1. Introduction
Cardiovascular diseases (CVDs) are a main cause of morbidity and mortality worldwide. It is estimated, according to World Health Organization, that almost 18 million of annual deaths are attributed to the disorders of heart and blood vessels. The risk of CVDs tends to increase in the middle- and low-income countries, due to the inadequacy of the healthcare system, absence of monitoring, and preventing measurements, all combined with an unhealthy lifestyle. CVDs include hypertension, ischaemic heart disease, coronary heart disease, cardiomyopathies, congenital heart disease, peripheral vascular disease, valvular disease, and others. Most of them involve a stage of compensation in the heart, usually observed as compensatory hypertrophy and, in later stages, heart dilation and failure., Heart failure, the final stage, is a condition of poor prognosis with its prevalence increasing by age, according to the American Heart Association. At this stage, compensatory mechanisms fail to restore cardiac function, concluding in an imbalance between blood demand, and supply in the body. A common type of valvular disease, that includes defects of the aortic valve, is referred to as aortic stenosis (AS). In AS patients, a secondary effect of pressure overload due to the narrowing of aortic valve, is the hypertrophic response of the left ventricular (LV) myocardium, which progressively becomes decompensated resulting in a failing heart. While existing treatments can alleviate the symptoms, this, in most of the cases, non-reversible situation can only be confronted with surgical procedures, which are not always a favourable option. For this reason, a better understanding of the progress of the disease, in order to facilitate earlier interventions, and new research aiming at new therapeutic strategies are crucial.
CVDs, including left ventricular hypertrophy (LVH) secondary to AS, are characterized by excessive complexity and heterogeneity, influenced by genetic, as well as environmental factors. Taking this into account, relative research should be performed according to very strict criteria reassuring that the human condition is well considered. Since the whole human heart and cardiac tissue samples have limited availability, alternatives, such as in vivo animal experimentation, in vitro or in silico studies, should be adapted. The main criteria for those alternatives include the resemblance to the human physiology and the underlying pathology of each disease, the implication of the same cellular mechanisms and molecular networks, experimental reproducibility and, especially when it comes to drug discovery and testing, predictive validity. Efforts to direct the research to in vitro or in silico experiments, still have not achieved to represent the human physiology and pathophysiology in an adequate degree. Moreover, a major percentage of drugs developed, finally fail in clinical trials due to lack of safety and efficacy. Translational research, since decades, recruits large animal models in order to investigate the disease mechanisms, develop and test new drugs. Animal testing prior to human studies is essential, leading to the need of animal model development that can provide predictability, by mimicking as much as possible the human disease aspects.
The first part of this review, aims to summarize the main events observed and that are associated with cardiomyocyte hypertrophy at different (sub)cellular levels. Extensive description of cardiomyocyte mechanisms, such as polyploidy and multinucleation, metabolism alterations and the role of mitochondria, intracellular homeostasis, cell death and sarcomeric reorganization and contractile activity, are highlighting the differences between a growing immature, healthy mature, and pathologic stage. Moreover, tissue homeostasis and the involvement of different cell types in each of the processes is well mentioned, as it is already well established that cells obey, apart from autocrine, also to paracrine signalling. The endocrine system is also cited, as it plays an important role in cardiac regulation, function and dysfunction. AS, a disease well known to trigger hypertrophic responses, is later described. Finally, the most relevant large animal models mimicking the disease are acknowledged, in accordance with the main discoveries and contributions to the field of cardiac hypertrophy (Figure 1). The animal models are discussed in a size order and not chronological order or importance.
