Plain language summary available online
Author Video: https://youtu.be/8snIWOZ0CK0
What is already known about this topic?
Atopic dermatitis (AD) is the most common inflammatory skin disorder.
While strong T helper (Th)2 activation is universal in AD, an opportunity remains to specifically target different mechanisms.
The OX40–OX40 ligand (OX40L) axis is a secondary costimulatory pathway that promotes persistent immune responses in AD.
Under inflammatory conditions, OX40L is upregulated on antigen-presenting cells (APCs) following antigen presentation, contributing to the activation of antigen-specific Th2 and Th1/Th17/Th22 cells and secretion of proinflammatory cytokines.
What does this study add?
This phase IIa proof-of-concept study in adults is the first to evaluate the targeting of APCs via the OX40–OX40L axis in AD and to demonstrate that OX40L blockade with amlitelimab results in sustained and clinically meaningful improvement in the signs and symptoms of moderate-to-severe AD inadequately controlled by topical therapies.
The data support the hypothesis that targeted modulation of T-cell responses can be achieved by influencing costimulatory signalling during antigen presentation, leading to meaningful clinical improvement and the possibility of disease modification or long-term remission in AD.
Atopic dermatitis (AD) is the most common inflammatory skin disorder and leading cause of nonfatal disease burden conferred by skin conditions worldwide. Up to 50% of adult patients have moderate-to-severe disease; for most patients AD is a lifelong condition, with variable phenotypic expression and disease course., Acute AD skin lesions are dominated by T helper (Th) 2 and Th22 cytokine activation, both central mediators of disease pathogenesis, whereas chronic lesions also include contributions from other inflammatory pathways, including those mediated by Th1 and Th17 cytokines. While strong Th2 activation is universal, the contribution of other immune axes might vary across ethnicities and age groups, and novel treatments that target multiple immune axes may address unmet needs in these subpopulations.
Biologics targeting Th2 cytokines or oral Janus kinase inhibitors (JAKi) broadly affecting multiple signalling pathways have been approved for the treatment of moderate-to-severe AD. However, an opportunity remains specifically to target different mechanisms leading to the activation and perpetuation of AD inflammation., One target is the OX40–OX40 Ligand (OX40L) axis, a secondary costimulatory pathway that promotes persistent immune responses in AD, facilitating both acute and chronic disease courses. OX40L is inducibly expressed on antigen-presenting cells (APCs) such as dendritic cells, B cells and macrophages,, and interacts with OX40 expressed on activated T cells., Under inflammatory conditions, OX40L is upregulated on APCs following antigen presentation, contributing to the activation of antigen-specific Th2 and Th1/Th17/Th22 cells, and the secretion of proinflammatory cytokines.,, A 2023 study demonstrated that targeting OX40–OX40L signalling with an anti-OX40 monoclonal antibody resulted in significant reductions in Eczema Area and Severity Index (EASI) at week 16 compared with placebo, and was well tolerated, with an acceptable safety profile. These findings indicate that targeting the OX40–OX40L signalling pathway presents a viable therapeutic strategy in the treatment of moderate-to-severe AD.
Amlitelimab (SAR445229; KY1005) is a potentially first-in-class fully human nondepleting, noncytotoxic monoclonal antibody that blocks OX40–OX40L interactions with null effector function., Based on pharmacodynamic observations from a phase I study in healthy volunteers (NCT03161288), amlitelimab showed potential as a novel pharmacological treatment in immune-mediated disorders., This proof-of-concept phase IIa study aimed to explore the safety and efficacy of amlitelimab in patients with moderate-to-severe AD.
Patients and methods
Participants and study design
This phase IIa randomized double-blind placebo-controlled parallel-group multicentre study (NCT03754309) enrolled adults with moderate-to-severe AD at 19 investigational sites in Germany, Poland, Spain and the UK. Eligible patients were aged ≥ 18 to < 75 years with a history of AD for ≥ 1 year and AD involvement of ≥ 10% of body surface area (BSA) at baseline, and a documented history of inadequate response to topical treatments (or topical treatments were inadvisable) within the last 6 months. Patients had to have an EASI score of ≥ 12 at screening, and an EASI score of ≥ 16 and validated Investigator Global Assessment (vIGA) of 3/4 at baseline, consistent with moderate-to-severe AD. Patients must have had applied a stable dose of topical emollient at least twice daily for ≥ 7 consecutive days before the first dose of the investigational medicinal product (IMP). AD treatments were to be washed out ≥ 14 days prior to baseline for topical treatments, and between 3 weeks and 3 months prior for other systemic therapies. This study followed the CONSORT reporting guidelines.
Key exclusion criteria were prior prohibited treatments (Appendix S1; see Supporting Information); live (attenuated) immunization within 12 weeks; and anticipated initiation of prohibited medications or planned elective surgery < 3 months after the last dose of the IMP.
Randomization and masking
Patients were randomized at baseline to receive a low intravenous (IV) dose of amlitelimab (200 mg loading dose, 100 mg maintenance dose thereafter), a high IV dose of amlitelimab (500 mg loading dose, 250 mg thereafter) or placebo (1 : 1 : 1) at 0, 4, 8 and 12 weeks. Patients were centrally randomized, and randomization was stratified by a baseline EASI score of ≤ 21 (moderate disease) or > 21 (severe disease). Patients were allocated a randomization number using an interactive response technology system prepared by Trial Form Support (TFS) International (Lund, Sweden); patient randomization lists were generated using SAS version 9.4 (SAS Institute, Cary, NC, USA).
