Introduction
Anemia is a common complication of chronic kidney disease (CKD) [] and is associated with increased risk of mortality, cardiovascular events, blood transfusion, and decreased quality of life [-]. Iron deficiency is a frequent cause of anemia in nondialysis-dependent CKD (NDD-CKD) [, ] and can result from decreased absorption of iron, blood loss, or sequestration of iron due to inflammation or a combination of these factors [].
Current clinical management of anemia in NDD-CKD involves the use of oral or intravenous (IV) iron replacement therapy, erythropoiesis-stimulating agents (ESAs), and red blood cell transfusion as a last resort []. According to the Kidney Disease: Improving Global Outcomes 2012 clinical practice guidelines, correctable causes of anemia, including iron deficiency anemia (IDA) and inflammatory states, should be addressed before initiating ESAs []. In NDD-CKD patients, a 1–3-month trial of oral iron therapy is recommended if transferrin saturation (TSAT) is ≤30%, ferritin levels are ≤500 μg/mL, and increased hemoglobin (Hb) concentration is desired without using ESAs.
Ferric citrate (FC) is a US Food and Drug Administration-approved oral iron replacement therapy for treatment of IDA in adults with NDD-CKD and hyperphosphatemia in adults with dialysis-dependent CKD []. FC significantly improves Hb response and iron parameters, with low rates of serious adverse events (AEs) (like hypophosphatemia) and treatment-related discontinuations. FC demonstrated improvements in Hb levels and iron parameters, with good tolerability in NDD-CKD patients [, ]. For NDD-CKD patients with IDA, the recommended FC starting dose is one tablet (1 g; each 1-g tablet contains 210 mg ferric iron) three times daily (TID) with meals.
Clinical studies in NDD-CKD patients demonstrated that FC increased Hb, TSAT, and ferritin without causing hypophosphatemia [-]. FC was evaluated in a randomized, double-blind, placebo-controlled trial in adults with IDA and NDD-CKD. Patients were randomized to FC TID (n = 117) versus placebo (n = 116) during a 16-week period, followed by an 8-week open-label extension []. The FC dose was titrated at weeks 4, 8, and 12, with the goal of achieving an increase in Hb of >1.0 g/dL from baseline. FC, at an average daily dose of 5 g, effectively restored iron stores and partially corrected anemia; 52% of FC-treated patients achieved an increase in Hb ≥1.0 g/dL at any time point (vs. 19% with placebo; p < 0.001). Because the longest published continuous exposure to FC in clinical studies for treatment of adults with IDA and NDD-CKD was 16 weeks, and all dosing strategies consisted of TID dosing with meals, this phase 4 study expanded on available knowledge by evaluating the long-term efficacy (48 weeks) and safety of FC TID or twice daily (BID).
Materials and Methods
Study Design
This 48-week, phase 4, randomized, open-label, multicenter study evaluated the long-term efficacy and safety of TID and BID dosing of FC for treatment of anemia in adults with NDD-CKD. It consisted of a 24-week dose titration period followed by a 24-week dose maintenance period and included approximately 12 scheduled clinic visits. The study was conducted between August 2017 and August 2019 at 28 sites in the USA and was conducted in accordance with the Declaration of Helsinki. The protocol was approved by the institutional review board (Schulman Associates Institutional Review Board, Inc., now Advarra IRB, Cincinnati, OH, USA) at each site and registered as the COMPASS trial at ClinicalTrials.gov (NCT03236246; https://clinicaltrials.gov/ct2/show/NCT03236246). Written informed consent was obtained from patients before study enrollment.
Study Population
The study population consisted of male and female patients aged ≥18 years with NDD-CKD (estimated glomerular filtration rate [eGFR] ≥20 mL/min and <60 mL/min), anemia (defined as Hb ≤11.5 g/dL with serum ferritin ≤500 ng/mL and TSAT ≤25%), and a serum intact parathyroid level ≤600 pg/mL at screening. For safety, patients with Hb <8.5 g/dL or a serum phosphate level <3.0 mg/dL at screening were excluded. Patients were also excluded if they had received IV iron, ESAs, or blood transfusion within 4 weeks of screening or during the screening period. Exclusion criteria included symptomatic gastrointestinal bleeding within 12 weeks and a history of kidney transplant or a kidney transplant scheduled within 6 months of screening.