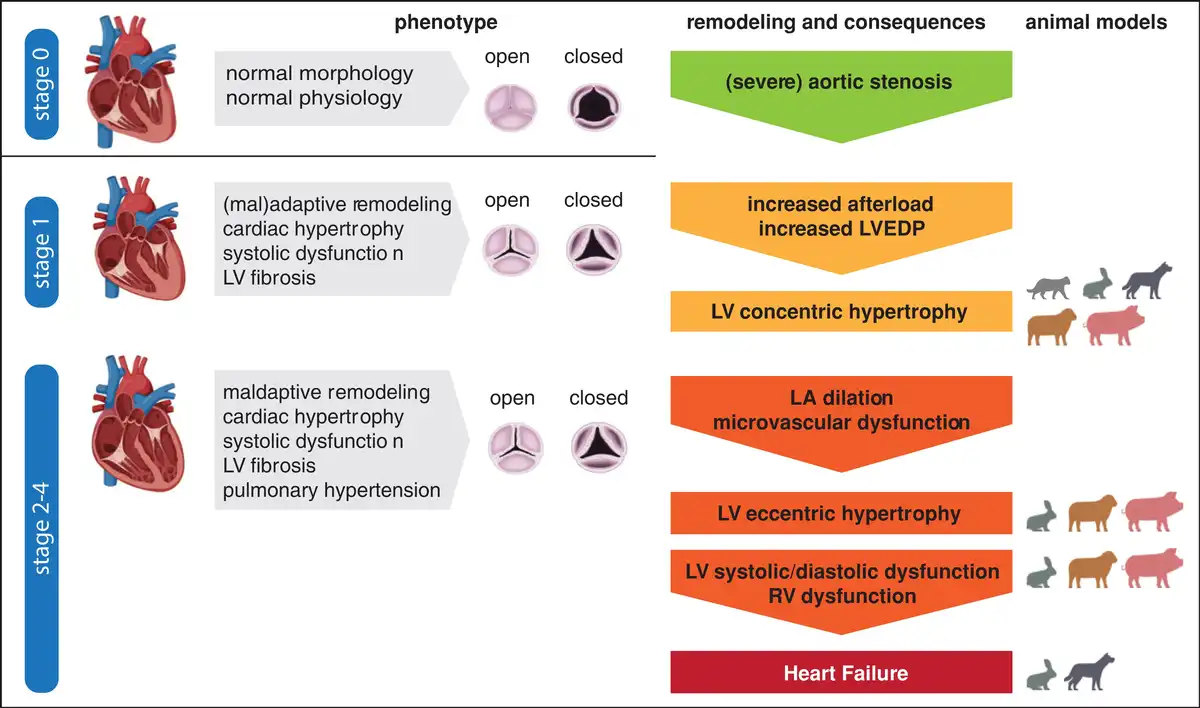
Figure 1
Cardiac pathophysiology of aortic stenosis. An illustration of the cardiac phenotypes is provided, including the respective animal models where the different stages of the disease were reached/observed as validation.
2. Pathologic hypertrophy
The heart, one of the first organs to be developed during early embryogenesis, serves a role of a pump, providing oxygenated blood in the whole body. In physiologic conditions, the heart is able to supply the body with oxygen by the circulation, while directing the non-oxygenated blood to the lungs. During physiologic growth, cardiac size increases in order to compensate the needs of the growing organism, in accordance with the workload., This is achieved by a hypertrophic cellular response, instead of hyperplastic, which is observed during embryonic and fetal development.,, The term hypertrophy refers to a cardiac cellular level, but corresponds to an increase in size and mass, also at the organ level. On the contrary, a hyperplastic reaction refers to cardiomyocyte proliferation, observed during early cardiogenesis. In case of organism development, a cellular hypertrophic reaction is considered physiological. Apart from maturation, physiological hypertrophy has been reported during pregnancy or exercise. This situation is shown to be reversible and adaptive, as a compensatory mechanism in which cardiac function is not compromised.
2.1 Molecular mechanisms of pathological hypertrophy
Upon various triggers, conditions of increased workload can occur. Genetic predisposition, pressure or volume overload or ischaemia in the cardiac muscle are some of those. Such stimuli, usually result from diseases such as, hypertrophic cardiomyopathy, hypertension, valvular heart disease (including AS, the most common valvular disease in the developed world), coronary artery disease and others, and are responsible for exposing the heart to chronic stress. The main consequence of this chronic stress is the initiation of an adaptive, with tendency to maladaptive, or, directly, maladaptive mechanism, in order to normalize the pump activity and maintain the balance between oxygen demand and supply.,, Thus, the cardiac muscle grows in mass, causing an increase in the size of the organ, mimicking a physiological hypertrophic response. During the process, wall thickening or thinning may appear, accompanied with unusual fibrosis, scar tissue formation, and immune infiltration. At this stage, a pro-inflammatory response triggers various molecular cascades, initiating a non-reversible procedure. Furthermore, fibrosis and extracellular matrix (ECM) deposition cause systolic and diastolic dysfunction, impeding electric conduction. This transition to a pathologic, maladaptive condition is characterized by compromised heart function.