All patients, investigators, the sponsor, TFS, clinical laboratories and the independent data monitoring committee were masked to treatment assignment except for monitors responsible for drug storage and preparation, and drug concentration analysis (EuroFins); the randomization statistician and the unblinded statistician required for the independent data monitoring committee; and safety personnel.
Procedures
Patients were assessed for study eligibility at screening, completed within 4 weeks before randomization. AD treatments were washed out for ≥ 2 weeks prior to baseline (except bland moisturizers) and patients were required to apply bland moisturizers (emollients) with no additives (e.g. urea) at least twice daily for 7 consecutive days before baseline and throughout the study. At baseline, patients received a low (200 mg) or high (500 mg) IV dose of amlitelimab, followed by three maintenance doses at 50% of the loading dose at 4-week intervals on weeks 4, 8 and 12, or matching placebo.
Efficacy, safety, pharmacokinetic and pharmacodynamic assessments were performed at 1-week intervals from week 0 through to week 16 (main study). All evaluations were performed in the clinic, except for adverse events (AEs) and concomitant medications, which could also be recorded via telephone. All patients continued in the study extension to week 36 with assessments up to the time that they relapsed or commenced drugs that have a significant impact on AD. While patients who had responded (vIGA 0/1) at the week-16 assessment were required to continue with all assessments, patients who did not respond (vIGA 2–4) were assessed for safety only.
Outcomes
The co-primary endpoints were the incidence of treatment-emergent AEs (TEAEs) and percentage change in EASI from baseline to 16 weeks. AEs were coded using Medical Dictionary for Regulatory Activities (MedDRA) version 21.1. Secondary endpoints included the proportion of patients with ≥ 50%, ≥ 75% and ≥ 90% reduction from baseline in EASI (EASI-50, EASI-75 and EASI-90, respectively); percentage of patients with a vIGA response of 0/1 at week 16 and over time; and change in vIGA, SCORing of Atopic Dermatitis (SCORAD) Index, affected BSA and pharmacokinetic/pharmacodynamic response to amlitelimab.
The percentage of patients with an improvement in pruritus numerical rating scale (NRS) of ≥ 4 was analysed post hoc. Serum samples were collected for pharmacokinetics, and antidrug antibody (ADA) and biomarker assessments throughout the study (Appendix S1).
Patients who completed the main study continued in the study extension through to 36 weeks.
Statistical analysis
This study was not prospectively powered; the aim was to include 20 evaluable patients per treatment group. All statistical testing was two-sided and performed using a 5% significance level. No adjustment was made for multiplicity. The primary endpoint and continuous secondary endpoints (SCORAD, BSA and pruritus NRS) were assessed using a mixed model for repeated measures with percentage change from baseline as the response (dependent variable); baseline value as a covariate; and treatment, day, day × treatment and day × baseline interaction as fixed effects (independent variables). The percentage of patients with vIGA 0/1 was analysed using a Cochran–Mantel–Haenszel test, with visit as the stratum for comparison of a high (250 mg Q4W) or low dose (100 mg Q4W) of amlitelimab vs. placebo. Missing results postbaseline were assumed as nonresponders for the calculation of responder endpoints. Efficacy analyses were performed on the full analysis set (FAS), defined as patients in the safety set (who received ≥ 1 dose of the IMP) who had ≥ 1 postbaseline efficacy measurement. The primary analysis was repeated using the per protocol set (all patients in the FAS with no major protocol deviations) analysed according to the treatment received.
All statistical analyses were performed with SAS software (version 9.4 or higher for Windows; SAS Institute). The study was registered with ClinicalTrials.gov (NCT03754309) and EudraCT (registration number 2018-002299-41).
Results
Eighty-nine patients were randomized between 13 December 2018 and 12 May 2020; 88 patients [37 women (42%), 51 men (58%); mean (SD) age 33.6 (11.9) years] received the IMP (low-dose amlitelimab, n = 29; high-dose amlitelimab, n = 30; placebo, n = 29); one patient did not receive treatment due to a protocol deviation (Figure 1). Baseline disease characteristics were typical for patients with moderate-to-severe AD and generally well matched across treatment groups (Table 1). The percentage of patients with a vIGA score of 4 (severe) was higher in the placebo (62%) group than in either of the amlitelimab groups (37% and 33% in the low- and high-dose groups, respectively). No other notable differences were observed between the groups. Overall, 59 (67%) patients completed the 16-week main study: 20 (69%) in the low-dose amlitelimab group, 22 (73%) in the high-dose amlitelimab group and 17 (59%) in the placebo group. Fifty patients (57%) continued to the 36-week study extension, and 17 (59%) from the low-dose amlitelimab group, 19 (63%) from the high-dose amlitelimab group and 14 (48%) from the placebo group completed it successfully.
