CVOs were important for fever in response to IL-1β injected into the brain ventricles (). However, apart from differences between the mouse lines regarding MyD88 deletion in fenestrated capillaries, there were several other potential differences between these mouse lines, such as recombination efficacy in the brain endothelium and peripheral immune cells that may have influenced the results. Furthermore, since the IL-1β was given intracerebroventricularly (i.c.v.), it is not clear if the findings are relevant for IL-1-signaling from the periphery to the brain.
The Blood-Brain Barrier as Transducer of Immune Signals to the Brain
In vitro studies in the late 1980s demonstrated the presence of PGE2 production in the brain microvasculature (; ), but the critical evidence for blood-brain barrier cells as transducers of immune signals to the brain came with the in vivo demonstration of LPS-elicited expression of immunoreactivity for PGE2 () and of the inducible prostaglandin synthesizing enzyme cyclooxygenase-2 (Cox-2) in these cells (; ). However, immediately after the latter discovery the identity of the prostaglandin producing cells became a matter of controversy, since some studies suggested that they were endothelial cells whereas other studies identified them as perivascular cells, immune cells located in the vessel wall on the parenchymal side of the endothelial cells, enveloped by the two sheets of the basal lamina (for a review of the literature, see ). While most investigators now agree on that the vast majority of the brain vascular cells that express Cox-2 in response to moderate and high doses of LPS, independently of route of administration, are endothelial cells, there are still different opinions with regard to the response to IL-1β and to low doses of LPS, with one laboratory reporting that perivascular cells are the main source of PGE2 under these conditions (; ).
Although the findings demonstrating induced Cox-2 expression in the brain vasculature strongly indicated that blood-brain barrier cells were the site of PGE2 production, the definite conclusion was hampered by the lack of evidence that these cells in fact produced PGE2, because Cox-2 catalyzes the formation of PGH2, which in turn can be transformed into several other prostanoids. The critical and final step came with the demonstration that the inducible terminal PGE2 synthase, microsomal prostaglandin E synthase 1 (mPGES-1) (), was expressed by brain vascular cells (; ; ). These studies demonstrated that mPGES-1 was only minimally expressed in the naïve brain of the species examined (rat) but was strongly induced in the brain vasculature following i.v. injection of a low dose IL-1β () and intraperitoneal (i.p.) injection of moderate to high doses of LPS (; ), respectively. The vascular expression of mPGES-1 was localized to cells that expressed IL-1 type 1 receptor (IL-1R1), Cox-2 and an endothelial cell marker (; ; ; see also ). Subsequent studies, including studies using more natural disease models such as arthritis, burn injury, and inflammation in the paw (; ; ; ; ; ), as well as studies in mice (; ), also found mPGES-1 induction and expression in brain endothelial cells but not in perivascular cells (Fig. 2). There is, to our knowledge, only a single study, performed in rats, that reports mPGES-1 induced expression in perivascular cells ().
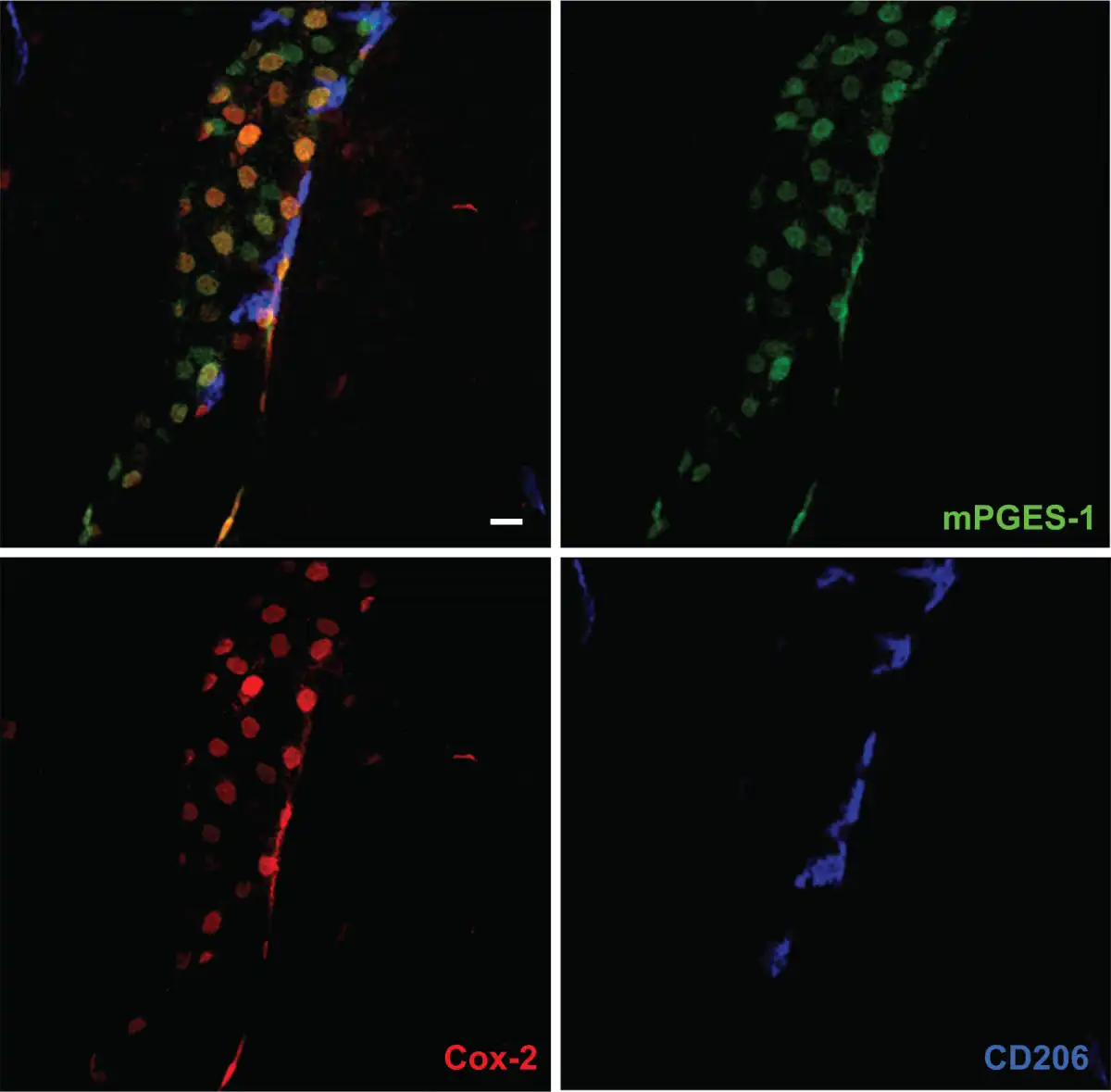
Figure 2
Confocal micrographs of blood vessel in the mouse brain, stained with antibodies against the prostaglandin E2 synthesizing enzymes cyclooxygenase-2 (Cox-2) and microsomal prostaglandin E synthase-1 (mPGES-1), and CD206, a macrophage marker expressed by perivascular cells. Upper left panel shows triple labeling for these proteins, and the other panels single labeling for each protein. Note that most cells that express Cox-2 also express mPGES-1 and vice versa. Note also that none of the Cox-2/mPGES-1 expressing cells stain for CD206, implying that this population does not include perivascular cells. Scale bar = 20 µm.
Studies of animals with global deletion of Cox-2 and mPGES-1, which corroborated the idea that these enzymes were critical for fever (; ; ; ), were subsequently followed by functional studies of the role of the prostaglandin synthesis in the brain vasculature for the febrile response. The data obtained so far, show a critical but maybe not exclusive role for the brain endothelial cells. The first functional evidence came in a study by , showing that knockdown of the IL-1R1 in brain endothelial cells abolished sickness symptoms (fever and reduced locomotor activity) elicited by i.v. injected IL-1β as well as the induced blood-brain barrier Cox-2 expression and paraventricular hypothalamic Fos expression, hence demonstrating the critical role of endothelial IL-1R1 for these phenomena. Subsequently, demonstrated that brain endothelial specific deletion of the MAP (mitogen-activated protein) kinase kinase kinase TAK1, which is an important component of IL-1β signaling upstream of the transcription factors nuclear factor–κB (NF-κB) and c-Jun that control Cox-2 gene transcription, resulted in a blunted febrile response to i.v. injected IL-1β. Using chimeric mice that expressed the terminal PGE2-synthesizing enzyme mPGES-1 in either hematopoietic cells (including perivascular macrophages) or non-hematopoietic cells (including brain endothelial cells), Engström and coworkers showed that mPGES-1 in non-hematopoietic cells was sufficient for eliciting a febrile response to a peripheral immune challenge, whereas expression of mPGES-1 restricted to hematopoietic cells, including perivascular cells, resulted in ablated fever () (Fig. 3). Finally, provided direct evidence that PGE2 synthesis in endothelial cells was critically involved in the febrile response. Using the same tissue specific Cre-mediated recombination as in the study by , Wilhelms and collaborators demonstrated that brain endothelial specific deletion of Cox-2 and mPGES-1 resulted in blunted fever after i.p. injection of IL-1β and LPS () (Fig. 4), whereas deletion in other cell types, such as nerve cells and myeloid cells, has been shown to have no effect (). The attenuation of fever in the study of was not due to unspecific effects on the inflammatory process, since IL-1β levels in the blood after LPS challenge were unaffected as well as the brain endothelial induction of inflammatory genes (Cxcl10, Ccl2, and Lcn2). This specificity is also demonstrated by the findings that many other symptoms of systemic inflammation are unaffected by the same endothelium-specific manipulations that attenuate fever. Thus inflammation-induced anorexia, inactivity, and hypothalamic-pituitary-adrenal axis activation have been shown to be intact in mice with endothelial manipulations attenuating fever, whereas inflammation-induced place aversion also requires brain endothelial signaling (; ; ; ; ). Adding to the finding by Wilhelms and coworkers, it was also demonstrated that deletion of IL-6 receptor alpha (IL-6Rα) and the IL-1R1 on brain endothelial cells attenuated the febrile response to LPS (; ; see below). Taken together, the findings from the functional studies using cell-type specific genetic manipulation strongly support a role of the brain endothelium in the generation of fever. Since cell-type specific manipulations are not always 100% specific or selective, it is important to note that the role of the brain endothelium has been shown using two different endothelium specific promoters, that is, the Slco1c-promoter and the Tie2-promoter. It is hence very unlikely that any off-target effects would explain the effects seen in these studies. It is more likely that the importance of the brain endothelium was underestimated, because the Cre-lines used may not target all brain endothelial cells (see, for example, ).