2.2 Types of pathological hypertrophy
Two main types of pathological hypertrophy have been observed and are correlated to the stimuli that causes them. In the first case, an eccentric response appears when increased blood volume enters the heart ventricle. Volume overload triggers cardiomyocytes to stretch longitudinally. This results in an elongation of the cardiac muscle. Wall thinning and dilation characterizes those hearts, in which the contractility and overall function progressively attenuates. On the other side, even though it is known that the heart has the ability to calibrate small increases in the ventricular pressure by stretching of the cardiac muscle and higher force of contraction, according to Frank Starling law, when constant pressure overload occurs, due to chronic hypertension, valvular disease and other pathologic conditions, different cardiomyocyte molecular mechanisms are activated. This results in increased cellular mass, at a cross-sectional point of view, which is responsible for an increase in heart mass, observed as extended wall thickening, representing an effort to produce more tension. Wall thickening, often in parallel to a decrease in ventricular chamber volume, is accompanied with a wall stiffening, decreasing the elasticity of the muscle, and the functionality of the organ. This is the case of a concentric hypertrophic response. In summary, small cumulative cellular reactions and alterations are causing a changing effect in the organ architecture, in a concentric or eccentric way, which will finally result in cardiac dysfunction.,, Other triggers, such as inherited diseases or myocardial infarction may also cause local or general hypertrophic responses, but without leading specifically to one of those two classifications.
2.3 Dynamic cell population state under pathologic conditions
A normal cardiac ventricular muscle consists of many cell types, such as mural cells, including pericytes and vascular smooth muscle cells, fibroblasts, endothelial cells (ECs), immune cells, surrounding and assisting the cardiomyocytes, the essential cells for cardiac contractile function., Due to the pathology of a maladaptive hypertrophic heart, this analogy changes, with differentiated percentages of cell types, having an effect on the features of hypertrophic response. The cellular composition turns into a more fibrotic and inflammatory content, surrounded by altered ECM. The percentage of cardiomyocytes tends to decrease, due to increased non-cardiomyocyte migration, as well as their number, due to cell death. Cell proliferation from cardiac progenitors does not have an important effect in the adult heart, as their percentage is very low and the whole organ renewal capacity attenuated. Mechanical triggers not only initiate intracellular alterations in cardiomyocytes but they also stimulate cardiomyocytes to start a neurohumoral reaction, attracting multiple cell types, with a protective intention. However, chronic stimulation and subsequent drastic changes in cardiomyocytes and cellular processes such as inflammation, fibrosis, and angiogenesis will ultimately lead to a dilated and failing heart.
3. Cardiomyocyte events
3.1 Ploidy and multinucleation
Cardiomyocytes are the main regulators of a hypertrophic response. They suffer alterations in their structure and architecture at multiple levels, which, will cause humoral reactions with, apart from autocrine, also paracrine and endocrine-stimulating properties. Initially, polyploidy and multinucleation are characteristics of adult cardiomyocytes, not common in most of the other cell types. Human adult cardiomyocytes, which are known for their low proliferative capacity, appear to be mononuclear but polyploid., This happens in a postnatal period, during maturation, as in the fetal period, normal cell cycle and hyperplasia characterizes the cardiac cells. During this postnatal period, normal heart stiffening is observed with alterations in ECM, accompanied by cardiomyocyte maturation and cell cycle exit. Upon a hypertrophic stimulus, in disease conditions, ploidy seems to increase, as a mechanism to facilitate additional protein transcription due to high metabolic demands. Ploidy in hypertrophic cardiomyocytes, and especially right ventricular cardiomyocytes, was positively correlated with cellular hypertrophic response. Hypertrophy does not seem to have an effect in multinucleation in human, in contrast to observations in animal models, which appear to have binucleated or multinucleated cardiomyocytes. This may be a limitation in the translation of animal experimentation to humans.
3.2 Metabolic modifications and mitochondria
Energy metabolism of cardiomyocytes differs from most of other cell types due to their increased demand of energy, required for contractility. Fetal cardiomyocytes are known to mainly use glucose, as their main substrate for energy production, which switches after birth to fatty acid β-oxidation. During hypertrophy, cardiomyocyte metabolic demands increase, as stated above, corresponding to an excessive need for anabolic products, like nucleic acids, proteins and lipids, as well as energy. Hypertrophied cardiomyocytes are subjected to a metabolic switch, similar to the embryonic condition, from fatty acids to glucose.,, Interestingly, it was suggested that this switch exists independently of the energy demand, probably correlating to biosynthetic/anabolic demands of the cell. Furthermore, fructolysis, fructose metabolism, provides has been implicated in the pathology of hypertrophy., Mitochondria, the responsible organelles for energy production, undergo changes during the transition to pathologic hypertrophy and energy metabolism switch. Normally, they are characterized by high density, occupying in approximately 30% of the cardiomyocyte mass and located in the perinuclear area. During hypertrophy, they are subjected to morphological and functional alterations, resulting in an impaired function. Their dimensions increase, in contrary to their volume density, while their morphology transforms from ovate to elongated-filamentous, with ruptured membranes and structures., Quality control mechanisms such as mitochondrial biogenesis, mitophagy, fusion and fission appear to be imbalanced, while mitochondrial Ca2+ uptake, important for ATP production and cardiac contractility, is abnormal. Finally, increased release of Reactive Oxygen Species (ROS) from the damaged mitochondria, exceeding the antioxidant capacity of the cell, leads to oxidative stress, which may further stimulate apoptotic cascades and maladaptive extracellular remodelling.,