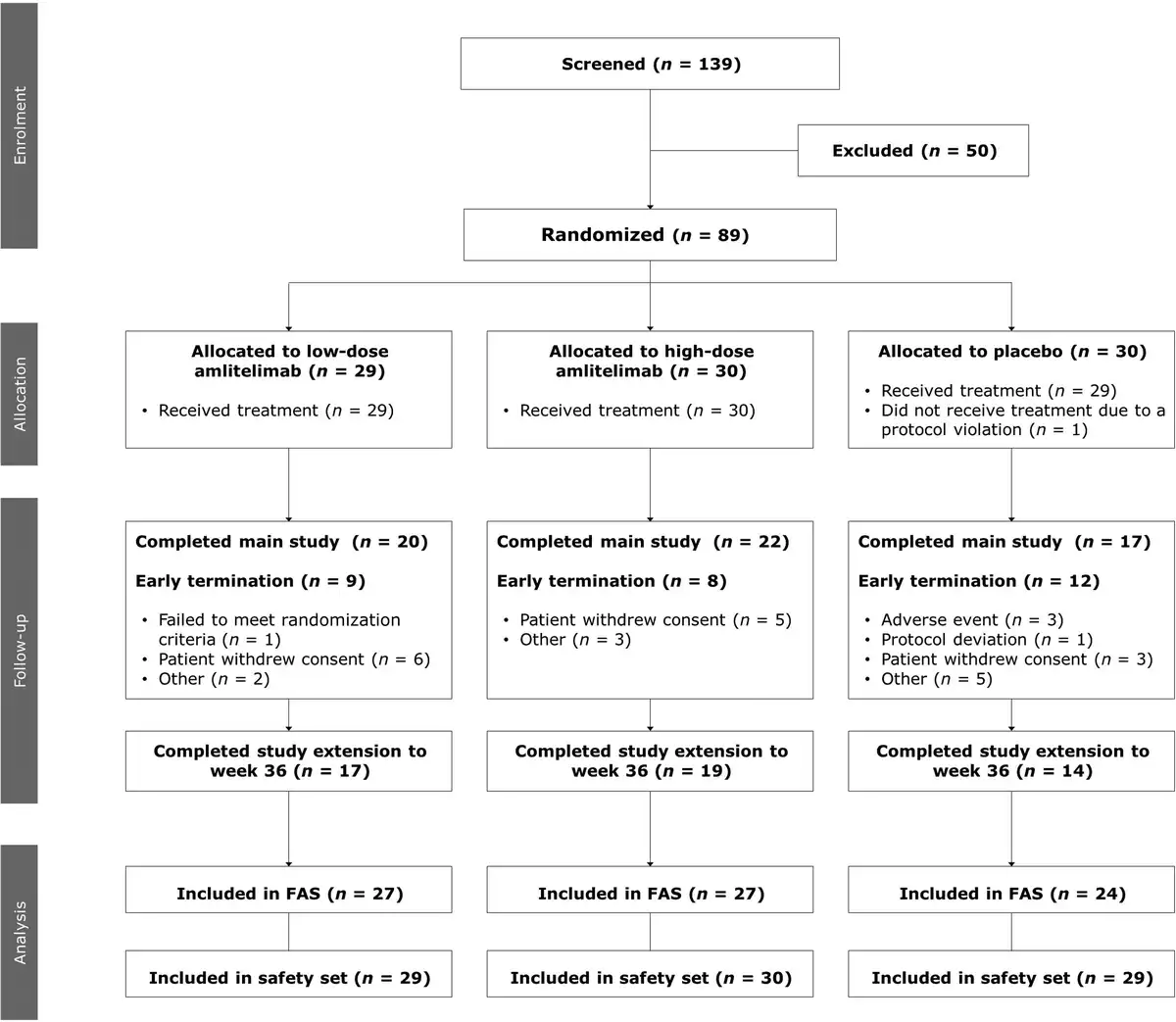
Figure 1
Patient flowchart. ‘Other’ reasons for early termination from the study pertain to issues with data from one investigation site and a failure to meet randomization criteria. FAS, full analysis set.
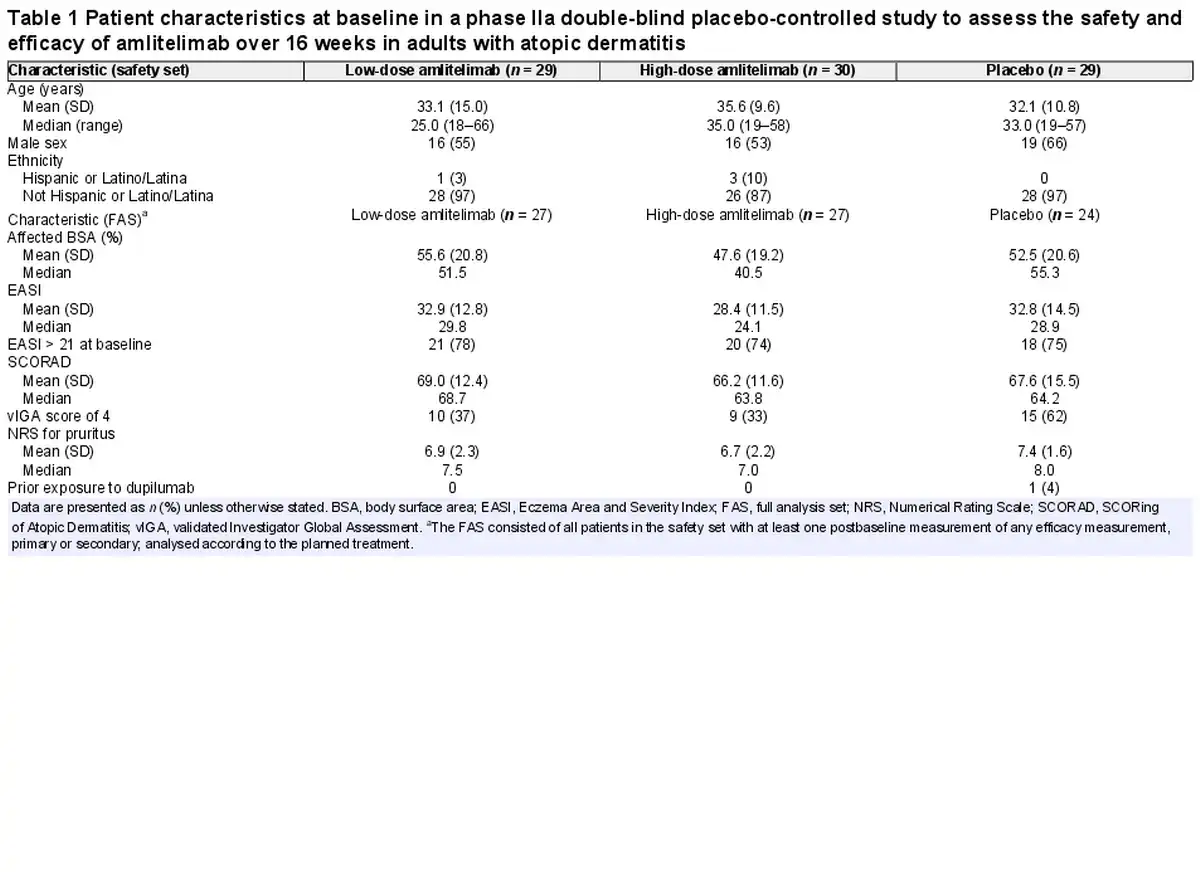
Amlitelimab was generally well tolerated, with 18 (62%) patients in the low-dose amlitelimab group, 14 (47%) in the high-dose amlitelimab group and 20 (69%) in the placebo group reporting ≥ 1 TEAE up to week 16 (Table 2). Headache, hyperhidrosis, upper respiratory tract infection, pyrexia, increased aspartate aminotransferase and iron deficiency anaemia were more prevalent in the amlitelimab groups than in the placebo group up to week 16 (difference of ≥ 5%) (Table S1; see Supporting Information). Two conjunctivitis events were reported: one case of allergic conjunctivitis in the high-dose amlitelimab group and one case of infective conjunctivitis in the low-dose amlitelimab group – both cases were mild to moderate in intensity, deemed unrelated to treatment by the investigator and both patients completed dosing. One serious TEAE of an infected dermal cyst was reported in the low-dose amlitelimab group, in a patient