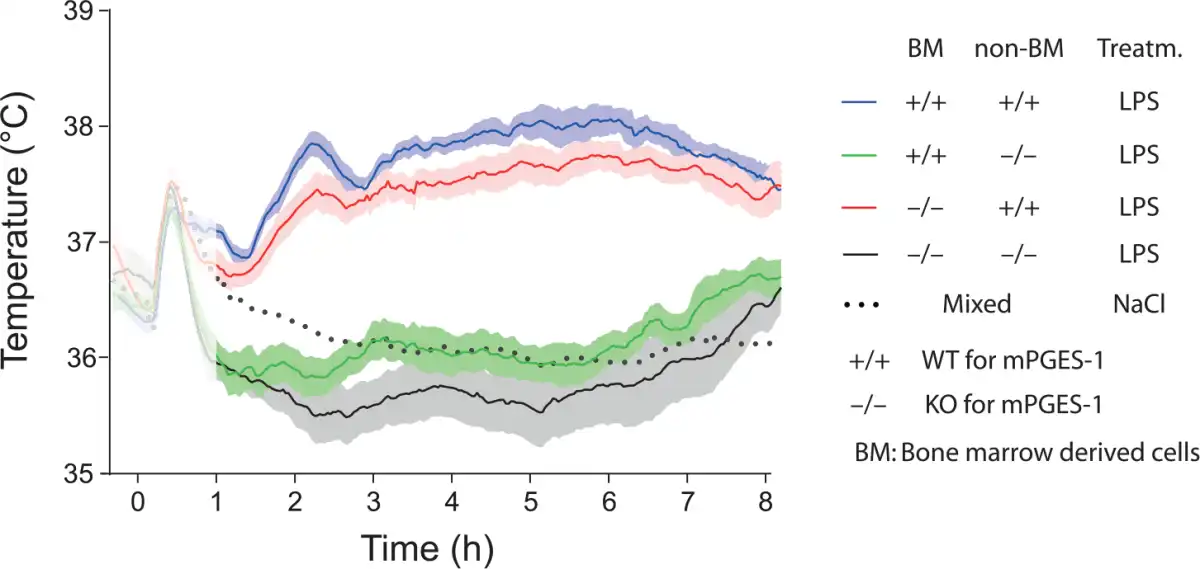
Figure 3
Temperature responses to intraperitoneal injection of bacterial wall lipopolysaccharide (LPS) in wild type (WT) and mPGES-1 knockout (KO) mice that were subjected to whole body irradiation and then transplanted with either WT (+/+) or KO (−/−) bone marrow. Note that WT mice (non-BM +/+) transplanted with WT or KO bone marrow display a prominent febrile response (two top fever curves), whereas KO mice (non-BM −/−) transplanted with WT or KO bone marrow are afebrile (lower fever curves). The initial temperature peak (shadowed) in all groups is handling stress-induced hyperthermia. Replacement of native hematopoietically derived cells was in these experiments about 90% among white blood cells and brain macrophages (perivascular cells), and around 70% among liver (Kupffer cells) and lung macrophages. For the NaCl treated group mean is shown, whereas for the other traces mean and SEM (standard error of the mean) are shown. Adapted from .
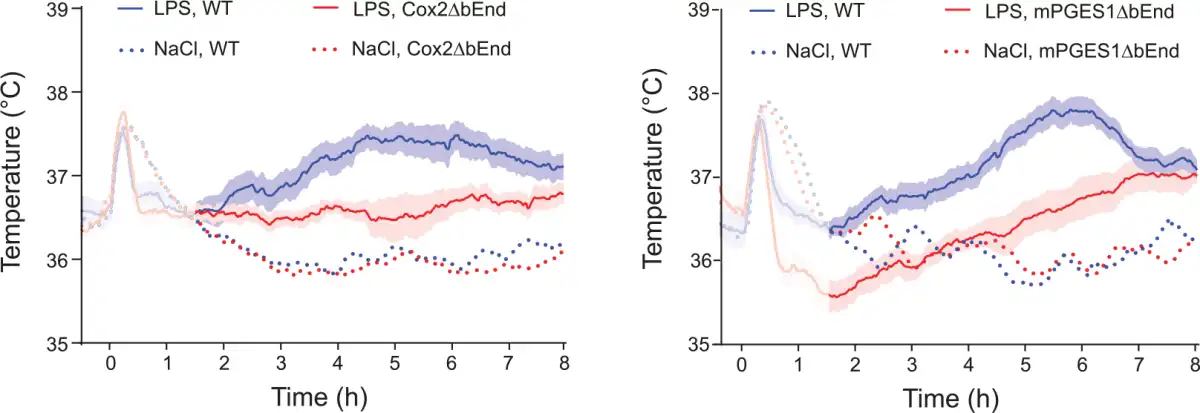
Figure 4
Blunted febrile response to intraperitoneally injected bacterial wall lipopolysaccharide (LPS) in mice with deletion selectively in brain endothelial cells of the prostaglandin E2 synthesizing enzymes cyclooxygenase-2 (Cox2ΔbEnd) and microsomal prostaglandin E synthase-1 (mPGES1ΔbEnd). WT, wild type mice. For the NaCl-treated groups mean is shown, whereas for the LPS treated groups mean and SEM (standard error of the mean) are shown. Adapted from .
Role of Perivascular Cells for the Febrile Response
While the studies using genetic deletions of inflammatory signaling molecules in endothelial cells hence provide convincing evidence for a critical role of these cells in the febrile response, the contribution of brain perivascular macrophages to fever seems limited. It has been suggested that perivascular cells on the one hand are critical for some aspects of central nervous system activation by IL-1β, but that these cells, on the other hand, subserve an inhibitory action on the PGE2 synthesis in endothelial cells and the concomitant acute phase responses elicited by this PGE2 production. Using a model in which perivascular cells were ablated by intracerebral injection of chlodronate liposomes (), Serrats and coworkers reported that such ablation attenuated the cerebral Cox-2 induction as well as the ACTH and corticosterone release in response to i.v. injection of IL-1β, but augmented the same responses following i.v. injection of LPS (). In contrast, the febrile response was intact or moderately enhanced, both to LPS and IL-1β. Thus, although the methodology in this study leaves many open questions regarding the relationship between Cox-2 induction in perivascular cells and the hypothalamic-pituitary-adrenal axis activation, it seems clear that perivascular cells are not important for eliciting the febrile response.
Role of IL-1 and IL-6 Signaling Across the Blood-Brain Barrier for the Febrile Response
As described above, IL-1 signaling in brain endothelial cells is critical for the febrile response to exogenously administered IL-1β. However, the contribution of this pathway for the febrile response to a more natural stimulus, such as LPS, is less clear. Thus, mice with deletion of IL-1β not only show intact acute phase responses (), but even display exaggerated fever () when immune challenged with LPS. Furthermore, early studies on mice with global deletion of the IL-1R1 and studies on mice that were treated with an IL-1 receptor antagonist reported that these mice displayed intact or only slightly reduced fever in response to LPS (; ; ; ; ), implying that the IL-1-signaling pathway may not be critical for the febrile response to this stimulus. We recently reexamined this issue, and found that global deletion of IL-1R1, as well as treatment of mice with an IL-1 receptor antagonist, attenuated but did not abolish the febrile response to i.p. injected LPS (). Furthermore, using mice with cell specific deletions of IL-1R1s, we observed attenuation of the febrile response following deletion of IL-1R1 in brain endothelial cells, but not after deletion of this receptor in neural cells or peripheral nerves. In the global knock-out mice, the remaining/attenuated fever was a delayed response, seen from about 5 to 6 hours after the LPS injection, compared with about 3 hours in wild-type mice (Fig. 5). Similar findings were reported by Ching et al. using IL-1R1 knock-down in endothelial cells. While this procedure completely abolished the response to i.v. and i.c.v. injected IL-1β, it only attenuated and delayed the febrile response to i.p. injection of IL-1β (). Taken together, these observations seem to suggest that i.p. injection of LPS or IL-1β elicits fever that in part is independent of brain endothelial IL-1R1 signaling.
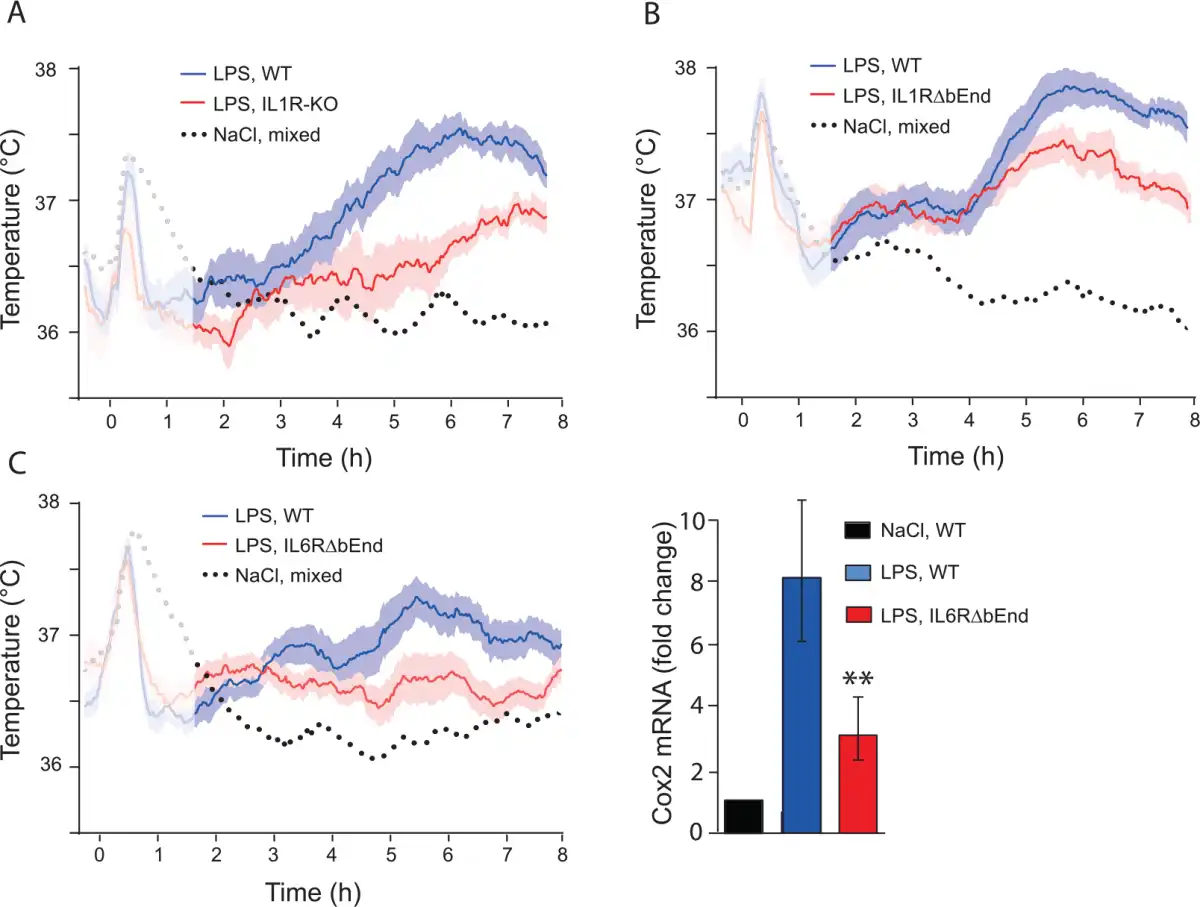
Figure 5
Febrile response to bacterial wall lipopolysaccharide (LPS) in mice with deletion of cytokine receptors. (A) Mice with global deletion of the interleukin-1 type 1 receptor (IL1R-KO) show attenuated fever, however note the late appearing fever in these mice. (B) Attenuated fever, seen after about 5 h, in mice with deletion of the IL-1R1 selectively in brain endothelial cells (IL1RΔbEnd). (C) Attenuated fever in mice with deletion of the interleukin-6 receptor alpha selectively in brain endothelial cells (IL6RΔbEnd) (left). This response was associated with attenuated induction of cyclooxygenase-2 (Cox-2) in the hypothalamus (right). ** indicates P < 0.01. Adapted from and Eskilsson and others (2014).
