Introduction
Acquired amegakaryocytic thrombocytopenia (AAMT) is a rare disorder which is characterized by severe thrombocytopenia and clinically significant bleeding. Although AAMT is often initially misdiagnosed as idiopathic thrombocytopenic purpura (ITP), the clinical manifestation of AAMT is generally more severe and a differential diagnosis can be achieved by a bone marrow assessment. The bone marrow of patients with AAMT patients shows a severe reduction or absence of megakaryocytes whereas the hematopoietic cells of other lineages are relatively well preserved [, ].
The hematopoietic growth factor thrombopoietin (TPO) is a ligand for c-mpl, a receptor of TPO, which is present on the surface of megakaryocytes. TPO contributes to the growth and differentiation of megakaryocytes by binding to c-mpl [-]. Previous reports have suggested that autoantibodies to TPO or c-mpl are associated with acquired amegakaryocytosis [, -].
Here, we report a case of AAMT with TPO-related antibodies (Abs) which responded to an immunosuppressive treatment. We also determined the presence of autoantibodies in this AAMT patient, enabling a comparison with ITP patients.
Case Report
A 60-year-old female patient presented at our hospital with gingival hemorrhage and extensive petechiae that had developed 1 week earlier. An initial assessment found her platelet count to be 6,000/mm3, hemoglobin level 6.6 g/dL, and white blood cell (WBC) count 6,280/mm3 (neutrophils 74.4% and lymphocytes 22.1%). Serum creatinine levels were 0.48 mg/dL, aspartate transaminase/alanine transaminase 65/66 IU, bilirubin 1.15 mg/dL, and lactic dehydrogenase 507 IU (reference range 240–480 IU). Her coagulation function was normal. Tests for hepatitis virus B and C were negative. Antiplatelet and antinuclear Ab testing was positive at a concentration of 1:80, while anti-dsDNA Ab was negative.
Physical examination showed no lymph node enlargement or hepatosplenomegaly. Other than hypertension, the patient had no preexisting conditions. Initially, she was diagnosed with ITP and intravenous (i.v.) methylprednisolone (1 mg/kg) was initiated on the day of admission.
However, after 1 week of the steroid treatment, her platelet count had not improved, and hematochezia had developed. No definite hemorrhagic focus was found on colonoscopy, but feces mixed with fresh blood were observed throughout the intestine. The patient required a large volume of transfused blood each day to compensate for the gastrointestinal bleeding. At this point, the bleeding symptoms were considered to be more severe than those usually associated with ITP, and a bone marrow examination was performed 8 days after methylprednisolone had been initiated to identify other causes of thrombocytopenia. The patient’s platelet count was now 4,000/mm3. The bone marrow assessment showed normal cellularity of approximately 50%. Hematopoietic cells of the myeloid lineage showed a normal shape and number; the number of cells of the erythroid lineage was increased, possibly as a result of blood loss. However, megakaryocyte numbers were significantly reduced and were rarely observed (Fig. 1a, b). No abnormal or malignant cells were present, and a conventional chromosome analysis revealed a normal karyotype.
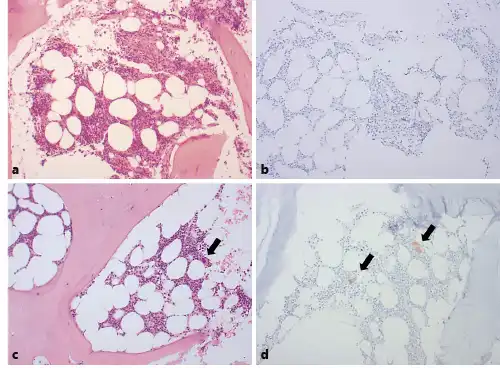
Fig. 1
a Bone marrow biopsy at the time of the initial diagnosis. ×100. b Immunohistochemical staining for CD61 of bone marrow at initial diagnosis. c Bone marrow biopsy 3 months after the initial diagnosis. ×100. d Immunohistochemical staining for CD61 of bone marrow 3 months after the initial diagnosis. c, d Black arrows indicate megakaryocytes.
The patient’s diagnosis was changed from ITP to AAMT and oral cyclosporine (200 mg/day) was initiated on day 9 of the methylprednisolone treatment, which was continued. Immunoglobulin (i.v. 1 g/kg/day; total 2 g/kg) was administered on days 13 and 14. The platelet count began to rise from day 13 and the gastrointestinal bleeding improved (Fig. 2). Seventeen days after administration, her complete blood count was as follows: WBC 10,690 /mm3, hemoglobin 8.0 g/dL, and platelets 225,000/mm3. At this time, i.v. methylprednisolone was replaced by the same dose of oral prednisolone.