with a history of recurrent dermal cysts. It was managed via surgical intervention and the patient completed the study. Other severe events reported were three TEAEs of AD (two patients receiving high-dose amlitelimab and one patient receiving placebo) and one TEAE each of insomnia and neck pain (in the same patient receiving high-dose amlitelimab), none of which was considered by the investigator to be related to treatment. Four patients experienced TEAEs leading to study discontinuation: two instances of exacerbation of the underlying disease, one instance of drug ineffectiveness in the placebo group and one instance of nasopharyngitis in the low-dose amlitelimab group. No trends in clinical laboratory parameters were observed in any treatment group, and no specific safety concerns were raised.
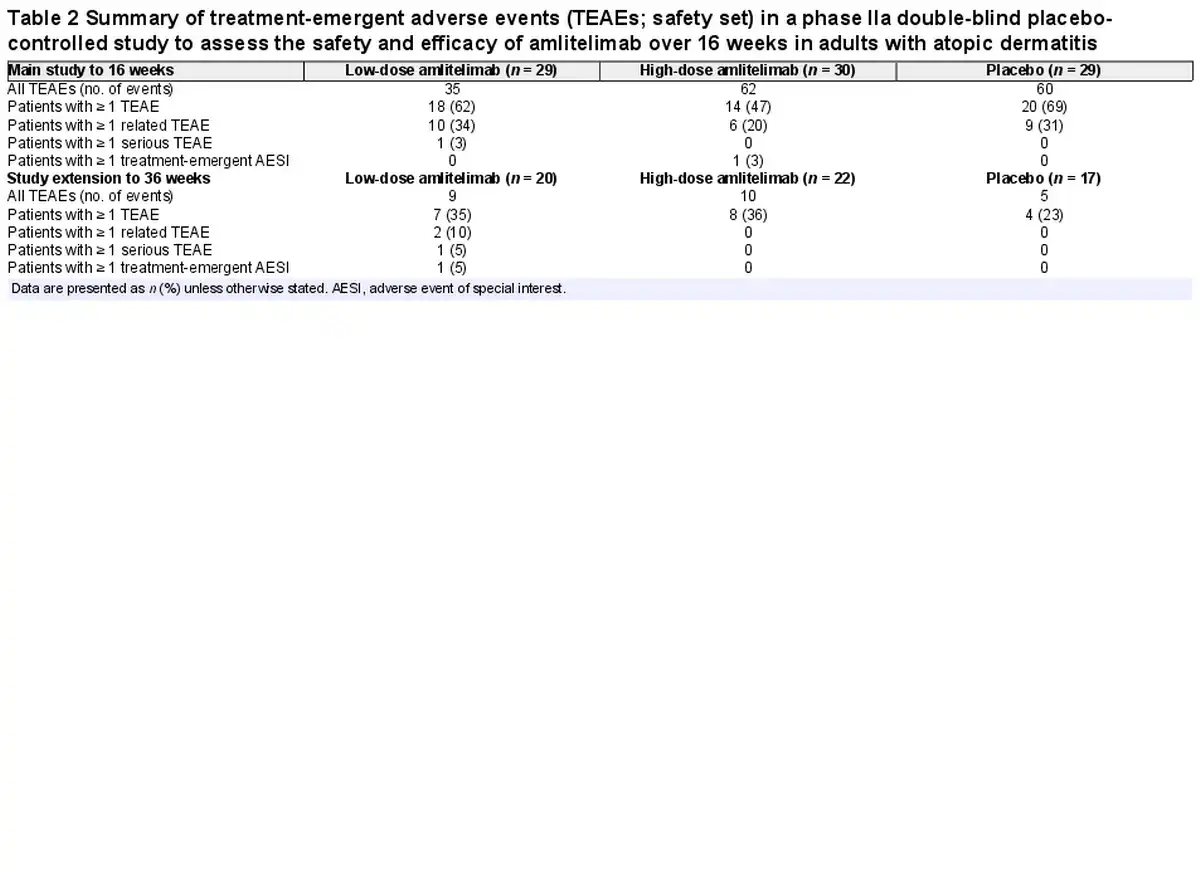
In the study extension period, one death was reported in a 44-year-old man 3 months after the last administration of a low dose of amlitelimab. The cause of death was unknown but was considered unrelated to treatment by the investigator and the independent data monitoring committee. No postmortem examination was performed due to COVID-19 restrictions at the time.