Study Intervention and Dosing
Following screening, eligible patients were randomized 1:1, with an open-label design, to receive either one FC tablet (1 g including 210 mg ferric iron) TID with meals (total of 3 g FC per day), or 2 FC tablets BID with meals (total of 4 g FC per day). At week 12, patients with Hb increased <0.5 g/dL from baseline or Hb <10 g/dL had their dosage increased to 6 tablets (FC 6 g/day). Patients in the FC TID group received 2 tablets TID (3 → 6 g/day) and patients in the FC BID group received 3 tablets BID (4 → 6 g/day) (Fig. 1). Only patients completing the 24-week dose titration period were eligible for the 24-week dose maintenance period.
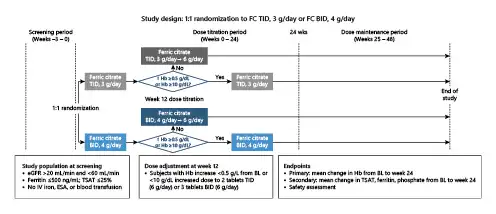
Fig. 1
Study design. BID, twice daily; BL, baseline; eGFR, estimated glomerular filtration rate; ESA, erythropoiesis-stimulating agents; FC, ferric citrate; Hb, hemoglobin; IV, intravenous; TID, three times daily; TSAT, transferrin saturation.
During the maintenance period, patients continued using the dose determined during the titration period. The maximum dose was assigned during week 12. However, dose adjustments or interruptions could be made at any point to address laboratory abnormalities (e.g., serum phosphate <2.5 mg/dL, TSAT ≥50%) or AEs, per prespecified guidance and dosing algorithms (see online suppl. Fig. 1; for all online suppl. material, see http://www.karger.com/doi/10.1159/000516012) intended to assist investigators with decision making.
ESA use was permitted only as rescue therapy in patients with 2 consecutive Hb values <8.0 g/dL more than 3 days apart if the investigator deemed it appropriate (as per standard of care) and for the patient to continue the study. Additionally, the patient had to continue using FC throughout the study. Patients who had 2 consecutive Hb values <8.0 g/dL more than 3 days apart but did not meet the other criteria for ESA rescue therapy were discontinued from the study.
Other prohibited interventions during the trial included use of IV iron, blood transfusion, oral iron therapy other than FC, phosphate binders, commercial FC, aluminum-containing therapies, and other investigational therapies. Use of multivitamins containing iron was permitted.
Study Endpoints
The primary efficacy endpoint was change in Hb from baseline to week 24. Key secondary efficacy endpoints included change in Hb from baseline at week 48, time from randomization to first increase from baseline in Hb of ≥0.5 g/dL, and changes from baseline to weeks 24 and 48 in TSAT, serum phosphate, eGFR, and intact fibroblast growth factor 23 (iFGF23). Changes from baseline to week 48 in Work Productivity and Activity Impairment questionnaire score (adapted for CKD-related anemia) and the Functional Assessment of Chronic Illness Therapy (FACIT) scale were also evaluated [, ]. Safety variables assessed included incidence, seriousness, intensity, duration, and type of AEs, plus clinically significant changes in vital signs and laboratory test results.
Laboratory Measurements
Hematology and chemistry studies were performed by Clinical Central Laboratory Services, PPD® Laboratories. Fibroblast growth factor 23 (intact [iFGF23] and C terminus [cFGF23]) were measured by commercial enzyme-linked immunosorbent assay (ELISA). cFGF23 was measured in plasma using the second-generation carboxyterminal ELISA (Immutopics, San Clemente, CA, USA), which recognizes iFGF23 and cFGF23. iFGF23 was measured in serum using an ELISA against the intact protein (Kainos, Tokyo, Japan). iFGF23 units are expressed in pg/mL, and cFGF23 units are expressed in RU/mL.
Statistical Analysis
Results from a prior phase 3 study in a similar population reported a standard deviation (SD) on the change in Hb from baseline at week 16 of 0.9 g/dL. Assuming a population SD of 0.9 g/dL, with a sample size of 140 patients, the 95% confidence intervals (CIs) for mean change in Hb from baseline to week 24 were estimated, allowing for a CI half-width of 0.149 g/dL. It was assumed that 30% of patients would drop out or be excluded from the per-protocol (PP) population. Thus, 200 patients were planned to be randomized to ensure a sufficient number would be included in the longitudinal analysis.