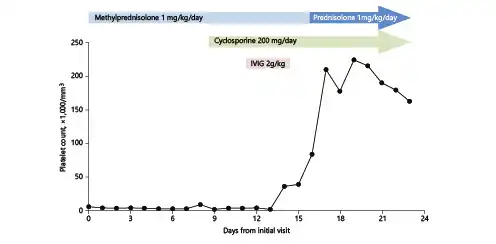
Fig. 2
Changes in the platelet count over the course of treatment. IVIG, intravenous immunoglobulin.
From day 30 after administration, the dose of prednisolone was reduced. At 6 weeks after diagnosis, neutropenia (WBC count 940/mm3 and absolute neutrophil count 440/mm3) and increased blood urea nitrogen (BUN, 62.8 mmol/L) were detected. Cyclosporine was discontinued at this time due to the emergence of neutropenia and uremia, which were considered to be a side effect of this drug. At week 7 (1 week after discontinuation of cyclosporine), her leukocyte count normalized and BUN decreased. At 12 weeks after initial diagnosis, the platelet count was 147,000/mm3 and the steroid therapy was discontinued.
A follow-up bone marrow examination was performed at 3 months after the initial diagnosis, at which point the cellularity was 40% and the megakaryocyte shape and numbers were normal (Fig. 1c, d). At 1 year after presentation, the patient was found to have maintained a platelet count ≥100,000/mm3 without the need for additional medication.
Detection of Autoantibodies
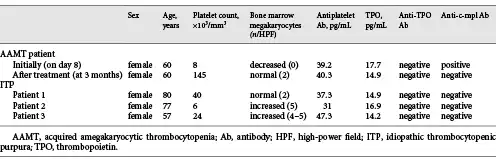
Serum levels of antiplatelet Ab, TPO, anti-TPO Ab, and anti- c-mpl Ab were assessed 8 days and 3 months after admission using enzyme-linked immunosorbent assay (ELISA) kits (MyBioSource. Inc., San Diego, CA, USA). For comparison, serum samples from 3 typical ITP patients with normal or increased bone marrow megakaryocytes were also tested.
The results of peripheral blood tests, the bone marrow biopsy and the ELISAs are summarized in Table 1. Concentrations of antiplatelet Abs did not differ significantly between the samples. TPO levels on day 8 and at 3 months for the AAMT patient were 17.7 and 14.9 pg/mL, respectively, similar to the levels in the 3 ITP patients (14.9, 16.9, and 14.2 pg/mL, respectively). Anti-TPO Ab was negative in all samples. The serum of our patient on day 8 was positive for anti-c-mpl Ab, while the serum sample taken at 3 months was negative. The sera of the 3 ITP patients were also negative for anti-c-mpl Ab.
Discussion
Although the pathogenesis of AAMT has not yet been fully established, some researchers have reported that it may be caused by an inherent defect in the megakaryocyte itself []. However, there are cases where AAMT is accompanied by diseases that induce a disturbance in the immune function [, ], and AAMT can occur with various autoimmune diseases [, ]. Therefore, it is generally presumed that an autoimmune mechanism inhibits the maturation and differentiation of megakaryocytes and is associated with the development of AAMT.
TPO regulates the differentiation and maturation of megakaryocytes and promotes platelet production. The TPO receptor, also called c-mpl, is present on the surface of hematopoietic cells including megakaryocytes, and it plays a role in regulating the concentration of TPO in the body [, ]. When the action of TPO is disrupted, the maturation of megakaryocytes is inhibited and their numbers are reduced. It has been reported that a decrease in c-mpl expression is due to the mutation of the c-mpl gene and the main cause of congenital AAMT []. It is also reported that TPO is elevated in the serum of patients with AAMT []. It is therefore believed that the mechanisms that interfere with the action of TPO on megakaryocytes play an important role in the pathogenesis of AAMT.