Serum concentrations of amlitelimab increased rapidly following dosing, with a median tmax of 43 min for low and high doses after the first and fourth infusions, and with an approximately dose-proportional relationship for Cmax and area under the curve (AUC)0–28. Following each dose, serum concentration steadily decreased in a biphasic manner, with similar estimated t1/2 values for low and high doses, and minimal accumulation; no target mediated disposition was observed.
At week 16, ADAs to amlitelimab were detected in 10 patients (50%) in the low-dose group, six of whom had vIGA 0/1 at week 16, and none in the high-dose amlitelimab group (Tables S2, S3; see Supporting Information). No TEAEs were associated with ADAs and no impact on amlitelimab clearance or treatment response was seen, indicating that ADAs were weakly or non-neutralizing.
In the FAS, the co-primary endpoint of the least square mean percentage change in EASI from baseline to week 16 was –80.12% [95% confidence interval (CI) –95.55 to –64.68; P = 0.009 vs. placebo] and –69.97% (95% CI –85.04 to –54.60; P = 0.07 vs. placebo) for the low- (n = 27) and high-dose (n = 27) amlitelimab groups vs. –49.37% (95% CI –66.02 to –32.72) for placebo (n = 24) (Figure 2). Mean baseline and week-16 data, along with other Harmonising Outcome Measures for Eczema outcomes, can be found in Table S4 (see Supporting Information). The primary analysis for the per-protocol set and for the sensitivity analysis provided similar results. Numerically greater reductions were observed in patients in the amlitelimab groups vs. the placebo group at all timepoints from week 2 to week 16. Progressive reductions in EASI score from baseline to week 16 were observed across both amlitelimab groups.

Figure 2
Effect of amlitelimab on Eczema Area and Severity Index (EASI). Percentage change in EASI least square (LS) mean score from baseline over time with 95% confidence interval for the full analysis set. Results presented are nominal P-values. ns, P = 0.07 (not significant) high-dose amlitelimab vs. placebo at week 16. **P = 0.009 (low-dose amlitelimab vs. placebo at week 16); ***P ≤ 0.001 (low- and high-dose amlitelimab vs. placebo at week 12).
More patients receiving amlitelimab achieved EASI-75 (59% receiving low-dose amlitelimab; 52% receiving high-dose amlitelimab) and EASI-90 (33% receiving low-dose amlitelimab; 30% receiving high-dose amlitelimab) by week 16 vs. placebo (25% and 13%, respectively). Nominally statistically significant improvements at week 16 in the amlitelimab groups vs. placebo were observed for vIGA (low- and high-dose amlitelimab P < 0.001), SCORAD (low-dose amlitelimab P = 0.01; high-dose amlitelimab P = 0.02) and affected BSA (low-dose amlitelimab P = 0.001; high-dose amlitelimab P = 0.005) (Figure 3).
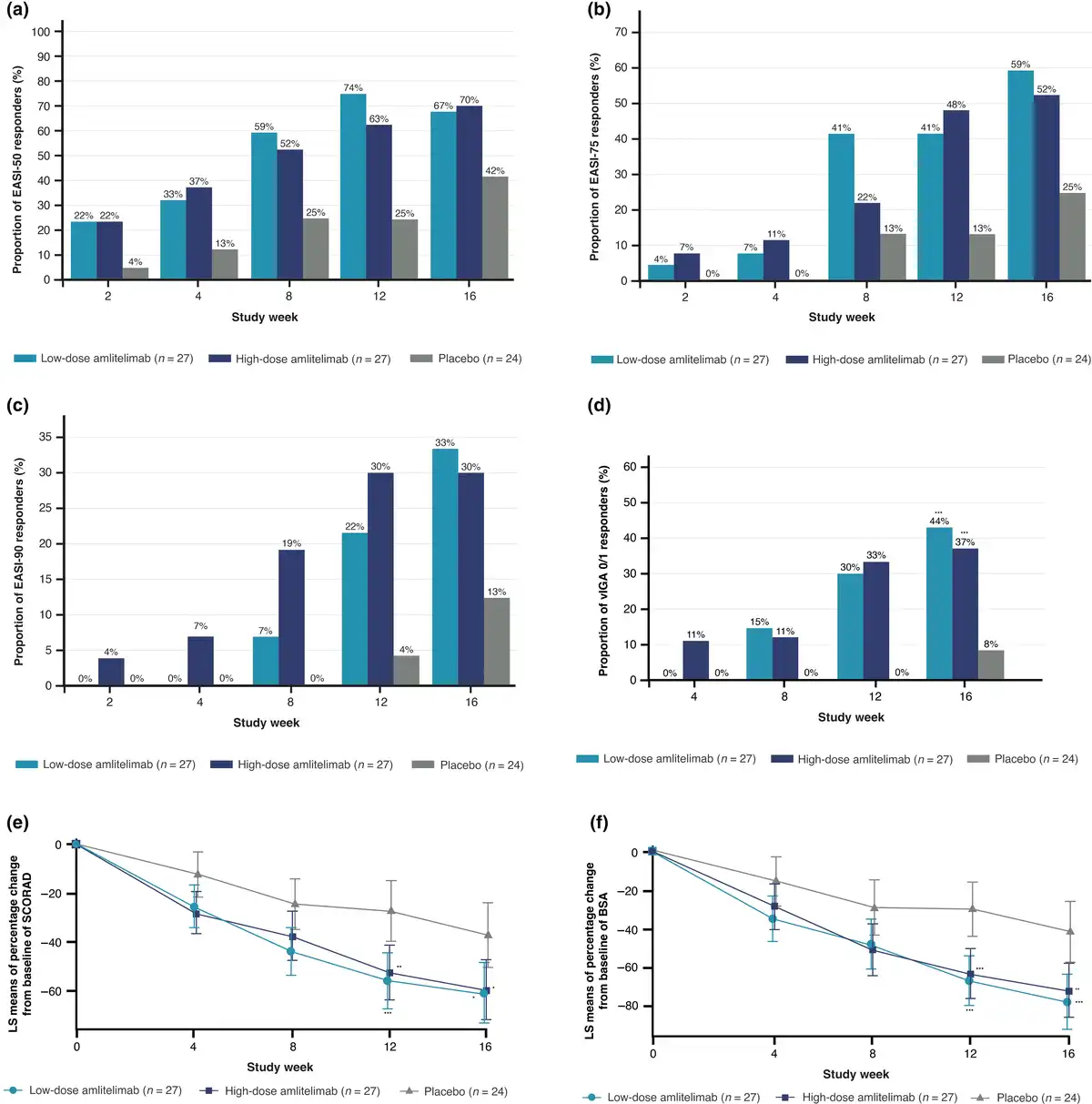