In contrast to IL-1β, IL-6, which is also released on, for example, LPS challenge, seems to be critical for LPS-induced fever. IL-6 knock-out animals, as well as animals given neutralizing antibodies against IL-6, cannot mount a febrile response (; ; ), in spite of the fact that IL-6 by itself is not or only weakly pyrogenic (; ; ; ). The action of IL-6 seems to be exerted via signaling in brain endothelial cells, because mice with endothelial specific deletion of the IL-6Rα showed, as reported above, strongly attenuated fever to peripherally administered LPS, and the same was found for mice with deletion of the downstream signaling molecule STAT3. The IL-6Rα deletion also strongly reduced LPS-induced Cox-2 expression in the brain endothelial cells (Fig. 5) (). An intriguing, yet unanswered question is how the IL-6 signaling pathway interacts with the IL-1-signaling pathway (Fig. 6), and perhaps also with Toll-like receptor 4-signaling, in brain endothelial cells, considering that both cytokines seem to be necessary for the febrile response to LPS. It should be noted that endogenous TNFα, which similar to IL-1β and IL-6 is released by LPS, is a cryogen, although it evokes fever when administered exogenously, probably by releasing other cytokines (; ).
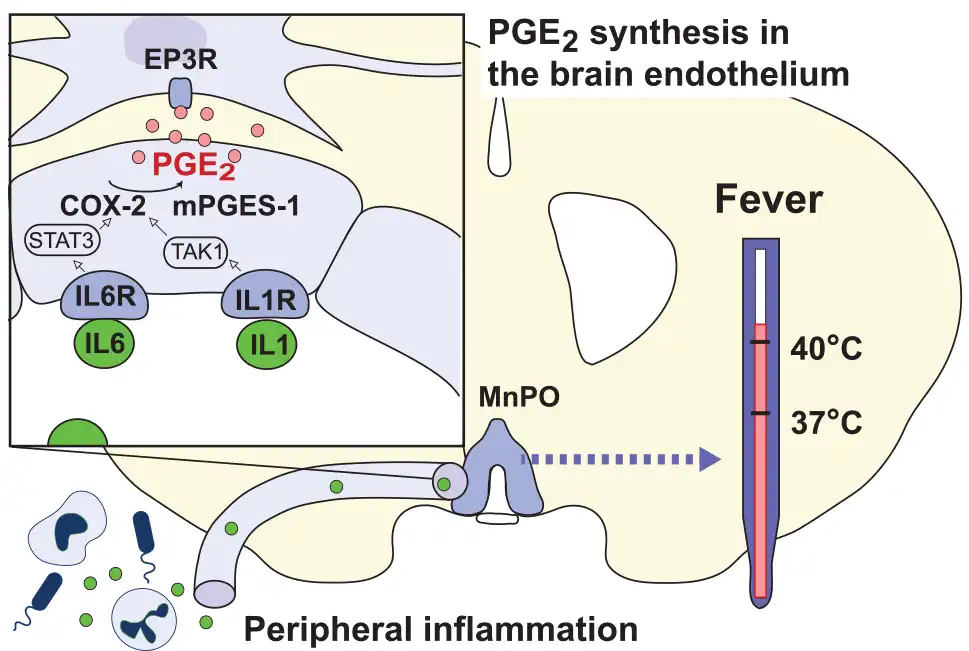
Figure 6
Transduction mechanisms in the blood-brain barrier elicited by peripherally released inflammatory mediators. The cytokines IL-1β and IL-6 (green circles) bind to receptors (IL1R, IL6R) on brain endothelial cells in the preoptic hypothalamus resulting in transcription of cyclooxygenase-2 (COX-2) and microsomal prostaglandin E synthase-1 (mPGES-1) via TAK1 and STAT3, respectively. The subsequent binding of neosynthesized PGE2 (pink circles) to PGE2 EP3 receptor (EP3R) expressing cells in the median preoptic nucleus (MnPO) of the hypothalamus elicits fever.
A Febrile Response to Blood-Borne PGE2?
When mice and rats are given an i.p. injection of LPS, the initial temperature peak induced by the handling stress is followed by a biphasic febrile response (). However, if injections instead are given via an indwelling venous catheter during conditions that do not involve handling of the animals, a triphasic febrile response is seen, with the first phase present during the first 60 minutes after injection, and hence obscured by the handling stress that occurs when injections are given i.p. (; ). The mechanism behind this first febrile phase has been the subject of much debate and has been suggested to involve activation of peripheral nerves, in particular the vagus nerve, as will be discussed below (see section “Role of Peripheral Nerves for Fever”). However, a prevailing idea, suggested by Romanovsky and collaborators, is that the first phase of fever is elicited by the release of PGE2 from lung macrophages into the circulation, and that the blood-borne PGE2 enters the brain in the preoptic area, there to elicit the febrile response (). This hypothesis is based mainly on two observations. First, the first phase of fever was found to precede the induction of prostaglandin synthesizing enzymes in the hypothalamus as determined by Western blot, whereas it was concomitant to cyclooxygenase induction seen in peripheral tissues, and in particular that in lung macrophages (; ). Second, i.v. injection of neutralizing antibodies against PGE2 attenuated (but did not abolish) the initial febrile response to peripherally administered LPS (). Both observations need to be interpreted with some caution. Induced synthesis of, for example, Cox-2 in a few but critically located blood vessel (i.e., in the preoptic hypothalamus) may not have been detected by protein analysis of the entire hypothalamus. It has been shown that blood vessels in key autonomic regions display more pronounced expression of IL-1 receptors and downstream intracellular signaling molecules than blood vessels in other regions (). Furthermore, recent work in this laboratory has demonstrated strong Cox-2 mRNA induction in the hypothalamus associated with high levels of PGE2 in the cerebral fluid already 30 minutes after i.v. injection of LPS in mice, as well as Cox-2 protein expression in hypothalamic blood vessels (Eskilsson and others, unpublished). As for the experiments with the neutralizing antibodies, it should be noted that the effect was only partial. While this observation could be due to a failure of the antibodies to completely neutralize all circulating PGE2, it also opens for the possibility that an additional mechanism for the initiation of fever, independent of circulating PGE2, exists. Yet another possibility that cannot be excluded is that a small proportion of the injected antibodies partially penetrated the blood-brain barrier and there neutralized PGE2 produced by endothelial cells. By demonstrating that a small amount of antibodies injected i.c.v. did not affect the initial phase of fever, Steiner and others (2006) tried to control for the possibility that minute amounts of the systemically injected antibodies had penetrated the brain and exerted their action in the brain parenchyma. However, this experiment may not be conclusive, because it is not clear that the concentration of antibodies in the extracellular fluid surrounding the EP3 receptor expression neurons in the preoptic area was high enough to block locally produced prostaglandins.
Most important, however, the idea that PGE2 produced by peripheral macrophages gives rise to the initial phase of fever has been directly tested and seemed disproved. In the previously mentioned study using mice chimeric for mPGES-1, the terminal isomerase in which absence there is no fever in response to LPS (; ; ), showed that mice with deletion of mPGES-1 in hematopoietic cells displayed a normal first phase of fever, whereas mice that expressed mPGES-1 only in hematopoietic cells showed no fever (but instead hypothermia) (Fig. 7). Notably, the latter mice showed significantly elevated levels of PGE2 metabolites in plasma, but not of PGE2 in the cerebrospinal fluid (CSF), 40 minutes after LPS injection, implying that the transplanted hematopoietic cells were functional with respect to their prostaglandin producing capacity. Thus, while this experiment confirmed that at least part of the circulating PGE2 seen early after LPS injection is of hematopoietic origin, it indicates that this PGE2 does not elicit a febrile response, and, furthermore, that it does not seem to enter the brain, at least not to an extent that is reflected in elevated PGE2 levels in the CSF. A similar finding was reported in a study on tumor-bearing mice (). In that study high levels of PGE2 as well as of PGE2 metabolites were recorded in plasma, yet there was no fever and no elevated PGE2 levels in the CSF; it should be noted, however, that conditions could be different when elevated PGE2 levels occur during inflammatory conditions that could change the permeability of the blood-brain barrier.
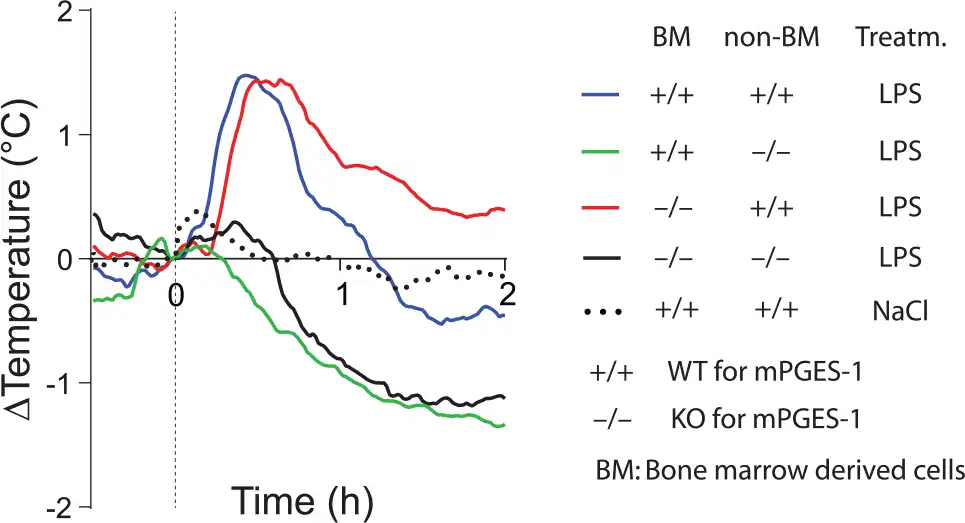
Figure 7
Fever response to intravenously injected lipopolysaccharide (LPS) in whole-body irradiated wild type (WT) and mPGES-1 knockout (KO) mice transplanted with WT (+/+) and KO (−/−) bone marrow. WT mice (non-BM +/+) show a first phase of fever, irrespective of whether they were transplanted with WT or KO bone marrow (upper two traces; cf. the temperature curve for mice injected with saline). In contrast, KO mice (non-BM −/−) transplanted with WT bone marrow instead show a hypothermic response, similar to KO mice transplanted with KO bone marrow (lower two traces). For all traces mean is shown. Dashed vertical line indicates time of injection. For further details of these experiments, see Figure 3. Adapted from .
Role of Peripheral Nerves for Fever
The role of peripheral nerves as sensors of peripheral inflammation has been the subject of extensive research, with the vagus nerves being most studied. Early studies using transection of the vagus nerve reported that the vagus nerve was involved in various sickness symptoms such as changes in pain sensitivity, feeding behavior, social exploration, sleep, and stress hormone release (; ; ; ; ; ; ). In support of these observations, the nodose ganglion was shown to express mRNA for the IL-1R1 as well as the EP3 receptor (; ), the PGE2 receptor subtype critical for the febrile response (), and i.v. or intraportal injection of IL-1β was shown to increase the discharge activity of vagal afferents (; ). The putative downstream signaling mechanisms have been little explored, but analysis of slice preparations of the lower brainstem has shown that PGE2 elicits an EP3-mediated synaptic depression of vagal synaptic transmission that involves Gi/o proteins coupled to adenylyl cyclase ().
As for the role of the vagus nerve in fever, the available data are conflicting. Early observations that surgical transection of the vagus nerve attenuated or abolished the febrile response to i.p. injected LPS or IL-1β (; ; ) were subsequently questioned (; ; ) because the vagotomy-produced malnutrition may render the animals incapable of mounting a febrile response (; ; ; ). Also, chemical denervation of visceral afferent fibers with capsaicin was reported to attenuate fever (), but this effect has later been suggested to depend on a non-neural mechanism, such as capsaicin-induced alteration of endotoxin action in the liver (; ).