Autoantibodies to c-mpl are often found in patients with AAMT. Kuwana et al. []reported that approximately 11.6% of patients with systemic lupus erythematosus (SLE) exhibit anti-c-mpl Ab and patients with SLE and anti-c-mpl Ab often have accompanying thrombocytopenia and a decrease in the number of bone marrow megakaryocytes. Hashimoto et al. []observed a complete loss of megakaryocytes in the bone marrow of a patient with rheumatoid arthritis who had thrombocytopenia and anti-c-mpl Ab; the megakaryocyte and platelet count of this patient recovered following cyclosporine treatment. Ichimata et al. []reported that a patient with hepatitis C who had thrombocytopenia had decreased megakaryocyte numbers along with plasma anti-c-mpl Ab. Anti-c-mpl Ab appears to be found at a higher frequency than expected in immune-related thrombocytopenia. In a recently published paper, anti-c-mpl Ab was found in about 30% (54/187) of ITP patients []. The authors also reported that the positivity of anti-c-mpl Ab in ITP patients was associated with lower platelet and megakaryocyte counts, higher serum TPO levels, and inferior responsiveness to a recombinant human TPO treatment than in patients without anti-c-mpl Ab. In our case study, we detected anti-c-mpl Ab in the serum at the time of the AAMT diagnosis, but this Ab was not detected in the sera of the ITP patients whose bone marrow megakaryocyte counts were normal or increased. These findings suggest that the presence of anti-c-mpl Ab prevents TPO from binding to its receptor, thereby inhibiting megakaryocyte maturation, which can be considered as one of the pathological mechanisms of AAMT.
Although the pathogenesis of AAMT can vary, the autoimmune mechanism is presumed to be the underlying cause and treatment generally involves the use of immunosuppressants. Unlike in cases of ITP, steroid therapy alone appears to be ineffective in patients with AAMT. Some researchers report cases of AAMT where a good response was achieved with cyclosporine treatment [, ]. One such study involved a patient with AAMT who failed steroid and immunoglobulin therapy but responded well to sequential antithymocyte globulin and cyclosporine []. Anti-CD20 monoclonal Ab (rituximab) and azathioprine can also be effective in the treatment of patients with AAMT [, ]; Cela et al. []reported the successful treatment of SLE-associated AAMT using the TPO receptor agonist eltrombopag. However, a standard treatment strategy for AAMT has not yet been established. In this report, the initial steroid monotherapy did not result in an improvement, but the platelet count started to increase after the addition of cyclosporine and immunoglobulin. In contrast to the management of ITP, this observation suggests that an intensive immunosuppressive regimen, including a combination of several immunosuppressants, should be considered for the treatment of AAMT. In addition, we performed a follow-up bone marrow evaluation 3 months after treatment, which confirmed that the megakaryocyte level had normalized. Anti-c-mpl Ab was also negative at this point, demonstrating that autoantibody clearance by immunosuppressive therapy contributes to the increase in megakaryocyte and platelet counts in patients with AAMT.
Assuming that anti-c-mpl Ab is likely to be involved in the loss of megakaryocytes, it can be assumed that thrombocytopenia is likely to recur after treatment if anti-c-mpl Ab is not completely removed. Therefore, with regard to thrombocytopenia in which anti-c-mpl Ab is positive and a loss of bone marrow megakaryocytes is observed, the goal of treatment may involve both an increased platelet count and the elimination of anti-c-mpl Ab. In previous studies, Anti-c-mpl Ab was reported to have decreased or disappeared after the use of immunosuppressants [, ]. We also found that anti-c-mpl Ab was negative when platelets and megakaryocytes were normalized after treatment. However, there has been no report of changes in anti-c-mpl Ab when these patients showed recurrent thrombocytopenia or other autoimmune diseases associated with AAMT. Further studies are needed to define the role of anti-c-mpl Ab in the treatment for AAMT.
This case report provides evidence of an association between anti-c-mpl Ab and the pathophysiology of AAMT. In addition, we suggested that clearance of autoantibodies using a combined immunosuppressive therapy is required for the treatment of AAMT. Further investigations will be necessary to establish best practice for the treatment of patients with AAMT.
Acknowledgement
The biospecimens for this study were provided by the Chungbuk National University Hospital, a member of the National Biobank of Korea, which is supported by the Ministry of Health, Welfare and Family Affairs.
Statement of Ethics
All samples derived from the National Biobank of Korea were obtained with informed consent under institutional review board-approved protocols.
Disclosure Statement
The authors declare that they have no conflict of interest.
References
- 1. Tristano AG. Acquired amegakaryocytic thrombocytopenic purpura: review of a not very well-defined disorder. Eur J Intern Med. 2005;16(7):477–81.
- 2. Agarwal N, Spahr JE, Werner TL, Newton DL, Rodgers GM. Acquired amegakaryocytic thrombocytopenic purpura. Am J Hematol. 2006;81(2):132–5.
- 3. Kaushansky K. Thrombopoietin: the primary regulator of megakaryocyte and platelet production. Thromb Haemost. 1995;74(1):521–5.
- 4. Broudy VC, Kaushansky K. Thrombopoietin, the c-mpl ligand, is a major regulator of platelet production. J Leukoc Biol. 1995;57(5):719–25.