Figure 3
Effect of amlitelimab on secondary efficacy measures. Proportion of patients over time by treatment (full analysis set) achieving (a) ≥ 50% reduction from baseline in Eczema Area and Severity Index (EASI-50); (b) ≥ 75% reduction from baseline in EASI (EASI-75); (c) ≥ 90% reduction from baseline in EASI (EASI-90); (d) percentage of patients defined as having a validated Investigator Global Assessment (vIGA) score 0/1 (***P < 0.001 low-dose amlitelimab vs. placebo at week 16; ***P ≤ 0.001 high-dose amlitelimab vs. placebo at week 16); and percentage change in least square (LS) mean score from baseline with 95% confidence interval in (e) SCORing of Atopic Dermatitis (SCORAD) index (***P = 0.001 low-dose amlitelimab vs. placebo at week 12; **P = 0.004 high-dose amlitelimab vs. placebo at week 12; *P = 0.011 low-dose amlitelimab vs. placebo at week 16; *P = 0.016 high-dose amlitelimab vs. placebo at week 16); and in (f) body surface area (BSA) (***P < 0.001 low-dose amlitelimab vs. placebo at week 12; ***P = 0.001 high-dose amlitelimab vs. placebo at week 12; ***P = 0.001 low-dose amlitelimab vs. placebo at week 16; **P = 0.005 high-dose amlitelimab vs. placebo at week 16). Results presented are nominal P-values.
By week 16, the percentage of patients with clear/almost clear skin (vIGA 0/1) was 44% (n = 12/27) and 37% (n = 10/27) in the low- and high-dose amlitelimab groups vs. 8% (n = 2/24) for placebo (P < 0.001 for both).
Post hoc analysis showed that more patients achieved an improvement in pruritus NRS ≥ 4 points with amlitelimab (47% in the low-dose amlitelimab group; 38% in the high-dose amlitelimab group) vs. placebo (19%) at week 16. Baseline and week 16 mean (SD) data can be found in Table S4. Results for patients in the EASI > 21 stratum (severe disease; n = 59) were similar to those in the FAS.
Total IgE, interleukin (IL)-13, IL-31 and IL-22 were assessed as biomarkers of allergy, Th2, pruritus and Th22 biology, respectively, for AD. Nominally statistically significant reductions in serum levels of total IgE, IL-13, IL-31 and IL-22 at week 16 compared with baseline were observed in the low- (P < 0.0001 for IgE, IL-13 and IL-22; P < 0.001 for IL-31) and high-dose (P < 0.0001 for IgE; P < 0.001 for IL-13 and IL-22; P < 0.01 for IL-31) amlitelimab groups but not in the placebo group (Figure S1; see Supporting Information).
Twenty-four patients had vIGA 0/1 at week 16 (12, 10 and 2 patients in the low-dose amlitelimab, high-dose amlitelimab and placebo groups, respectively). In total, 67% in the low-dose group maintained vIGA 0/1 24 weeks after the last amlitelimab dose vs. 70% in the high-dose group and 50% in the placebo group. Amlitelimab-dependent reductions in total IgE, IL-13, IL-22 and IL-31 were also largely maintained during the study extension (Figure S1).