The analysis populations included the safety population, full analysis set, and PP population. The safety population included all patients who took ≥1 dose of study medication, and the full analysis set included all randomized patients administered ≥1 dose of study medication who had ≥1 post-randomization efficacy assessment. The PP population included all randomized patients who took ≥1 dose of study medication, had ≥1 post-randomization efficacy measurement and corresponding baseline data, and had no major protocol deviations and did not receive additional treatment (e.g., ESA therapy) that affected the validity of the efficacy measurements. Efficacy analyses were based on the PP population for the 24-week analysis and the final analysis. An interim analysis was conducted at the 24-week dose titration period [].
Change in Hb from baseline was the primary endpoint of interest. A two-sided CI was constructed for the primary efficacy endpoint for each starting-dose treatment group in the PP population. A mixed-model repeated-measures approach was used, with baseline Hb, randomized dose group, time, and time-by-dose interactions included as fixed effects. Changes in TSAT, ferritin, serum phosphorus, eGFR, and iFGF23 were analyzed using a similar approach.
A Kaplan-Meier estimate was used to determine the time to first Hb increase from baseline of ≥0.5 g/dL for both starting-dose groups. Descriptive statistics were used to quantify changes in FACIT Fatigue Scale and Work Productivity and Activity Impairment questionnaire scores from baseline to weeks 24 and 48.
Safety analyses were conducted in the safety population. Data were summarized using descriptive statistics overall and by dose group.
Results
Patient Disposition and Demographics
Of 484 patients screened, 206 were randomized, and 205 received FC (Fig. 2), including 104 assigned to the TID group (3 g/day) and 101 assigned to the BID group (4 g/day). Although 155 patients (75.2%) completed the dose titration period, 51 (24.8%) prematurely discontinued from the study. Reasons for discontinuation were similar across treatment groups. No patient discontinued participation in the study because of lack of efficacy. One patient in the BID 4 g/day group received permitted ESA rescue medication, whereas 4 patients were discontinued for receiving prohibited therapies, including blood transfusion (one patient in the TID 3 g/day group) and ESA therapy outside the use defined in the protocol (one patient each in the TID 3 g/day and BID 4 g/day groups).
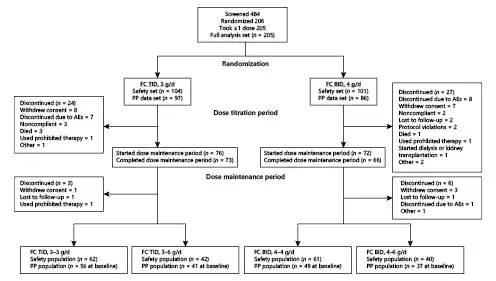
Fig. 2
Patient disposition. AEs, adverse events; BID, twice daily; FC, ferric citrate; PP, per protocol; TID, three times daily.
Baseline characteristics and clinical data for patients in the PP population are shown in Table 1. Baseline demographics by dosing sequence were generally similar across groups and dosing sequences, and in the dose titration and dose maintenance periods of the trial.
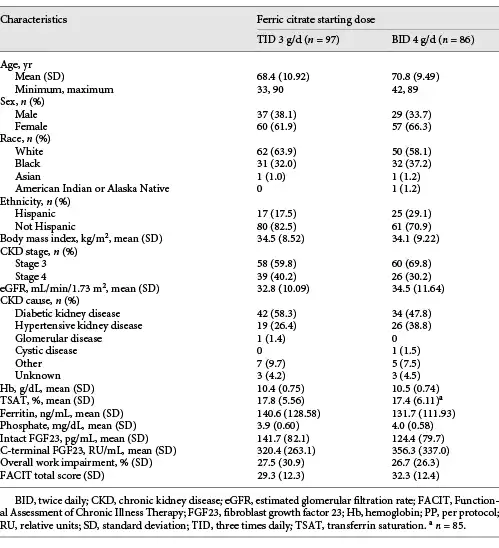
FC Dosing
At the end of titration, the mean daily dose of FC was 3.40 g/day over a mean duration of 144.1 days in the FC TID 3 g/day group and 4.27 g/day over a mean duration of 148.8 days in the FC BID 4 g/day group. Similar numbers of patients were on a maximum daily dose of 6 g/day after dose titration in both groups, 42 (40.4%) in the FC TID 3 g/day starting-dose group and 40 (39.6%) in the FC BID 4 g/day starting-dose group.