However, the possibility remains that monophasic fever, seen in response to threshold doses of IL-1β or LPS is mediated by the vagus nerve. Thus, it was reported that this response, a brief fever peak of less than 1°C that is seen when the immunogen is given i.v. under conditions not involving handling stress, is abolished following vagotomy also in rats in which malnutrition is prevented (; ). It seems to depend on the hepatic branch of the vagus nerve, but not on other vagus branches or on the splanchnic nerve (; ). Such as role for the hepatic branch of the vagus nerve fits into the suggestion that the first phase of polyphasic fever, which however may not be similar to the monophasic fever (), is mediated by activation of the vagus nerve (; ; ). This activation has been ascribed to a complement-induced release from liver Kupffer cells of PGE2, synthesized by constitutive cyclooxygenases and hence not requiring de novo protein synthesis. However, this idea has been challenged. Thus, both following subdiaphragmatic and cervical vagotomy (the latter in anesthetized rats), i.v. injection of PGE2, believed to mimic its endogenous peripheral release, still elicited fever (). Because the fever was abolished by inhibition of the presympathetic neurons in the medullary raphe nuclei, the thermogenic effect of the peripherally administrated PGE2 was ascribed to a direct action of PGE2 on the brain (see above) not involving the vagus nerve ().
In addition to the vagus nerve, somatic afferent fibers have also been suggested to be involved in the febrile response. Similar to the nodose ganglion, dorsal root ganglia have been reported to express IL-1R1 and EP3 receptors (; ), and peripheral nerves have been shown to respond to IL-1β and PGE2, although these inflammatory mediators were found to sensitize the nerves to other stimuli rather than to elicit a discharge per se (; ). In line with the idea that fine afferent fibers throughout the body sense the internal milieu and through their afferent discharge influence various autonomic relay structures to maintain homeostasis (), it is conceivable that somatic afferent fibers could mediate the febrile response to localized peripheral inflammation. However, in most models used, such as LPS injection into an air pouch or into an artificial subcutaneous chamber there is almost invariably leakage of cytokines, particularly IL-6, from the site of the local inflammation into the circulation (; ; ; ; ), and systemic treatment with an IL-6 antiserum abolishes the febrile response (), suggesting that a humoral rather than neural route is responsible for the fever. When a local anesthetic was injected together with LPS into a subcutaneous chamber, fever but not circulating IL-6 levels was attenuated, which was interpreted as evidence that a local neural activation was responsible for the fever (). However, as acknowledged by the authors, local anesthetics may interfere with the immune response (; ) and inhibition of, for example, IL-1β (), an important co-factor for the pyretic effect of IL-6 (), may have attenuated the febrile response.
A few studies using localized inflammation have reported that under some conditions no Cox-2 upregulation was seen in the brain despite the presence of fever. Rummel and colleagues injected a low, yet pyretic dose of LPS into an artificial subcutaneously implanted Teflon chamber in guinea pigs, but did not detect any hybridization signal for Cox-2 in the brain (), and Zhang and colleagues, employing casein injection into a subcutaneous air pouch in rats, likewise reported absence of Cox-2 induction in the hypothalamus, as determined by immunoassays and real-time PCR (). These observations hence suggest that even though there is a leakage of IL-6 into the circulation from the local inflammatory site, there is no activation of the central prostaglandin synthesis. While these data are intriguing and may be interpreted as pointing to the presence of a neural afferent pathway for fever, they also suggest that such a pathway might feed into the central thermoregulatory system without involving activation of EP3 receptors in the preoptic hypothalamus, that is, in the same way as peripheral thermoreceptors (; ). As discussed by Ross and collaborators, strong candidates obviously are cold sensitive fibers. It should be noted that the TRPA1 ion channel, a chemosensor that is activated by noxious cold in rats and mice () and that is expressed on nociceptive afferent fibers (), is activated by LPS, mediating acute neurogenic inflammation and pain (). With these observations in mind, we recently examined the involvement of TRPA1 in LPS-induced fever. However, TRPA1 KO mice displayed the same febrile response to i.p. LPS as did WT mice (unpublished).
Further support for the role of somatic afferent fibers for fever comes from two studies in which transection of peripheral nerves was reported to attenuate fever. In one of the studies fever elicited by LPS injection into a gingival pouch in the maxilla was shown to be attenuated after transection of all trigeminal nerve branches emerging from the infra-orbital foramen (as it was after local injection of a local anesthetic or a cyclooxygenase inhibitor) (). However, this report lacks confirmation that the surgical procedures, which seem likely to influence the animals’ ability to chew properly, had not affected their body weight and hence their thermoeffector capacity. In the other study, transection of the glossopharyngeal nerve attenuated the febrile response to LPS or IL-1β injected into the soft palate, but not when the pyrogens were injected i.p. (). However, the effect was small, the differences in temperature between the nerve-transected and sham operated groups amounting only to tenths of degrees, and the temperature responses obviously influenced by the general anesthesia during which the injections were performed.
A few studies so far have used genetic deletion techniques to examine the role of cytokine receptors on peripheral nerves for the febrile response. Mice with deletion of IL-6Rα in neural crest derivates that include peripheral nerves, or in vanilloid receptor expressing fine afferents (i.e. nociceptive C-fibers), displayed intact febrile response to i.p. injected LPS (), and the same was found for mice with similar deletions of the IL-1R1 (). In both studies, deletion of IL-6Rα and IL-1R1 in brain endothelial cells attenuated the febrile response (see above). While these data speak against a direct activation of peripheral nerves by peripherally released IL-6 and IL-1β, it should be noted that a neuronal route of immune-to-brain communication might play a role only when high levels of circulating cytokines are absent (; ; ). Because considerable amounts of cytokines are released into the circulation when LPS is injected in the dose used in the two studies reported above (), it is conceivable that a contribution of sensory nerves could have escaped detection.
In sum, the data on the role of peripheral nerves in fever are contradictory. An obvious weakness in all studies is that they examine whether peripheral nerves are necessary for fever, that is, whether interrupting the signaling will attenuate the febrile response. This approach implies, as noted above, that it be difficult to detect if neural signaling exists in parallel with, for example, humoral signaling. Studies in animals with e.g. expression of signaling molecules involved in immune-induced fever specifically on peripheral nerves would help resolve this issue. Such animal models are now becoming available (e.g., ).
Central Neurons Critical for the Febrile Response
It is well-established that PGE2, through the activation of EP3 receptors in the central nervous system, is the final mediator of fever induced by systemic immune challenge with LPS or cytokines (; ; ; ; ). Although peripheral inflammation results in strong transcriptional upregulation of pyrogenic cytokines not only in the periphery but also in the brain, brain-produced cytokines do not elicit fever in the absence of induced PGE2 synthesis (). It was early recognized that the preoptic hypothalamic area played a critical role for eliciting fever (), and this region was subsequently found to be the site that was most sensitive to the pyrogenic action of PGE2 (; ) and to express high concentration of EP3 receptor expressing neurons (; ; ). It is also the site at which local restoration of PGE2 production results in a temperature response to LPS in otherwise fever-refractive animals ().
But how does the preoptic hypothalamus generate fever? The prevailing idea is that neurons in the preoptic hypothalamus in the healthy animal provide a tonic inhibitory GABAergic input to thermogenic presympathetic neurons in the rostral medullary raphe nucleus (RMR) of the brain stem, and that the preoptic neurons are silenced on binding of PGE2 to their EP3 receptors, hence providing a disinhibition of the presympathetic neurons (Fig. 8). This idea is primarily based on the findings by Nakamura and collaborators in rat that (1) EP3 receptor expressing neurons in the preoptic region project to the RMR, (2) a large majority of the EP3 receptor expressing neurons in the preoptic region co-express transcripts for the GABA-synthesizing enzyme GAD67, and (3) injection of the GABA agonist muscimol in the RMR blocks the thermogenesis evoked by PGE2 injection into the preoptic hypothalamic region (). It is also supported by observations that ablation of the preoptic area or knife cuts caudal to this region generate hyperthermia (; ; ), hence suggesting that the preoptic region tonically inhibits caudally located thermogenic neuronal groups. However, as will be further detailed below, this description represents a highly simplified view. Here, it should be noted that only a minority of the preoptic cells that project to the RMR are EP3 receptor expressing. Most are not () and may instead be excitatory glutamatergic cells (). Furthermore, not all EP3 receptors in the preoptic region are connected to inhibitory G-proteins. Analysis of the EP3 receptor subtypes in the preoptic regions has shown that a considerable proportion represents a splice variant coupled to stimulatory G-protein (). Whether some cells exclusively express such receptor variants, or whether the different receptor variants are present on the same cells is not known, although preoptic neurons that are excited by PGE2 have been identified (). Finally, little is known about the molecular events downstream of EP3 activation in preoptic neurons (). The idea that a tonic inhibitory activity of these neurons is silenced by PGE2 is inferred from the release phenomena observed in their downstream targets on PGE2 administration (; ), and observations of the inhibitory role of these receptors in other cell types (e.g., ). As to direct observations so far, recordings from cultured anterior hypothalamic neurons showed that PGE2 decreased the firing rate in EP3 receptor-positive GABAergic neurons (). Preoptic neurons that are inhibited by PGE2 have been shown also to be warm sensitive (), and warm sensitive neurons are found to be GABAergic and to drive thermogenesis through descending projections (). However, it is not clear to what extent the populations of warm sensitive and PGE2-responsive neurons overlap.
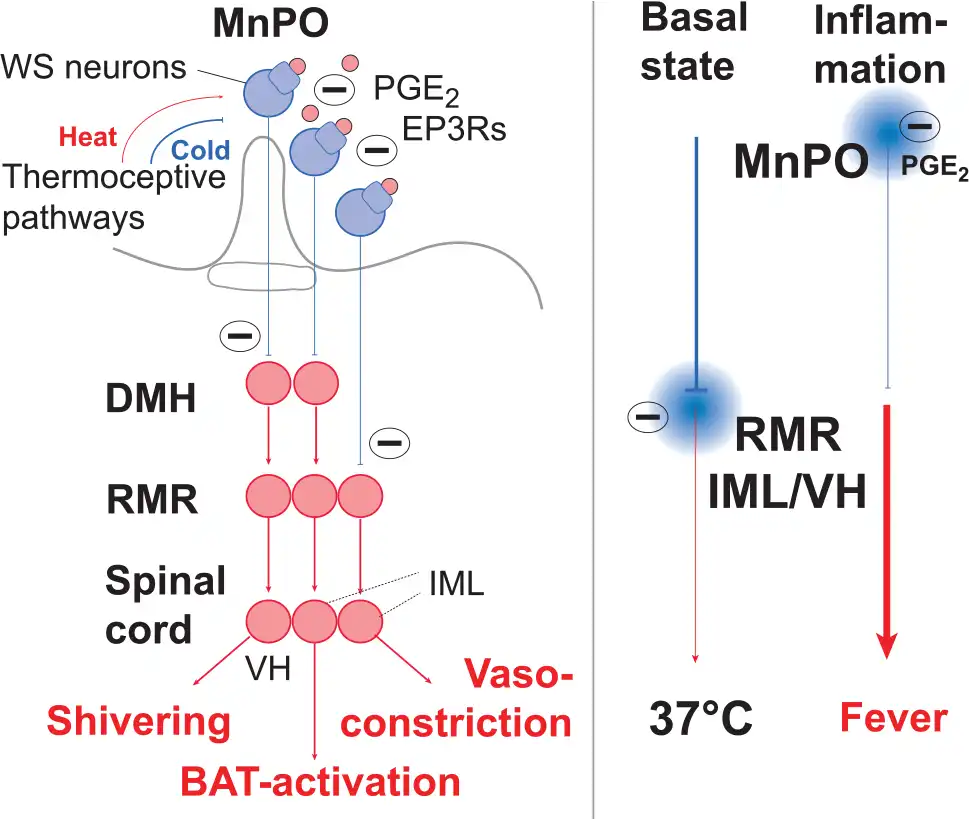
Figure 8
Circuitry of pyrogenic pathways from the preoptic hypothalamus. EP3 receptor expressing neurons in the median preoptic nucleus (MnPO) exert tonic inhibition on presympathetic neurons in the rostral medullary raphe nucleus (RMR) as well as on neurons in the dorsomedial hypothalamus (DMH), hence silencing, in the resting state, sympathetic, thermogenic output from the intermediolateral cell column (IML) as well as excitatory output to motorneurons in the ventral horn (VH) of the spinal cord responsible for shivering thermogenesis. On immune-induced PGE2 release (pink circles) and binding to the EP3 receptors (EP3Rs), the MnPO neurons are silenced, resulting in disinhibition of neurons in DMH and RMR and activation of the thermogenic circuitry. In addition to responding to PGE2, EP3 expressing neurons are warm-sensitive (WS), and hence activated by heat, inhibiting thermogenesis, and inhibited by cold, promoting thermogenesis. Note that the direct projection from MnPO to RMR controls vasoconstriction, whereas the indirect pathway over the DMH controls shivering and non-shivering (brown adipose tissue [BAT] activation) thermogenesis.