Discussion
This phase IIa proof-of-concept study is the first to evaluate the safety and efficacy of using an anti-OX40L antibody to target APCs via blockade of the OX40–OX40L axis in AD. In adult patients with moderate-to-severe AD inadequately controlled by topical therapies, amlitelimab – a fully human nondepleting, noncytotoxic anti-OX40L IgG4 monoclonal antibody – demonstrated no specific safety concerns and the treatment continued to be well tolerated for 24 weeks following the last injection. Notably, no hypersensitivity events were reported; ADAs to amlitelimab were only detected in 50% of patients in the low-dose amlitelimab group, while patients in the high-dose amlitelimab and placebo groups remained negative for ADA. No unexpected changes in pharmacokinetic profile or efficacy were observed in patients positive for ADAs indicating that, where present, ADAs were likely to be weak or non-neutralizing.
Treatment with amlitelimab resulted in sustained and clinically meaningful improvement in the signs and symptoms of AD. Despite the relatively limited patient population, the findings demonstrated a consistent and nominally statistically significant improvement from baseline with both high- and low-dose amlitelimab vs. placebo in secondary efficacy measures, including EASI (low-dose amlitelimab P = 0.009; high-dose amlitelimab P = 0.07), vIGA (P < 0.001 for both low- and high-dose amlitelimab), SCORAD (P = 0.011 for low-dose amlitelimab; P = 0.02 for high-dose amlitelimab) and BSA (P = 0.001 for low-dose amlitelimab; P = 0.005 for high-dose amlitelimab). While there was no discernible difference in response and change over time between dose groups, the sample size was too small to identify the most effective dose. Consequently, a phase IIb study is underway to further assess the optimal subcutaneous dose (NCT05131477).
Consistent differences in efficacy between amlitelimab and placebo were seen from 2 weeks, suggesting that targeting the APC–T-cell interface resulted in an early onset of effect, although larger studies may better differentiate when the effect significantly differs from placebo. Unlike anticytokine and JAKi therapies tested in AD, no plateauing in clinical change in response over time was apparent after treatment over 16 weeks, indicating that further improvement may be possible with continued treatment.
The difference in mean percentage change in EASI for amlitelimab vs. placebo was statistically significant in the low-dose amlitelimab group (P = 0.009). It should be noted that we observed a higher-than-expected placebo response rate of 49% improvement in EASI from baseline at week 16 vs. other studies.,, However, while there was no obvious reason for this response rate, rates for the more stringent EASI-75, EASI-90 and vIGA 0/1 endpoints were consistent with previous studies.,,,
It was hypothesized that blocking the inflammatory pathway at the APC–T-cell interface by targeting OX40L may generate a long and durable response. Indeed, clinical improvements and biomarker reductions were maintained up to 36 weeks for patients achieving vIGA 0/1 at week 16 for the exploratory endpoints of EASI, vIGA, ≥ 4-point improvement in pruritus NRS and selected biomarkers, indicating a sustained response up to 24 weeks following the last dose, while serum amlitelimab steadily decreased in most patients to concentrations below those expected to be pharmacologically active. These data suggest that in patients who achieve vIGA 0/1 following induction, there may be opportunities to explore extended dosing intervals with the goal of maintaining disease control with infrequent dosing.
OX40–OX40L pathway signalling sustains T-cell activation and the inflammatory (Th2/Th1/Th17/Th22) pathways implicated in AD and other immune-mediated diseases. By blocking OX40–OX40L interaction, amlitelimab may target antigen-specific Th2, Th22 and Th17 cytokine activation in skin lesions. In this study, amlitelimab was associated with a reduction in serum concentrations of the Th2 cytokines IL-13 and IL-31, and the Th22 cytokine IL-22, potentially providing benefits to patients with chronic-persistent AD or from groups that experience stronger Th1, Th17 and Th22 activation.,, In a primate model, OX40L blockade with amlitelimab showed significant control of CD4+ T effector cell proliferation while maintaining the regulatory/conventional T-cell ratio, indicating immune homeostasis restoration. Hence, amlitelimab may re-establish homeostasis in patients by inhibiting OX40–OX40L signalling without depleting T cells, which may result in a targeted and durable therapeutic response in immune-mediated conditions. By targeting APCs, amlitelimab could have a broad therapeutic effect and the potential to treat other immune-mediated diseases caused by immune dysregulation.
The study was limited by the relatively small sample size and limited overall patient exposure, with a total of approximately 30 patient-years exposure to amlitelimab. Of this small population, only 67% completed the main study up to week 16 and only a further 57% successfully completed the 36-week extension study. However, it should be noted that there were greater rates of study completion with amlitelimab vs. placebo in both treatment arms. In addition to limited patient diversity, a greater proportion of patients in the placebo group had a vIGA score of 4 vs. the amlitelimab groups. Finally, no neutralizing ADA assay was available and multiplicity in statistical testing was not controlled.
In conclusion, this phase IIa study provides preliminary evidence that amlitelimab, a nondepleting, noncytotoxic anti-OX40L IgG4 monoclonal antibody given as IV monotherapy once every 4 weeks, might be effective with an acceptable safety profile in patients with moderate-to-severe AD whose disease is not adequately controlled with topical therapies. These data support the hypothesis that targeted modulation of T-cell responses can be achieved by influencing costimulatory signalling during antigen presentation, leading to meaningful clinical improvement and the possibility of disease modification or long-term remission in AD. Further clinical studies of amlitelimab in AD and other immune-mediated conditions are ongoing.