Primary Efficacy Assessment
Mean change from baseline in Hb at week 24 was 0.77 g/dL (SD, 0.84) in the FC TID 3 g/day group and 0.70 g/dL (SD, 0.983) in the FC BID 4 g/day group (Fig. 3). Achieved Hb (Fig. 3; Table 2) was maintained at or above 11.0 g/dL in patients in both groups between weeks 24 and 48. Forty-one patients (42.3%) in the FC TID 3 g/day group and 37 (43.0%) in the FC BID 4 g/day group had Hb <10.0 g/dL at week 12 and required dose increases.
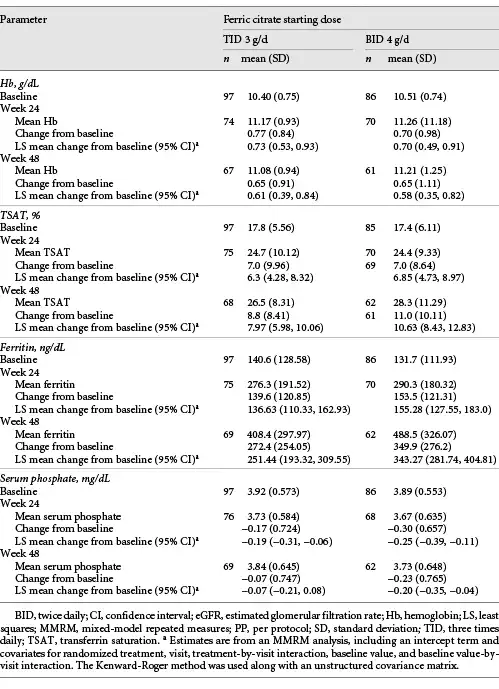
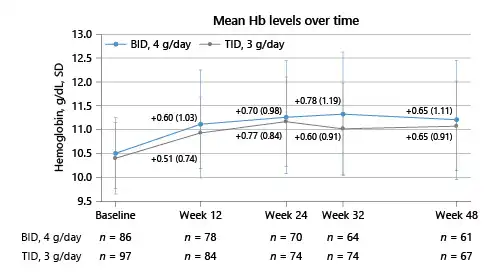
Fig. 3
Hb concentration over time, by starting dose of FC (PP population). BID, twice daily; BL, baseline; FC, ferric citrate; Hb, hemoglobin; PP, per-protocol; SD, standard deviation; TID, three times daily.
Secondary Efficacy Assessments
Mean change from baseline in Hb at week 48 was 0.65 g/dL (SD, 0.911) in the FC TID 3 g/day group and 0.65 g/dL (SD, 1.11) in the FC BID 4 g/day group (Table 2). In the dose titration period, 77 patients (77.8%) in the FC TID 3 g/day group and 64 (71.1%) in the FC BID 4 g/day group achieved an increase in Hb of ≥0.5 g/dL from baseline values. Median time to this increase was 84 and 57 days, respectively.
There were no clinically meaningful differences between dosing or dose-sequencing groups in TSAT, ferritin, and serum phosphate (Table 2; online suppl. Table 1). Least-squares (LS) mean changes from baseline in eGFR were −1.08 mL/min/1.73 m2 (95% CI, −2.73, 0.57) in the FC TID 3 g/day group and −1.53 mL/min/1.73 m2 (95% CI, −3.27, 0.21) in the FC BID 4 g/day group at week 24. At week 48, the respective LS mean changes in eGFR were −1.85 mL/min/1.73 m2 (95% CI, −3.78, 0.08) and −2.20 mL/min/1.73 m2 (95% CI, −4.24, −0.15).
LS mean changes from baseline in iFGF23 at week 24 were −0.23 pg/mL (95% CI, −16.27, 15.80) in the FC TID 3 g/day group and −2.01 pg/mL (95% CI, −18.40, 14.37) in the FC BID 4 g/day group; at week 48, the respective changes were 18.52 pg/mL (95% CI, −10.66, 47.70) and 10.15 pg/mL (95% CI, −21.69, 41.98). Changes from baseline in cFGF23 at week 24 were −94.78 RU/mL (95% CI, −146.99, −42.56) in the FC TID 3 g/day group and −63.22 RU/mL (95% CI, −115.44, −11.01) in the FC BID 4 g/day group; at week 48, the respective changes were −85.70 RU/mL (95% CI, −131.30, −40.10) and −80.34 RU/mL (95% CI, −128.60, −32.09).