In addition to the direct projection from the preoptic hypothalamus to the RMR, this structure also receives input from the preoptic hypothalamus via a relay in the dorsomedial hypothalamus (DMH) (Fig. 8), a critical node for various stress induced responses (), including cold induced thermogenesis (). Whereas the projection to the RMR from the preoptic hypothalamus is inhibitory, the input from the DMH is excitatory, albeit under inhibitory control from the preoptic hypothalamus (). Retrograde tracing experiments have demonstrated that the preoptic projections to the DMH and the RMR, respectively, largely constitute intercalated but distinct populations (; ). Furthermore, the projection neurons are found in two distinct nuclear groups in the preoptic hypothalamus, the median preoptic nucleus (MnPO) and the dorsolateral preoptic area (DLPO). Both groups provide tonic inhibitory input to their target neurons in the DMH and RMR, but only the MnPO is responsive to the action of PGE2, whereas the DLPO has been suggested to mediate thermosensory regulation in response to changes in skin temperature (). Data suggests that skin vasoconstriction on skin cooling or PGE2 delivery in the preoptic hypothalamus is primarily dependent on the inhibition of the direct projection from the preoptic area to the RMR, whereas thermogenesis through sympathetic activation of brown adipose tissue requires the disinhibition of the projection to the RMR from the DMH (). The latter structure has also been reported to mediate shivering thermogenesis, via its projection to the RMR () (Fig. 8).
Nakamura and Morrison, based on studies of cold-induced thermogenic responses as well as the thermogenic responses to local PGE2 injections, suggest that the PGE2 sensitive neurons that provide tonic inhibition onto the DMH are located in the medial preoptic nucleus (MPO) and not in the MnPO, and that the latter structure provides an ambient cold driven GABAergic inhibition of MPO (, ). However, as pointed out by , there is no known projection from the MPO to neither the DMH nor the presympathetic neurons in the RMR, and the dense concentration of EP3 receptor expressing neurons in the preoptic hypothalamus does not involve the MPO.
It is important to note that during fever, thermoregulatory threshold changes, including an initial upward shift of the threshold for both cold-defense and thermolytic responses, followed by a widening of the interthreshold temperature zone (). The increased interthreshold also makes the body temperature less stable, permitting the rapid changes in body temperature than can be seen following i.v. injection of LPS. It is also well-known that anesthesia produces a similar poikilothermic state (). This fact should be considered when evaluating some of the core publications on the efferent thermoregulatory pathways, because the findings in these publications were obtained from anesthetized preparations, kept artificially at normal body temperature (e.g., , ; ; ).
The close anatomical relationships between the pathways mediating the pyrogenic response to peripheral inflammation and the thermoregulatory responses to changes in ambient temperature is corresponded by a close functional relationship between these phenomena. It is well known that the febrile response is modulated by the ambient temperature. At low ambient temperatures, rats do not display fever, but hypothermia, when challenged with peripheral injection of LPS, especially when this immunogen is given at high doses (; ).
The febrile response is also modulated by the nutritional status of the animal: Starvation, which leads to depressed body temperature, strongly attenuates the febrile response and/or produces hypothermia when animals that are kept at subneutral ambient temperature are challenged with LPS (; ; ), probably through a leptin dependent mechanism (; see also ; ). Both the influences of ambient temperature and nutritional status on the febrile response may be mediated by the DMH. DMH is critical both for LPS-induced thermoregulation and behavioral cold-seeking in response to sepsis (; ), and leptin receptor expressing neurons in DMH have been shown to be activated by cold exposure and to be connected with brown adipose tissue via synaptic connections with the RMR ().
The attenuated fever during starvation does not seem to involve attenuated brain PGE2 production, because fasting does not alter the febrigenic signaling from the periphery to the brain, that is important for central PGE2 synthesis or PGE2 levels in the brain. However, fasting attenuates the response to intracerebrally injected PGE2 (). It should also be noted that the mechanism behind immune-induced hypothermia seems to be PGE2 independent, probably being mediated by a different prostanoid synthesis pathway (; ), although this hypothermia also has been suggested to be a consequence of the poikilothermic state and behavioral thermoregulation (). It is conceivable that the mechanism promoting hypothermia coexists with the pyrogenic response, but generally is masked or suppressed by the latter, as seen in mice lacking mPGES-1 (Fig. 7). However, episodes with rapid, active temperature fall can be seen also during normal fever, such as following the initial temperature peak in the 3-phasic fever that occurs after i.v. injection of LPS. Hypothetically, the DMH, or with the DMH connected structures, could serve as an integrator that evaluates the strength of the immune stimulus (i.e., the severity the immune challenge) in relation to the metabolic status and the ambient temperature, and that determines whether the available energy resources are sufficient for eliciting fever, or whether hypothermia will be the more adaptive response ().
Conclusions
Twenty years of research since the review article by has yielded deep insight into the mechanism of fever. Yet, several important outstanding questions remain. These relate particularly to the afferent limb of the febrile response. Thus, of the potential pathways for transmitting information about ongoing peripheral inflammation to the brain, pyrogen action on the OVLT, activation of peripheral nerves by immune signals, transport of peripherally synthetized PGE2 into the brain, and activation of cells in the blood-brain barrier, only humoral signaling via the blood-brain barrier is supported by unequivocal observations. These observations are obtained in studies in which either cytokine receptors or enzymes for PGE2 synthesis were selectively deleted in brain endothelial cells and demonstrate that such deletion attenuates the febrile response to peripherally injected immunogen. However, it should be noted that although fever was attenuated in these studies, fever was never completely abolished, at least not when the immunogen (LPS) was given i.p. Hence, additional pathways exist that are responsible for the residual fever. With regard to studies on the efferent pyrogenic pathways, there is yet little understanding on the mechanisms by which factors such a nutritional status and ambient temperature shape the response to a peripheral immune challenge and how the neuronal circuits that determines whether the response will be fever or hypothermia are organized. The fact that most of the functional studies on the efferent pathways have been performed in anesthetized preparation, in which normal thermoregulation is impaired, is also a caveat in our understanding of these pathways.
Declaration of Conflicting Interests The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work in the authors’ laboratories referred to in this article were supported by grants from Swedish Medical Research Council (#07879 to AB, #20725 to DE), the European Research Council (ERC-starting grant to DE), the Knut and Alice Wallenberg foundation (DE), the Swedish Brain Foundation (DE and AB), the Swedish Cancer Foundation (#213/692 to AB), and the County Council of Östergötland (DE and AB).
Anders Blomqvist
https://orcid.org/0000-0002-6928-4473
References
- Abbott SBG, Machado NLS, Geerling JC, Saper CB. 2016. Reciprocal control of drinking behavior by median preoptic neurons in mice. J Neurosci 36(31):8228–37.
- Alheim K, Chai Z, Fantuzzi G, Hasanvan H, Malinowsky D, Di Santo E, and others. 1997. Hyperresponsive febrile reactions to interleukin (IL) 1α and IL-1β, and altered brain cytokine mRNA and serum cytokine levels, in IL-1β-deficient mice. Proc Natl Acad Sci U S A 94(6):2681–6.
- Almeida MC, Steiner AA, Branco LG, Romanovsky AA. 2006. Neural substrate of cold-seeking behavior in endotoxin shock. PLoS One 1:e1.
- Atkins E. 1982. Fever: Its history, cause, and function. Yale J Biol Med 55(3–4):283–9.
- Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L, and others. 2008. Nociceptors are interleukin-1β sensors. J Neurosci 28(52):14062–73.
- Bishai I, Dinarello CA, Coceani F. 1987. Prostaglandin formation in feline cerebral microvessels: Effect of endotoxin and interleukin-1. Can J Physiol Pharmacol 65(11):2225–30.
- Blatteis CM. 2007. The onset of fever: new insights into its mechanism. Prog Brain Res 162:3-14.
- Blatteis CM, Bealer SL, Hunter WS, Llanos QJ, Ahokas RA, Mashburn TA Jr. 1983. Suppression of fever after lesions of the anteroventral third ventricle in guinea pigs. Brain Res Bull 11(5):519–26.
- Bluthé R-M, Layé S, Michaud B, Combe C, Dantzer R, Parnet P. 2000. Role of interleukin-1β and tumour necrosis factor-α in lipopolysaccharide-induced sickness behaviour: a study with interleukin-1 type I receptor-deficient mice. Eur J Neurosci 12(12):4447–56.
- Bluthe RM, Walter V, Parnet P, Laye S, Lestage J, Verrier D, and others. 1994. Lipopolysaccharide induces sickness behaviour in rats by a vagal mediated mechanism. C R Acad Sci III 317(6):499–503.
- Boltana S, Rey S, Roher N, Vargas R, Huerta M, Huntingford FA, and others. 2013. Behavioural fever is a synergic signal amplifying the innate immune response. Proc Biol Sci 280(1766):20131381.
- Borner T, Arnold M, Ruud J, Breit SN, Langhans W, Lutz TA, and others. 2017. Anorexia-cachexia syndrome in hepatoma tumour-bearing rats requires the area postrema but not vagal afferents and is paralleled by increased MIC-1/GDF15. J Cachexia Sarcopenia Muscle 8(3):417–27.
- Breder CD, Saper CB. 1996. Expression of inducible cyclooxygenase mRNA in the mouse brain after systemic administration of bacterial lipopolysaccharide. Brain Res 713(1–2):64–9.
- Bret-Dibat JL, Bluthe RM, Kent S, Kelley KW, Dantzer R. 1995. Lipopolysaccharide and interleukin-1 depress food-motivated behavior in mice by a vagal-mediated mechanism. Brain Behav Immun 9(3):242–6.
- Breyer MD, Breyer RM. 2001. G protein-coupled prostanoid receptors and the kidney. Annu Rev Physiol 63(1):579–605.
- Caldwell FT Jr, Graves DB, Wallace BH. 1999. Humoral versus neural pathways for fever production in rats after administration of lipopolysaccharide. J Trauma 47(1):120–9.
- Cao C, Matsumura K, Yamagata K, Watanabe Y. 1995. Induction by lipopolysaccharide of cyclooxygenase-2 mRNA in rat brain; its possible role in the febrile response. Brain Res 697(1–2):187–96.