Acknowledgements
The authors would like to thank Xiaodan Wei PhD for providing statistical review and support; Manisha Brahmachary PhD for quality control checks of biomarker data; and Richard Sainson for his contributions to the biomarker strategy. Medical writing and editorial assistance in preparation of this article was provided by Lisa Buttle PhD for Insight Medical Writing (a Certara Company), and Cam Hubert PhD of Fishawack Communications Ltd, part of Fishawack Health, and funded by Kymab Ltd (a Sanofi company).
References
- 1. Weidinger S, Beck LA, Bieber T et al Atopic dermatitis. Nat Rev Dis Primers2018; 4:1.
- 2. Zhang J, Loman L, Voorberg AN et al Prevalence of adult atopic dermatitis in the general population, with a focus on moderate-to-severe disease: results from the Lifelines Cohort Study. J Eur Acad Dermatol Venereol2021; 35:e787-90.
- 3. Abuabara K, Yu AM, Okhovat JP et al The prevalence of atopic dermatitis beyond childhood: a systematic review and meta-analysis of longitudinal studies. Allergy2018; 73:696–704.
- 4. Garmhausen D, Hagemann T, Bieber T et al Characterization of different courses of atopic dermatitis in adolescent and adult patients. Allergy2013; 68:498–506.
- 5. Leung DY, Guttman-Yassky E. Deciphering the complexities of atopic dermatitis: shifting paradigms in treatment approaches. J Allergy Clin Immunol2014; 134:769–79.
- 6. Brunner PM, Guttman-Yassky E. Racial differences in atopic dermatitis. Ann Allergy Asthma Immunol2019; 122:449–55.
- 7. Ariëns LFM, Bakker DS, van der Schaft J et al Dupilumab in atopic dermatitis: rationale, latest evidence and place in therapy. Ther Adv Chronic Dis2018; 9:159–70.
- 8. Duggan S. Tralokinumab: first approval. Drugs2021; 81:1657–63.
- 9. Solimani F, Meier K, Ghoreschi K. Emerging topical and systemic JAK inhibitors in dermatology. Front Immunol2019; 10:2847.
- 10. Bieber T. Atopic dermatitis: an expanding therapeutic pipeline for a complex disease. Nat Rev Drug Discov2022; 21:21–40.
- 11. Croft M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu Rev Immunol2010; 28:57–78.
- 12. Webb GJ, Hirschfield GM, Lane PJ. OX40, OX40L and autoimmunity: a comprehensive review. Clin Rev Allergy Immunol2016; 50:312–32.
- 13. Furue M, Furue M. OX40L–OX40 signaling in atopic dermatitis. J Clin Med2021; 10:2578.
- 14. Fu N, Xie F, Sun Z et al The OX40/OX40L axis regulates T follicular helper cell differentiation: implications for autoimmune diseases. Front Immunol2021; 12:670637.
- 15. Guttman-Yassky E, Simpson EL, Reich K et al An anti-OX40 antibody to treat moderate-to-severe atopic dermatitis: a multicentre, double-blind, placebo-controlled phase 2b study. Lancet2023; 401:204–14.
- 16. Lé AM, Torres T. OX40–OX40L inhibition for the treatment of atopic dermatitis-focus on rocatinlimab and amlitelimab. Pharmaceutics2022; 14:2753.
- 17. Tkachev V, Furlan SN, Watkins B et al Combined OX40L and mTOR blockade controls effector T cell activation while preserving T(reg) reconstitution after transplant. Sci Transl Med2017; 9:eaan3085.
- 18. Saghari M, Gal P, Gilbert S et al OX40L inhibition suppresses KLH-driven immune responses in healthy volunteers: a randomized controlled trial demonstrating proof-of-pharmacology for KY1005. Clin Pharmacol Ther2022; 111:1121–32.
- 19. Schulz KF, Altman DG, Moher D. CONSORT 2010 statement: updated guidelines for reporting parallel group randomized trials. Ann Intern Med2010; 152:726–32.
- 20. Renert-Yuval Y, Thyssen JP, Bissonnette R et al Biomarkers in atopic dermatitis – a review on behalf of the International Eczema Council. J Allergy Clin Immunol2021; 147:1174–90.
- 21. Guttman-Yassky E, Blauvelt A, Eichenfield LF et al Efficacy and safety of lebrikizumab, a high-affinity interleukin 13 inhibitor, in adults with moderate to severe atopic dermatitis: a phase 2b randomized clinical trial. JAMA Dermatol2020; 156:411–20.
- 22. Guttman-Yassky E, Thaçi D, Pangan AL et al Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J Allergy Clin Immunol2020; 145:877–84.
- 23. Silverberg JI, Simpson EL, Thyssen JP et al Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol2020; 156:863–73.
- 24. Simpson EL, Bieber T, Guttman-Yassky E et al Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med2016; 375:2335–48.
- 25. Wollenberg A, Howell MD, Guttman-Yassky E et al Treatment of atopic dermatitis with tralokinumab, an anti-IL-13 mAb. J Allergy Clin Immunol2019; 143:135–41.
- 26. Simpson EL, Lacour JP, Spelman L et al Baricitinib in patients with moderate-to-severe atopic dermatitis and inadequate response to topical corticosteroids: results from two randomized monotherapy phase III trials. Br J Dermatol2020; 183:242–55.
- 27. Wang YH, Liu YJ. Thymic stromal lymphopoietin, OX40-ligand, and interleukin-25 in allergic responses. Clin Exp Allergy2009; 39:798–806.
- 28. Czarnowicki T, He H, Krueger JG et al Atopic dermatitis endotypes and implications for targeted therapeutics. J Allergy Clin Immunol2019; 143:1–11.