There was no change from baseline in percentage of work time missed at week 48 in the FC TID 3 g/day group (0%; SD, 0) or the FC BID 4 g/day group (−3.48%; SD, 7.412). The change in overall percent activity impairment at week 48 was −16.7% (SD, 36.9) in the FC TID group and −6.72% (SD, 36.2) in the FC BID group. At week 48, mean FACIT scores were 37.8 (SD, 11.6) in the FC TID group and 35.8 (SD, 13.0) in the FC BID group; changes from baseline in FACIT total score at week 48 were 7.5 and 3.7, respectively.
Post Hoc Analysis
Patients in the FC TID 3 g/day group who did not require dose titration (n = 56) had baseline Hb of 10.27 g/dL (SD, 0.69), TSAT of 16.8% (SD, 5.3), and ferritin of 129.4 ng/dL (SD, 128.1); those who required dose titration (n = 41) had baseline Hb of 10.57 g/dL (SD, 0.81), TSAT of 19.2% (SD, 5.7), and ferritin of 155.9 ng/dL (SD, 129.3). Patients in the FC BID 4 g/day group who did not require dose titration (n = 49) had baseline Hb of 10.40 g/dL (SD, 0.67), TSAT of 15.3% (SD, 5.7), and ferritin of 96.7 ng/dL (SD, 87.9), whereas those who required dose titration (n = 37) had baseline Hb of 10.65 g/dL (SD, 0.81), TSAT of 20.1% (SD, 5.6), and ferritin of 177.9 ng/dL (SD, 124.1).
Safety Assessments
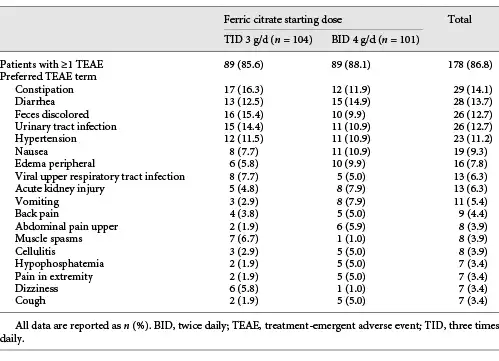
Throughout the study, 89 patients in the FC TID 3 g/day group (85.6%) and 89 in the FC BID 4 g/day group (88.1%) reported treatment-emergent AEs, the most common of which were diarrhea, stool discoloration, and constipation (Table 3). Twenty-six patients in the FC TID 3 g/day group (25.0%) and 25 in the FC BID 4 g/day group (24.8%) reported serious AEs. Twenty patients in the FC TID 3 g/day group (19.2%) and 9 in the FC BID 4 g/day group (8.9%) experienced AEs leading to dose alterations.
Five patients died during the study (3 [2.9%] in the FC TID 3 g/day group; 2 [2.0%] in the FC BID 4 g/day group). Reported causes of death included cardiac arrest, acute coronary syndrome, motor vehicle accident, aspiration pneumonia, and acute respiratory failure. None of these deaths were considered related to FC treatment per the investigators.
Discussion
In this phase 4 study of adults with NDD-CKD, long-term treatment of anemia with FC dosed with meals in either the currently approved TID schedule or BID was associated with similar increases in Hb at 24 and 48 weeks. Data from this study support using FC in a BID dosing regimen with meals if desired to correct anemia in this population. At 24 and 48 weeks, mean changes in Hb, TSAT, ferritin, and phosphate were similar in both regimens.
On average, Hb increased by approximately 0.7–0.8 g/dL by week 24, which was maintained at week 48 in the PP population (0.65 g/dL). Peak Hb responses occurred between 57 and 113 days of FC treatment. Because FC therapy helped participants achieve adequate erythropoiesis in this population, practitioners should be aware of the time course of this response and not abandon oral iron treatment with FC prematurely unless otherwise indicated. In fact, the time course of this response is similar to other interventions that increase erythropoiesis, like ESA or IV iron [, ]. Both the magnitude of the change in Hb and time to peak responses were similar to those seen with IV iron therapy [, ]. As noted, only a small number of patients whose Hb decreased below a safety threshold during the study were either given rescue therapy with an ESA or terminated from the study to allow additional interventions. This is despite an average entry eGFR between 32 and 34 mL/min/1.73 m2 and worsening of kidney function during the study.