- Cao C, Matsumura K, Yamagata K, Watanabe Y. 1998. Cyclooxygenase-2 is induced in brain blood vessels during fever evoked by peripheral or central administration of tumor necrosis factor. Brain Res Mol Brain Res 56(1–2):45–56.
- Cartmell T, Poole S, Turnbull AV, Rothwell NJ, Luheshi GN. 2000. Circulating interleukin-6 mediates the febrile response to localised inflammation in rats. J Physiol 526(Pt 3):653–61.
- Chai Z, Gatti S, Toniatti C, Poli V, Bartfai T. 1996. Interleukin (IL)-6 gene expression in the central nervous system is necessary for fever response to lipopolysaccharide or IL-1β: a study on IL-6-deficient mice. J Exp Med 183(1):311–6.
- Chen J, Kang D, Xu J, Lake M, Hogan JO, Sun C, and others. 2013. Species differences and molecular determinant of trpa1 cold sensitivity. Nat Commun 4:2501.
- Chen X-M, Hosono T, Yoda T, Fukuda Y, Kanosue K. 1998. Efferent projection from the preoptic area for the control of non-shivering thermogenesis in rats. J Physiol 512(3):883–92.
- Ching S, Zhang H, Belevych N, He L, Lai W, Pu XA, and others. 2007. Endothelial-specific knockdown of interleukin-1 (IL-1) type 1 receptor differentially alters CNS responses to IL-1 depending on its route of administration. J Neurosci 27(39):10476–86.
- Cooper KE, Cranston WI, Honour AJ. 1967. Observations on the site and mode of action of pyrogens in the rabbit brain. J Physiol 191(2):325–37.
- Craig AD. 2002. How do you feel? Interoception: the sense of the physiological condition of the body. Nat Rev Neurosci 3(8):655–66.
- Derow A, Izydorczyk I, Kuhn A, Reeh PW, Petho G. 2007. Prostaglandin E(2) and I(2) facilitate noxious heat-induced spike discharge but not iCGRP release from rat cutaneous nociceptors. Life Sci 81(25–26):1685–93.
- Díaz M, Becker DE. 2010. Thermoregulation: physiological and clinical considerations during sedation and general anesthesia. Anesth Prog 57(1):25–33.
- DiMicco JA, Samuels BC, Zaretskaia MV, Zaretsky DV. 2002. The dorsomedial hypothalamus and the response to stress: part renaissance, part revolution. Pharmacol Biochem Behav 71(3):469–80.
- Dinarello CA. 2015. The history of fever, leukocytic pyrogen and interleukin-1. Temperature (Austin) 2(1):8–16.
- Dogan MD, Kulchitsky VA, Patel S, Petervari E, Szekely M, Romanovsky AA. 2003. Bilateral splanchnicotomy does not affect lipopolysaccharide-induced fever in rats. Brain Res 993(1–2):227–9.
- Dogan MD, Patel S, Rudaya AY, Steiner AA, Székely M, Romanovsky AA. 2004. Lipopolysaccharide fever is initiated via a capsaicin-sensitive mechanism independent of the subtype-1 vanilloid receptor. Br J Pharmacol 143(8):1023–32.
- Ek M, Arias C, Sawchenko P, Ericsson-Dahlstrand A. 2000. Distribution of the EP3 prostaglandin E(2) receptor subtype in the rat brain: relationship to sites of interleukin-1-induced cellular responsiveness. J Comp Neurol 428(1):5–20.
- Ek M, Engblom D, Saha S, Blomqvist A, Jakobsson PJ, Ericsson-Dahlstrand A. 2001. Inflammatory response: pathway across the blood-brain barrier. Nature 410(6827):430–1.
- Ek M, Kurosawa M, Lundeberg T, Ericsson A. 1998. Activation of vagal afferents after intravenous injection of interleukin-1β: role of endogenous prostaglandins. J Neurosci 18(22):9471–9.
- Engblom D, Ek M, Andersson IM, Saha S, Dahlstrom M, Jakobsson PJ, and others. 2002a. Induction of microsomal prostaglandin E synthase in the rat brain endothelium and parenchyma in adjuvant-induced arthritis. J Comp Neurol 452(3):205–14.
- Engblom D, Ek M, Saha S, Ericsson-Dahlstrand A, Jakobsson PJ, Blomqvist A. 2002b. Prostaglandins as inflammatory messengers across the blood-brain barrier. J Mol Med 80(1):5–15.
- Engblom D, Saha S, Engstrom L, Westman M, Audoly LP, Jakobsson PJ, and others. 2003. Microsomal prostaglandin E synthase-1 is the central switch during immune-induced pyresis. Nat Neurosci 6(11):1137–8.
- Engström L, Ruud J, Eskilsson A, Larsson A, Mackerlova L, Kugelberg U, and others. 2012. Lipopolysaccharide-induced fever depends on prostaglandin E2 production specifically in brain endothelial cells. Endocrinology 153(10):4849–61.
- Ericsson A, Liu C, Hart RP, Sawchenko PE. 1995. Type 1 interleukin-1 receptor in the rat brain: distribution, regulation, and relationship to sites of IL-1-induced cellular activation. J Comp Neurol 361(4):681–98.
- Eskilsson A, Matsuwaki T, Shionoya K, Mirrasekhian E, Zajdel J, Schwaninger M, and others. 2017. Immune-induced fever is dependent on local but not generalized prostaglandin E2 synthesis in the brain. J Neurosci 37(19):5035–5044.
- Eskilsson A, Mirrasekhian E, Dufour S, Schwaninger M, Engblom D, Blomqvist A. 2014a. Immune-induced fever is mediated by IL-6 receptors on brain endothelial cells coupled to STAT3-dependent induction of brain endothelial prostaglandin synthesis. J Neurosci 34(48):15957–61.
- Eskilsson A, Tachikawa M, Hosoya K, Blomqvist A. 2014b. The distribution of microsomal prostaglandin E synthase-1 in the mouse brain. J Comp Neurol 522(14):3229–44.
- Evans SS, Repasky EA, Fisher DT. 2015. Fever and the thermal regulation of immunity: the immune system feels the heat. Nat Rev Immunol 15(6):335–49.
- Faggioni R, Moser A, Feingold KR, Grunfeld C. 2000. Reduced leptin levels in starvation increase susceptibility to endotoxic shock. Am J Pathol 156(5):1781–7.
- Fantuzzi G, Dinarello CA. 1996. The inflammatory response in interleukin-1β-deficient mice: Comparison with other cytokine-related knock-out mice. J Leukoc Biol 59(4):489–93.
- Feldberg W, Saxena PN. 1971. Fever produced by prostaglandin E1. J Physiol 217(3):547–56.
- Ferguson AV. 2014. Frontiers in neuroscience. Circumventricular organs: integrators of circulating signals controlling hydration, energy balance, and immune function. In: De Luca LA Jr, Menani JV, Johnson AK, editors. Neurobiology of body fluid homeostasis: transduction and integration. Boca Raton, FL: CRC Press/Taylor & Francis.
- Fritz M, Klawonn AM, Jaarola M, Engblom D. 2018. Interferon-mediated signaling in the brain endothelium is critical for inflammation-induced aversion. Brain Behav Immun 67:54–8.
- Fritz M, Klawonn AM, Nilsson A, Singh AK, Zajdel J, Wilhelms DB, and others. 2016. Prostaglandin-dependent modulation of dopaminergic neurotransmission elicits inflammation-induced aversion in mice. J Clin Invest 126(2):695–705.
- Gaykema RP, Dijkstra I, Tilders FJ. 1995. Subdiaphragmatic vagotomy suppresses endotoxin-induced activation of hypothalamic corticotropin-releasing hormone neurons and ACTH secretion. Endocrinology 136(10):4717–20.
- Goldman EE. 1913. Vitalfarbung am Zentralnervensystem. Berlin, Germany: Eimer.
- Hansen MK, Daniels S, Goehler LE, Gaykema RP, Maier SF, Watkins LR. 2000. Subdiaphragmatic vagotomy does not block intraperitoneal lipopolysaccharide-induced fever. Auton Neurosci 85(1–3):83–7.
- Hansen MK, Krueger JM. 1997. Subdiaphragmatic vagotomy blocks the sleep- and fever-promoting effects of interleukin-1β. Am J Physiol 273(4 Pt 2):R1246–53.
- Harden LM, Kent S, Pittman QJ, Roth J. 2015. Fever and sickness behavior: friend or foe? Brain Behav Immun 50:322–33.
- Hoffman-Goetz L, Kluger MJ. 1979. Protein deprivation: its effects on fever and plasma iron during bacterial infection in rabbits. J Physiol 295:419–30.
- Ibuki T, Matsumura K, Yamazaki Y, Nozaki T, Tanaka Y, Kobayashi S. 2003. Cyclooxygenase-2 is induced in the endothelial cells throughout the central nervous system during carrageenan-induced hind paw inflammation; its possible role in hyperalgesia. J Neurochem 86(2):318–28.
- Inoue W, Luheshi GN. 2010. Acute starvation alters lipopolysaccharide-induced fever in leptin-dependent and -independent mechanisms in rats. Am J Physiol Regul Integr Comp Physiol 299(6):R1709–19.
- Inoue W, Matsumura K, Yamagata K, Takemiya T, Shiraki T, Kobayashi S. 2002. Brain-specific endothelial induction of prostaglandin E(2) synthesis enzymes and its temporal relation to fever. Neurosci Res 44(1):51–61.
- Inoue W, Somay G, Poole S, Luheshi GN. 2008. Immune-to-brain signaling and central prostaglandin E2 synthesis in fasted rats with altered lipopolysaccharide-induced fever. Am J Physiol Regul Integr Comp Physiol 295(1):R133–43.
- Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. 1999. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci U S A 96(13):7220–5.
- Julius D. 2013. TRP channels and pain. Annu Rev Cell Dev Biol 29(1):355–84.
- Kent S, Bret-Dibat JL, Kelley KW, Dantzer R. 1996. Mechanisms of sickness-induced decreases in food-motivated behavior. Neurosci Biobehav Rev 20(1):171–5.
- King MK, Wood WB Jr. 1958. Studies on the pathogenesis of fever. IV. The site of action of leucocytic and circulating endogenous pyrogen. J Exp Med 107(2):291–303.
- Kluger MJ. 1991. Fever: role of pyrogens and cryogens. Physiol Rev 71(1):93–127.
- Knoll JG, Krasnow SM, Marks DL. 2017. Interleukin-1β signaling in fenestrated capillaries is sufficient to trigger sickness responses in mice. J Neuroinflamm 14(1):219.
- Konsman JP, Vigues S, Mackerlova L, Bristow A, Blomqvist A. 2004. Rat brain vascular distribution of interleukin-1 type-1 receptor immunoreactivity: relationship to patterns of inducible cyclooxygenase expression by peripheral inflammatory stimuli. J Comp Neurol 472(1):113–29.
- Kozak W, Kluger MJ, Soszynski D, Conn CA, Rudolph K, Leon LR, and others. 1998. IL-6 and IL-1β in fever. Studies using cytokine-deficient (knockout) mice. Ann N Y Acad Sci 856:33–47.
- Krall CM, Yao X, Hass MA, Feleder C, Steiner AA. 2010. Food deprivation alters thermoregulatory responses to lipopolysaccharide by enhancing cryogenic inflammatory signaling via prostaglandin D2. Am J Physiol Regul Integr Comp Physiol 298(6):R1512–21.