- 5. Kaushansky K, Broudy VC, Lin N, Jorgensen MJ, McCarty J, Fox N, et al Thrombopoietin, the Mp1 ligand, is essential for full megakaryocyte development. Proc Natl Acad Sci USA. 1995;92(8):3234–8.
- 6. Shiozaki H, Miyawaki S, Kuwaki T, Hagiwara T, Kato T, Miyazaki H. Autoantibodies neutralizing thrombopoietin in a patient with amegakaryocytic thrombocytopenic purpura. Blood. 2000;95(6):2187–8.
- 7. Hashimoto A, Kanisawa Y, Fujimi A, Nakajima C, Hayasaka N, Yamada S, et al Thrombocytopenia and Anemia with Anti-c-Mpl antibodies Effectively Treated with Cyclosporine in a Patient with Rheumatoid Arthritis and Chronic Renal Failure. Intern Med. 2016;55(6):683–7.
- 8. Kuwana M, Okazaki Y, Kajihara M, Kaburaki J, Miyazaki H, Kawakami Y, et al Autoantibody to c-Mpl (thrombopoietin receptor) in systemic lupus erythematosus: relationship to thrombocytopenia with megakaryocytic hypoplasia. Arthritis Rheum. 2002;46(8):2148–59.
- 9. Ichimata S, Kobayashi M, Honda K, Shibata S, Matsumoto A, Kanno H. Acquired amegakaryocytic thrombocytopenia previously diagnosed as idiopathic thrombocytopenic purpura in a patient with hepatitis C virus infection. World J Gastroenterol. 2017;23(35):6540–5.
- 10. Lu D, Chen Y, Ding R. [Study on the pathogenesis of acquired pure amegakaryocytic thrombocytopenic purpura]. Zhonghua Xue Ye Xue Za Zhi. 1999;20(3):124–6.
- 11. King JA, Elkhalifa MY, Latour LF. Rapid progression of acquired amegakaryocytic thrombocytopenia to aplastic anemia. South Med J. 1997;90(1):91–4.
- 12. Slater LM, Katz J, Walter B, Armentrout SA. Aplastic anemia occurring as amegakaryocytic thrombocytopenia with and without an inhibitor of granulopoiesis. Am J Hematol. 1985;18(3):251–4.
- 13. Cela I, Miller IJ, Katz RS, Rizman A, Shammo JM. Successful treatment of amegakaryocytic thrombocytopenia with eltrombopag in a patient with systemic lupus erythematosus (SLE). Clin Adv Hematol Oncol. 2010;8(11):806–9.
- 14. Ergas D, Tsimanis A, Shtalrid M, Duskin C, Berrebi A. T-gamma large granular lymphocyte leukemia associated with amegakaryocytic thrombocytopenic purpura, Sjögren’s syndrome, and polyglandular autoimmune syndrome type II, with subsequent development of pure red cell aplasia. Am J Hematol. 2002;69(2):132–4.
- 15. Ballmaier M, Germeshausen M, Schulze H, Cherkaoui K, Lang S, Gaudig A, et al c-mpl mutations are the cause of congenital amegakaryocytic thrombocytopenia. Blood. 2001;97(1):139–46.
- 16. Mukai HY, Kojima H, Todokoro K, Tahara T, Kato T, Hasegawa Y, et al Serum thrombopoietin (TPO) levels in patients with amegakaryocytic thrombocytopenia are much higher than those with immune thrombocytopenic purpura. Thromb Haemost. 1996;76(5):675–8.
- 17. Jing FM, Zhang XL, Meng FL, Liu XM, Shi Y, Qin P, et al Anti-c-Mpl antibodies in immune thrombocytopenia suppress thrombopoiesis and decrease response to rhTPO. Thromb Res. 2018;170:200–6.
- 18. Niparuck P, Atichartakarn V, Chuncharunee S. Successful treatment of acquired amegakaryocytic thrombocytopenic purpura refractory to corticosteroids and intravenous immunoglobulin with antithymocyte globulin and cyclosporin. Int J Hematol. 2008;88(2):223–6.
- 19. Mirzania M, Khalili S, Hasanpoor A, Shamshiri AR. Anti-CD20 Antibody is Effective in the Patient with Refractory Amegakaryocytic Thrombocytopenia, 25 Months Follow up. Int J Hematol Oncol Stem Cell Res. 2014;8(2):41–4.
- 20. Chang H, Tang TC. Successful treatment of amegakaryocytic thrombocytopenia with azathioprine. Acta Haematol. 2011;126(3):135–7.