Approximately 40% of patients in both the FC TID 3 g/day and BID 4 g/day groups required an increased dose at 12 weeks. As shown previously [], patients with lower baseline TSAT and serum ferritin values in either group showed the best response to FC and did not require an FC dose increase. In contrast, those who required dose titration had smaller increases in Hb in response to FC therapy, with higher baseline Hb and less obvious iron deficiency, as indicated by higher TSAT and ferritin levels. Regardless of whether patients required FC dose increases, most maintained Hb above 10 g/dL (and on average ≥11.0 g/dL) without use of ESA or blood transfusion. Additionally, for most patients, TSAT and ferritin values tended toward levels compatible with iron repletion, thus allowing the addition of ESA if needed. Moreover, changes in eGFR observed across all dose-sequence groups were consistent with published observations in patients with CKD, suggesting no apparent detrimental effect of long-term use of FC on kidney function [].
The initial dose of FC used for this study was lower than that required at the end of the blinded 16 weeks of therapy in the registrational trial of FC in patients with NDD-CKD and IDA (5.0 g/day) []. Nonetheless, the titration scheme used for this study, when needed, allowed patients to reach and exceed the average seen in that study. We show here that FC 3–4 g/day is sufficient for most patients with NDD-CKD to achieve an adequate Hb response (58 and 57% of patients in the FC TID 3 g/day and FC BID 4 g/day groups, respectively).
This phase 4 study confirmed prior results showing that serum phosphate remained within normal limits even in the context of dose titration and FC administration with meals. Although patients were excluded if they had a serum phosphate level <3.0 mg/dL at screening, no patient discontinued the study because of hypophosphatemia, and the frequency of serum phosphorus determinations seemed adequate to assess possible complications related to FC phosphate-binding properties. Evidence suggests that patients with NDD-CKD have a measurable decrease in urinary excretion of phosphorus during FC use that maintains serum phosphorus level within normal limits [].
Long-term treatment with FC resulted in no change in iFGF23 levels and significant reductions in cFGF23 that were sustained through week 48. Previous analysis of data from a phase 3 study of patients with IDA and CKD suggested FC acts via changes in serum phosphate to reduce iFGF23 and via changes in TSAT to reduce cFGF23, consistent with the expected effects of hyperphosphatemia and iron deficiency on iFGF23 and cFGF23, respectively []. Thus, the observed reduction in cFGF23 was likely related to the correction of iron deficiency in this population. The lack of change in iFGF23 may be related to the normal levels of serum phosphate at baseline.
Limitations of this study include lack of a placebo or oral iron comparator, a focus on laboratory rather than clinical outcomes, and a lack of patients receiving background ESA therapy at baseline (a population that frequently has concomitant iron deficiency and could potentially benefit from treatment with FC). Also not studied was FC administration outside of meals; although it could be safe and effective, dosing in this study was guided by the approved FC label. Strengths of this study include its long duration, providing the longest exposure to FC without use of ESA rescue, and its flexible design, allowing investigators to titrate dosing similar to real-world practice.
FC was effective and well tolerated for treatment of anemia in NDD-CKD patients for up to 48 weeks. Mean changes in Hb, TSAT, ferritin, and phosphate, and AE profiles were similar with the TID and BID regimens and in the 3 and 4 g/day dosing groups, supporting potential dosing flexibility with FC. Overall, the safety profiles of the 2 dosing regimens were similar and consistent with the known safety profile of FC. Constipation and diarrhea were the most commonly reported AEs. This study demonstrated that long-term use of FC administered twice a day provides a safe, clinically meaningful increase in Hb and may be a useful option for iron replacement therapy in NDD-CKD patients.
Acknowledgments
The authors thank the participants for volunteering and the study investigators and staff for conducting this study. Medical writing assistance was provided by Syneos Health and funded by Akebia Therapeutics, Inc.
Statement of Ethics
The study was conducted between August 2017 and August 2019 at 28 sites in the USA and was conducted in accordance with the Declaration of Helsinki. The protocol was approved by the institutional review board (Schulman Associates Institutional Review Board, Inc., now Advarra IRB, Cincinnati, OH, USA) at each site and registered as the COMPASS trial at ClinicalTrials.gov (NCT03236246; https://clinicaltrials.gov/ct2/show/NCT03236246). Written informed consent was obtained from patients before study enrollment.