- Labow M, Shuster D, Zetterstrom M, Nunes P, Terry R, Cullinan EB, and others. 1997. Absence of IL-1 signaling and reduced inflammatory response in IL-1 type I receptor-deficient mice. J Immunol 159(5):2452–61.
- Lacroix S, Rivest S. 1997. Functional circuitry in the brain of immune-challenged rats: partial involvement of prostaglandins. J Comp Neurol 387(2):307–24.
- Laflamme N, Rivest S. 2001. Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J 15(1):155–63.
- Lazarus M, Yoshida K, Coppari R, Bass CE, Mochizuki T, Lowell BB, and others. 2007. EP3 prostaglandin receptors in the median preoptic nucleus are critical for fever responses. Nat Neurosci 10(9):1131–3.
- Lee HY, Whiteside MB, Herkenham M. 1998. Area postrema removal abolishes stimulatory effects of intravenous interleukin-1beta on hypothalamic-pituitary-adrenal axis activity and c-fos mRNA in the hypothalamic paraventricular nucleus. Brain Res Bull 46(6):495–503.
- LeMay LG, Vander AJ, Kluger MJ. 1990. Role of interleukin 6 in fever in rats. Am J Physiol Regul Integr Comp Physiol 258(3):R798–803.
- Leon LR, Conn CA, Glaccum M, Kluger MJ. 1996. IL-1 type 1 receptor mediates acute phase response to turpentine, but not lipopolysaccharide, in mice. Am J Physiol 271(6 Pt 2):R1668–75.
- Li S, Wang Y, Matsumura K, Ballou LR, Morham SG, Blatteis CM. 1999. The febrile response to lipopolysaccharide is blocked in cyclooxygenase-2(−/−), but not in cyclooxygenase-1(−/−) mice. Brain Res 825(1–2):86–94.
- Li Z, Perlik V, Feleder C, Tang Y, Blatteis CM. 2006. Kupffer cell-generated PGE2 triggers the febrile response of guinea pigs to intravenously injected LPS. Am J Physiol Regul Integr Comp Physiol 290(5):R1262–70.
- Liu X, Yamashita T, Chen Q, Belevych N, McKim DB, Tarr AJ, and others. 2015. Interleukin 1 type 1 receptor restore: a genetic mouse model for studying interleukin 1 receptor-mediated effects in specific cell types. J Neurosci 35(7):2860–70.
- Luheshi G, Miller AJ, Brouwer S, Dascombe MJ, Rothwell NJ, Hopkins SJ. 1996. Interleukin-1 receptor antagonist inhibits endotoxin fever and systemic interleukin-6 induction in the rat. Am J Physiol Endocrinol Metab 270(1):E91–5.
- Luheshi GN, Bluthe RM, Rushforth D, Mulcahy N, Konsman JP, Goldbach M, and others. 2000. Vagotomy attenuates the behavioural but not the pyrogenic effects of interleukin-1 in rats. Auton Neurosci 85(1–3):127–32.
- Marty V, El Hachmane M, Amédée T. 2008. Dual modulation of synaptic transmission in the nucleus tractus solitarius by prostaglandin E2 synthesized downstream of IL-1β. Eur J Neurosci 27(12):3132–50.
- Matsuwaki T, Shionoya K, Ihnatko R, Eskilsson A, Kakuta S, Dufour S, and others. 2017. Involvement of interleukin-1 type 1 receptors in lipopolysaccharide-induced sickness responses. Brain Behav Immun 66:165–76.
- Meseguer V, Alpizar YA, Luis E, Tajada S, Denlinger B, Fajardo O, and others. 2014. TRPA1 channels mediate acute neurogenic inflammation and pain produced by bacterial endotoxins. Nat Commun 5:3125.
- Miller AJ, Luheshi GN, Rothwell NJ, Hopkins SJ. 1997. Local cytokine induction by LPS in the rat air pouch and its relationship to the febrile response. Am J Physiol 272(3 Pt 2):R857–61.
- Milton AS, Wendlandt S. 1970. A possible role for prostaglandin E1 as a modulator for temperature regulation in the central nervous system of the cat. J Physiol 207(2):76p–77p.
- Moore SA, Spector AA, Hart MN. 1988. Eicosanoid metabolism in cerebromicrovascular endothelium. Am J Physiol 254(1 Pt 1):C37–44.
- Morrison SF. 2016. Central neural control of thermoregulation and brown adipose tissue. Auton Neurosci 196:14-24.
- Nadeau S, Rivest S. 1999a. Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: a view from the blood-brain barrier. Neuroscience 93(4):1449–64.
- Nadeau S, Rivest S. 1999b. Regulation of the gene encoding tumor necrosis factor–α (TNF-α) in the rat brain and pituitary in response in different models of systemic immune challenge. J Neuropathol Exp Neurol 58(1):61–77.
- Nakamura K, Kaneko T, Yamashita Y, Hasegawa H, Katoh H, Negishi M. 2000. Immunohistochemical localization of prostaglandin EP3 receptor in the rat nervous system. J Comp Neurol 421(4):543–69.
- Nakamura K, Matsumura K, Kaneko T, Kobayashi S, Katoh H, Negishi M. 2002. The rostral raphe pallidus nucleus mediates pyrogenic transmission from the preoptic area. J Neurosci 22(11):4600–10.
- Nakamura K, Morrison SF. 2008. Preoptic mechanism for cold-defensive responses to skin cooling. J Physiol 586(10):2611–20.
- Nakamura K, Morrison SF. 2011. Central efferent pathways for cold-defensive and febrile shivering. J Physiol 589(Pt 14):3641–58.
- Nakamura Y, Nakamura K, Matsumura K, Kobayashi S, Kaneko T, Morrison SF. 2005. Direct pyrogenic input from prostaglandin EP3 receptor-expressing preoptic neurons to the dorsomedial hypothalamus. Eur J Neurosci 22(12):3137–46.
- Nakamura Y, Nakamura K, Morrison SF. 2009. Different populations of prostaglandin EP3 receptor-expressing preoptic neurons project to two fever-mediating sympathoexcitatory brain regions. Neuroscience 161(2):614–20.
- Navarro VP, Iyomasa MM, Leite-Panissi CRA, Almeida MC, Branco LGS. 2006. New role of the trigeminal nerve as a neuronal pathway signaling brain in acute periodontitis: participation of local prostaglandins. Pflügers Arch 453(1):73–82.
- Niijima A. 1996. The afferent discharges from sensors for interleukin 1β in the hepatoportal system in the anesthetized rat. J Auton Nerv Syst 61(3):287–91.
- Nilsberth C, Elander L, Hamzic N, Norell M, Lonn J, Engstrom L, and others. 2009a. The role of interleukin-6 in lipopolysaccharide-induced fever by mechanisms independent of prostaglandin E2. Endocrinology 150(4):1850–60.
- Nilsberth C, Hamzic N, Norell M, Blomqvist A. 2009b. Peripheral lipopolysaccharide administration induces cytokine mRNA expression in the viscera and brain of fever-refractory mice lacking microsomal prostaglandin E synthase-1. J Neuroendocrinol 21(8):715–21.
- Nilsson A, Wilhelms DB, Mirrasekhian E, Jaarola M, Blomqvist A, Engblom D. 2017. Inflammation-induced anorexia and fever are elicited by distinct prostaglandin dependent mechanisms, whereas conditioned taste aversion is prostaglandin independent. Brain Behav Immun 61:236–43.
- Oka T, Oka K, Scammell TE, Lee C, Kelly JF, Nantel F, and others. 2000. Relationship of EP(1-4) prostaglandin receptors with rat hypothalamic cell groups involved in lipopolysaccharide fever responses. J Comp Neurol 428(1):20–32.
- Oka Y, Ibuki T, Matsumura K, Namba M, Yamazaki Y, Poole S, and others. 2007. Interleukin-6 is a candidate molecule that transmits inflammatory information to the CNS. Neuroscience 145(2):530–8.
- Ootsuka Y, Blessing WW, Steiner AA, Romanovsky AA. 2008. Fever response to intravenous prostaglandin E2 is mediated by the brain but does not require afferent vagal signaling. Am J Physiol Regul Integr Comp Physiol 294(4):R1294–303.
- Ozaki-Okayama Y, Matsumura K, Ibuki T, Ueda M, Yamazaki Y, Tanaka Y, and others. 2004. Burn injury enhances brain prostaglandin E2 production through induction of cyclooxygenase-2 and microsomal prostaglandin E synthase in cerebral vascular endothelial cells in rats. Crit Care Med 32(3):795–800.
- Petervari E, Garami A, Pakai E, Szekely M. 2005. Effects of perineural capsaicin treatment of the abdominal vagus on endotoxin fever and on a non-febrile thermoregulatory event. J Endotoxin Res 11(5):260–6.
- Quan N. 2014. In-depth conversation: spectrum and kinetics of neuroimmune afferent pathways. Brain Behav Immun 40:1–8.
- Quan N, Stern EL, Whiteside MB, Herkenham M. 1999. Induction of pro-inflammatory cytokine mRNAs in the brain after peripheral injection of subseptic doses of lipopolysaccharide in the rat. J Neuroimmunol 93(1–2):72–80.
- Ranels HJ, Griffin JD. 2003. The effects of prostaglandin E2 on the firing rate activity of thermosensitive and temperature insensitive neurons in the ventromedial preoptic area of the rat hypothalamus. Brain Res 964(1):42–50.
- Rathner JA, Madden CJ, Morrison SF. 2008. Central pathway for spontaneous and prostaglandin E2-evoked cutaneous vasoconstriction. Am J Physiol Regul Integr Comp Physiol 295(1):R343–54.
- Ridder DA, Lang M-F, Salinin S, Röderer J-P, Struss M, Maser-Gluth C, and others. 2011. TAK1 in brain endothelial cells mediates fever and lethargy. J Exp Med 208(13):2615–23.
- Rivest S. 1999. What is the cellular source of prostaglandins in the brain in response to systemic inflammation? Facts and controversies. Mol Psychiatry 4(6):500–7.
- Romanovsky AA, Almeida MC, Aronoff DM, Ivanov AI, Konsman JP, Steiner AA, and others. 2005. Fever and hypothermia in systemic inflammation: recent discoveries and revisions. Front Biosci 10:2193–216.
- Romanovsky AA, Ivanov AI, Szekely M. 2000. Neural route of pyrogen signaling to the brain. Clin Infect Dis 31(Suppl 5):S162–7.
- Romanovsky AA, Kulchitsky VA, Simons CT, Sugimoto N. 1998. Methodology of fever research: Why are polyphasic fevers often thought to be biphasic? Am J Physiol 275(1 Pt 2):R332–8.
- Romanovsky AA, Kulchitsky VA, Simons CT, Sugimoto N, Szekely M. 1997a. Febrile responsiveness of vagotomized rats is suppressed even in the absence of malnutrition. Am J Physiol 273(2 Pt 2):R777–83.
- Romanovsky AA, Simons CT, Szekely M, Kulchitsky VA. 1997b. The vagus nerve in the thermoregulatory response to systemic inflammation. Am J Physiol 273(1 Pt 2):R407–13.
- Romanovsky AA, Steiner AA, Matsumura K. 2006. Cells that trigger fever. Cell Cycle 5(19):2195–7.
- Romanovsky AA, Sugimoto N, Simons CT, Hunter WS. 2003. The organum vasculosum laminae terminalis in immune-to-brain febrigenic signaling: a reappraisal of lesion experiments. Am J Physiol Regul Integr Comp Physiol 285(2):R420–8.