Conflict of Interest Statement
P.E.P., D.B., P.C., and M.M. served as clinical investigators in this study and currently serve as study consultants for Akebia Therapeutics, Inc., W.L. is an employee of Akebia Therapeutics, Inc. A.G.-R., and Y.M.K.F. were employees of Akebia Therapeutics, Inc., when the study was conducted. P.E.P. has received consulting fees from Akebia, AstraZeneca, Bayer, Reata, Gilead, Corvidia, Fibrogen, Tricida, and Ardelyx advisory board/speaker bureaus, and research support provided by Renal Associates, P.A. P.C., and has received grants from Keryx Biopharmaceuticals, Inc. The results presented in this study have not been published previously in whole or part, except in abstract format.
Funding Sources
The study was funded by Akebia Therapeutics, Inc. (Previously Keryx Biopharmaceuticals).
Author Contributions
P.E.P. contributed toward study design. W.L. led statistical analysis of data. P.E.P., D.B., P.C., M.M., W.L., A.G.-R., and Y.M.K.F. contributed toward data interpretation, drafting of and revising the article, and providing intellectual content of critical importance to the work described, and approved the final version of the article to be published.
Data Availability Statement
Requests for data submitted to [email protected] will be reviewed by an independent review board, with final approval by Akebia. All approved proposals are subject to a research agreement; data will be made available through third-party vendor software.
References
- 1. Stauffer ME, Fan T. Prevalence of anemia in chronic kidney disease in the United States. PLoS One. 2014;9(1):e84943.http://dx.doi.org/10.1371/journal.pone.0084943.
- 2. Kovesdy CP, Trivedi BK, Kalantar-Zadeh K, Anderson JE. Association of anemia with outcomes in men with moderate and severe chronic kidney disease. Kidney Int. 2006;69(3):560–4.http://dx.doi.org/10.1038/sj.ki.5000105.
- 3. Rao M, Pereira BJ. Optimal anemia management reduces cardiovascular morbidity, mortality, and costs in chronic kidney disease. Kidney Int. 2005;68(4):1432–8.http://dx.doi.org/10.1111/j.1523-1755.2005.00554.x.
- 4. Regidor DL, Kopple JD, Kovesdy CP, Kilpatrick RD, McAllister CJ, Aronovitz J, et al. Associations between changes in hemoglobin and administered erythropoiesis-stimulating agent and survival in hemodialysis patients. J Am Soc Nephrol. 2006;17(4):1181–91.http://dx.doi.org/10.1681/ASN.2005090997.
- 5. National Kidney Foundation. KDIGO clinical practice guideline for anemia in chronic kidney disease. Kidney Int Suppl. 2012;3(1):279–335.
- 6. Lee WC, Fang HY, Chen HC, Chen CJ, Yang CH, Hang CL, et al. Anemia: a significant cardiovascular mortality risk after ST-segment elevation myocardial infarction complicated by the comorbidities of hypertension and kidney disease. PLoS One. 2017;12(7):e0180165.http://dx.doi.org/10.1371/journal.pone.0180165.
- 7. Fishbane S, Pollack S, Feldman HI, Joffe MM. Iron indices in chronic kidney disease in the National Health and Nutritional Examination Survey 1988–2004. Clin J Am Soc Nephrol. 2009;4(1):57–61.http://dx.doi.org/10.2215/CJN.01670408.
- 8. Babitt JL, Lin HY. Mechanisms of anemia in CKD. J Am Soc Nephrol. 2012;23(10):1631–4.http://dx.doi.org/10.1681/ASN.2011111078.
- 9. Pergola PE, Fishbane S, Ganz T. Novel oral iron therapies for iron deficiency anemia in chronic kidney disease. Adv Chronic Kidney Dis. 2019;26(4):272–91.http://dx.doi.org/10.1053/j.ackd.2019.05.002.
- 10. Ganz T, Bino A, Salusky IB. Mechanism of action and clinical attributes of Auryxia® (ferric citrate). Drugs. 2019;79(9):957–68.