- Romeo HE, Tio DL, Rahman SU, Chiappelli F, Taylor AN. 2001. The glossopharyngeal nerve as a novel pathway in immune-to-brain communication: relevance to neuroimmune surveillance of the oral cavity. J Neuroimmunol 115(1):91–100.
- Ross G, Hubschle T, Pehl U, Braun HA, Voigt K, Gerstberger R, and others. 2003. Fever induction by localized subcutaneous inflammation in guinea pigs: the role of cytokines and prostaglandins. J Appl Physiol 94(4):1395–402.
- Ross G, Roth J, Störr B, Voigt K, Zeisberger E. 2000. Afferent nerves are involved in the febrile response to injection of LPS into artificial subcutaneous chambers in guinea pigs. Physiol Behav 71(3):305–13.
- Rudaya AY, Steiner AA, Robbins JR, Dragic AS, Romanovsky AA. 2005. Thermoregulatory responses to lipopolysaccharide in the mouse: dependence on the dose and ambient temperature. Am J Physiol Regul Integr Comp Physiol 289(5):R1244–52.
- Rummel C, Barth SW, Voss T, Korte S, Gerstberger R, Hubschle T, and others. 2005. Localized vs. systemic inflammation in guinea pigs: a role for prostaglandins at distinct points of the fever induction pathways? Am J Physiol Regul Integr Comp Physiol 289(2):R340–7.
- Rummel C, Inoue W, Sachot C, Poole S, Hubschle T, Luheshi GN. 2008. Selective contribution of interleukin-6 and leptin to brain inflammatory signals induced by systemic LPS injection in mice. J Comp Neurol 511(3):373–95.
- Rummel C, Matsumura K, Luheshi GN. 2011. Circulating IL-6 contributes to peripheral LPS-induced mPGES-1 expression in the rat brain. Brain Res Bull 86(5–6):319–25.
- Rummel C, Sachot C, Poole S, Luheshi GN. 2006. Circulating interleukin-6 induces fever through a STAT3-linked activation of COX-2 in the brain. Am J Physiol Regul Integr Comp Physiol 291(5):R1316–26.
- Ruud J, Nilsson A, Engstrom Ruud L, Wang W, Nilsberth C, Iresjo BM, and others. 2013. Cancer-induced anorexia in tumor-bearing mice is dependent on cyclooxygenase-1. Brain Behav Immun 29:124–35.
- Saha S, Engstrom L, Mackerlova L, Jakobsson PJ, Blomqvist A. 2005. Impaired febrile responses to immune challenge in mice deficient in microsomal prostaglandin E synthase-1. Am J Physiol Regul Integr Comp Physiol 288(5):R1100–7.
- Saper CB, Breder CD. 1994. The neurologic basis of fever. N Engl J Med 330(26):1880–6.
- Scammell TE, Elmquist JK, Griffin JD, Saper CB. 1996. Ventromedial preoptic prostaglandin E2 activates fever-producing autonomic pathways. J Neurosci 16(19):6246–54.
- Scammell TE, Griffin JD, Elmquist JK, Saper CB. 1998. Microinjection of a cyclooxygenase inhibitor into the anteroventral preoptic region attenuates LPS fever. Am J Physiol 274(3 Pt 2):R783–9.
- Schiltz JC, Sawchenko PE. 2002. Distinct brain vascular cell types manifest inducible cyclooxygenase expression as a function of the strength and nature of immune insults. J Neurosci 22(13):5606–18.
- Schmidt W, Schmidt H, Bauer H, Gebhard MM, Martin E. 1997. Influence of lidocaine on endotoxin-induced leukocyte-endothelial cell adhesion and macromolecular leakage in vivo. Anesthesiology 87(3):617–24.
- Sehic E, Blatteis CM. 1996. Blockade of lipopolysaccharide-induced fever by subdiaphragmatic vagotomy in guinea pigs. Brain Res 726(1):160–6.
- Serrats J, Schiltz JC, Garcia-Bueno B, van Rooijen N, Reyes TM, Sawchenko PE. 2010. Dual roles for perivascular macrophages in immune-to-brain signaling. Neuron 65(1):94–106.
- Shido O, Nagasaka T, Watanabe T. 1989. Blunted febrile response to intravenous endotoxin in starved rats. J Appl Physiol 67(3):963–9.
- Simons CT, Kulchitsky VA, Sugimoto N, Homer LD, Szekely M, Romanovsky AA. 1998. Signaling the brain in systemic inflammation: which vagal branch is involved in fever genesis? Am J Physiol 275(1 Pt 2):R63–8.
- Sinclair R, Eriksson AS, Gretzer C, Cassuto J, Thomsen P. 1993. Inhibitory effects of amide local anaesthetics on stimulus-induced human leukocyte metabolic activation, LTB4 release and IL-1 secretion in vitro. Acta Anaesthesiol Scand 37(2):159–65.
- Siso S, Jeffrey M, Gonzalez L. 2010. Sensory circumventricular organs in health and disease. Acta Neuropathol 120(6):689–705.
- Smith BK, Kluger MJ. 1992. Human IL-1 receptor antagonist partially suppresses LPS fever but not plasma levels of IL-6 in fischer rats. Am J Physiol 263(3 Pt 2):R653–5.
- Steiner AA, Dogan MD, Ivanov AI, Patel S, Rudaya AY, Jennings DH, and others. 2004. A new function of the leptin receptor: mediation of the recovery from lipopolysaccharide-induced hypothermia. FASEB J 18(15):1949–51.
- Steiner AA, Hunter JC, Phipps SM, Nucci TB, Oliveira DL, Roberts JL, and others. 2009. Cyclooxygenase-1 or -2—which one mediates lipopolysaccharide-induced hypothermia? Am J Physiol Regul Integr Comp Physiol 297(2):R485–94.
- Steiner AA, Ivanov AI, Serrats J, Hosokawa H, Phayre AN, Robbins JR, and others. 2006. Cellular and molecular bases of the initiation of fever. PLoS Biol 4(9):e284.
- Szekely M, Balasko M, Romanovsky AA. 1997. Peripheral neural inputs. Their role in fever development. Ann N Y Acad Sci 813(1):427–34.
- Tabarean IV, Behrens MM, Bartfai T, Korn H. 2004. Prostaglandin E2-increased thermosensitivity of anterior hypothalamic neurons is associated with depressed inhibition. Proc Natl Acad Sci U S A 101(8):2590–5.
- Takahashi Y, Smith P, Ferguson A, Pittman QJ. 1997. Circumventricular organs and fever. Am J Physiol 273(5 Pt 2):R1690–5.
- Takemiya T, Matsumura K, Sugiura H, Maehara M, Yasuda S, Uematsu S, and others. 2010. Endothelial microsomal prostaglandin E synthase-1 exacerbates neuronal loss induced by kainate. J Neurosci Res 88(2):381–90.
- Tan CL, Cooke EK, Leib DE, Lin Y-C, Daly GE, Zimmerman CA, and others. 2016. Warm-sensitive neurons that control body temperature. Cell 167(1):47–59.e15.
- Tsai VW, Manandhar R, Jorgensen SB, Lee-Ng KK, Zhang HP, Marquis CP, and others. 2014. The anorectic actions of the TGFβ cytokine MIC-1/GDF15 require an intact brainstem area postrema and nucleus of the solitary tract. PLoS One 9(6):e100370.
- Ushikubi F, Segi E, Sugimoto Y, Murata T, Matsuoka T, Kobayashi T, and others. 1998. Impaired febrile response in mice lacking the prostaglandin E receptor subtype EP3. Nature 395(6699):281–4.
- Vallieres L, Rivest S. 1997. Regulation of the genes encoding interleukin-6, its receptor, and GP130 in the rat brain in response to the immune activator lipopolysaccharide and the proinflammatory cytokine interleukin-1β. J Neurochem 69(4):1668–83.
- Van Dam AM, Brouns M, Man AHW, Berkenbosch F. 1993. Immunocytochemical detection of prostaglandin E2 in microvasculature and in neurons of rat brain after administration of bacterial endotoxin. Brain Res 613(2):331–6.
- Van Rooijen N. 1989. The liposome-mediated macrophage ‘suicide’ technique. J Immunol Methods 124(1):1–6.
- Vane JR. 1971. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol 231(25):232–5.
- Vasilache AM, Andersson J, Nilsberth C. 2007. Expression of PGE(2) EP(3) receptor subtypes in the mouse preoptic region. Neurosci Lett 423(3):179–83.
- Vasilache AM, Kugelberg U, Blomqvist A, Nilsberth C. 2013. Minor changes in gene expression in the mouse preoptic hypothalamic region by inflammation-induced prostaglandin E2. J Neuroendocrinol 25(7):635-43.
- Vasilache AM, Qian H, Blomqvist A. 2015. Immune challenge by intraperitoneal administration of lipopolysaccharide directs gene expression in distinct blood-brain barrier cells toward enhanced prostaglandin E2 signaling. Brain Behav Immun 48:31–41.
- Vybiral S, Szekely M, Jansky L, Cerny L. 1987. Thermoregulation of the rabbit during the late phase of endotoxin fever. Pflugers Arch 410(1–2):220–2.
- Wang J, Ando T, Dunn AJ. 1997. Effect of homologous interleukin-1, interleukin-6 and tumor necrosis factor-α on the core body temperature of mice. Neuroimmunomodulation 4(5–6):230–6.
- Wanner SP, Almeida MC, Shimansky YP, Oliveira DL, Eales JR, Coimbra CC, and others. 2017. Cold-induced thermogenesis and inflammation-associated cold-seeking behavior are represented by different dorsomedial hypothalamic sites: a three-dimensional functional topography study in conscious rats. J Neurosci 37(29):6956–71.
- Watkins LR, Goehler LE, Relton JK, Tartaglia N, Silbert L, Martin D, and others. 1995. Blockade of interleukin-1 induced hyperthermia by subdiaphragmatic vagotomy: evidence for vagal mediation of immune-brain communication. Neurosci Lett 183(1–2):27–31.
- Watkins LR, Wiertelak EP, Goehler LE, Mooney-Heiberger K, Martinez J, Furness L, and others. 1994. Neurocircuitry of illness-induced hyperalgesia. Brain Res 639(2):283–99.
- Wilhelms DB, Kirilov M, Mirrasekhian E, Eskilsson A, Kugelberg UÖ, Klar C, and others. 2014. Deletion of prostaglandin E2 synthesizing enzymes in brain endothelial cells attenuates inflammatory fever. J Neurosci 34:11684–90.
- Xu Y, Tao X, Shen B, Horng T, Medzhitov R, Manley JL, and others. 2000. Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature 408(6808):111–5.
- Yamagata K, Matsumura K, Inoue W, Shiraki T, Suzuki K, Yasuda S, and others. 2001. Coexpression of microsomal-type prostaglandin E synthase with cyclooxygenase-2 in brain endothelial cells of rats during endotoxin- induced fever. J Neurosci 21(8):2669–77.
- Yoshida K, Li X, Cano G, Lazarus M, Saper CB. 2009. Parallel preoptic pathways for thermoregulation. J Neurosci 29(38):11954–64.
- Zhang H, Ching S, Chen Q, Li Q, An Y, Quan N. 2008. Localized inflammation in peripheral tissue signals the CNS for sickness response in the absence of interleukin-1 and cyclooxygenase-2 in the blood and brain. Neuroscience 157(4):895–907.
- Zhang Y, Kerman IA, Laque A, Nguyen P, Faouzi M, Louis GW, and others. 2011. Leptin-receptor-expressing neurons in the dorsomedial hypothalamus and median preoptic area regulate sympathetic brown adipose tissue circuits. J Neurosci 31(5):1873–84.