- 11. Block GA, Fishbane S, Rodriguez M, Smits G, Shemesh S, Pergola PE, et al. A 12-week, double-blind, placebo-controlled trial of ferric citrate for the treatment of iron deficiency anemia and reduction of serum phosphate in patients with CKD stages 3–5. Am J Kidney Dis. 2015;65(5):728–36.http://dx.doi.org/10.1053/j.ajkd.2014.10.014.
- 12. Fishbane S, Block GA, Loram L, Neylan J, Pergola PE, Uhlig K, et al. Effects of ferric citrate in patients with nondialysis-dependent CKD and iron deficiency anemia. J Am Soc Nephrol. 2017;28(6):1851–8.http://dx.doi.org/10.1681/ASN.2016101053.
- 13. Lewis JB, Sika M, Koury MJ, Chuang P, Schulman G, Smith MT, et al. Ferric citrate controls phosphorus and delivers iron in patients on dialysis. J Am Soc Nephrol. 2015;26(2):493–503.http://dx.doi.org/10.1681/ASN.2014020212.
- 14. Acaster S, Dickerhoof R, DeBusk K, Bernard K, Strauss W, Allen LF. Qualitative and quantitative validation of the FACIT-fatigue scale in iron deficiency anemia. Health Qual Life Outcomes. 2015;13:60.http://dx.doi.org/10.1186/s12955-015-0257-x.
- 15. Reilly MC, Zbrozek AS, Dukes EM. The validity and reproducibility of a work productivity and activity impairment instrument. Pharmacoeconomics. 1993;4(5):353–65.http://dx.doi.org/10.2165/00019053-199304050-00006.
- 16. Pergola P, Belo D, Crawford P, Moustafa M, Vargo D, Luo W, et al. Different ferric citrate dose regimens in the treatment of iron deficiency anaemia in patients with non-dialysis-dependent CKD: the COMPASS trial. Nephrol Dial Transplant. 2020;35(Suppl_3):gfaa139.SO050.
- 17. Pergola PE, Gartenberg G, Fu M, Wolfson M, Rao S, Bowers P. A randomized controlled study of weekly and biweekly dosing of epoetin alfa in CKD patients with anemia. Clin J Am Soc Nephrol. 2009;4(11):1731–40.http://dx.doi.org/10.2215/CJN.03470509.
- 18. Macdougall IC, Bock AH, Carrera F, Eckardt KU, Gaillard C, Van Wyck D, et al. FIND-CKD: a randomized trial of intravenous ferric carboxymaltose versus oral iron in patients with chronic kidney disease and iron deficiency anaemia. Nephrol Dial Transplant. 2014;29(11):2075–84.http://dx.doi.org/10.1093/ndt/gfu201.
- 19. Batchelor EK, Kapitsinou P, Pergola PE, Kovesdy CP, Jalal DI. Iron deficiency in chronic kidney disease: updates on pathophysiology, diagnosis, and treatment. J Am Soc Nephrol. 2020;31(3):456–68.http://dx.doi.org/10.1681/ASN.2019020213.
- 20. Pergola PE, Fishbane S, LeWinter RD, Neylan JF, Uhlig K, Block GA, et al. Hemoglobin response to ferric citrate in patients with nondialysis-dependent chronic kidney disease and iron deficiency anemia. Am J Hematol. 2018;93(6):e154–6.http://dx.doi.org/10.1002/ajh.25088.
- 21. Xie Y, Bowe B, Xian H, Balasubramanian S, Al-Aly Z. Estimated GFR trajectories of people entering CKD stage 4 and subsequent kidney disease outcomes and mortality. Am J Kidney Dis. 2016;68(2):219–28.http://dx.doi.org/10.1053/j.ajkd.2016.02.039.
- 22. Isakova T, Wahl P, Vargas GS, Gutiérrez OM, Scialla J, Xie H, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79(12):1370–8.http://dx.doi.org/10.1038/ki.2011.47.
- 23. Block GA, Pergola PE, Fishbane S, Martins JG, LeWinter RD, Uhlig K, et al. Effect of ferric citrate on serum phosphate and fibroblast growth factor 23 among patients with nondialysis-dependent chronic kidney disease: path analyses. Nephrol Dial Transplant. 2019;34(7):1115–24.http://dx.doi.org/10.1093/ndt/gfy318.
A.G.-R. and Y.M.K.F. were affiliated with Akebia Therapeutics, Inc., at the time this study was conducted.
Alexander Goldfarb-Rumyantzev is deceased.