3.3 Subcellular homeostasis
Excessive ROS generation in cells can trigger mechanisms like autophagy. Autophagy is considered to be the housekeeping mechanism of the cell, recycling damaged macromolecules and organelles inside it, or occasionally driving the cell to death. This homeostatic mechanism can provide biosynthetic components to the cell and has been found to play a controversial role in cardiac hypertrophy. Initially, autophagy, triggered by metabolic demands, provides a compensatory mechanism to haemodynamic stressors facilitating the heart function and morphology alterations. During the passage to the pathologic state of a failing heart, autophagy has been found to be importantly overactivated in human, fact that may be associated with increased mitochondrial dysfunction and energy crisis, worsening progressively the prognosis of the disease., In contrast, some studies in mice strongly support that autophagy induction has a reversing effect in hypertroph, while autophagy deficiency is associated with hypertrophic reactions and dysfunction of mitochondria, calcium handling, and contractility mechanisms. This may be attributed to an early stage of the disease or organism-specific implicated mechanisms. Proteostasis, the mechanism responsible for protein balance inside the cells, plays an important role in cardiac pathology. A dysfunctional ubiquitin–proteasome system, the system that subcellularly invigilates the protein function, is suspected to be a hallmark of cardiomyopathies, especially during stress conditions, nevertheless, further investigation on the topic is required.,
3.4 Oxidative stress
It has been demonstrated that different cardiovascular abnormalities such as functional hypoxia, metabolic derangements, uncoupling of mitochondrial electron transport, and inflammation produce oxidative stress in the hypertrophied failing heart. A small amount of oxyradicals is generated at the onset of the hypertrophic process but these radicals are readily removed by the endogenous scavengers to maintain cardiac redox homeostasis and proper function. However, prolonged exposure of hypertrophied hearts to pathological stimuli promotes the development of oxidative stress as a consequence of functional hypoxia due to constriction of the coronary arteries, reduction in the capillary density, inflammation, metabolic alterations, and mitochondrial dysfunction. Oxidative stress has been suggested to cause Ca2+-handling abnormalities in association with subcellular remodelling, defect in energy production, inflammation, apoptosis, fibrosis, and loss of cardiomyocytes. These abnormalities are considered to result in cardiac dysfunction and heart failure (reviewed in). Regarding AS, oxidative stress is mainly associated with calcific AS by correlating with the degree of inflammation and calcification of human aortic valves. In fact, ROS mediate the increase in BMP2 expression and signalling, favouring osteogenesis (reviewed in).
3.5 Cell homeostasis and death
Cell death is another reparative process, removing damaged or dysfunctional cells, usually triggered by mechanical forces, oxidative stress, hypoxia, and others, common characteristics of a hypertrophic response. It is known that cardiac homeostasis is preserved with the balance of CM regeneration and cell death, including apoptosis, necrosis, and autophagy associated with the ubiquitin-proteasomal pathway. Necrosis, a non-regulated and non-physiological form of cell death, does not seem to play an important role in hypertrophic hearts. On the other side, apoptosis has a controversial role in the progress of a hypertrophic response. In the initial stages, hypertrophy promotes cell survival and evasion of apoptotic mechanisms. Large, aged cardiomyocytes are more susceptible to apoptosis, especially when they suffer of mitochondrial dysfunction, as a result of chronic growth signalling exposure. They are progressively led to atrophy and death, facilitating muscular remodelling., At late stages of the disease, cellular apoptosis increases due to the increasing number of damaged cells. Organ failure is then irreversible, with the causality relationship of failure and apoptosis being still unclear. Finally, in a study in AS patients, autophagic cell death appeared to be more significant than the programmed cell death via apoptosis. Further investigation of the homeostatic mechanisms of the heart could elucidate important time points indicative of the transition from compensatory to pathologic condition and failure.
3.6 Contractile activity and sarcomeric reorganization
Mechanotransduction is a machinery enabling cardiomyocytes to sense and respond to mechanical load. The main sensors of this system are the three cellular compartments: the sarcomere, the intercalated discs and the sarcolemma. During increased chronic workload, those sensors are triggered to promote changes that will facilitate a hypertrophic response. Sarcomeres are structures responsible for force generation in cardiomyocytes, that by actin and myosin filament interactions promote contraction reactions inside cardiomyocytes., In case of a workload stressor, a cellular response is initiated leading to an increase in the amount of connected sarcomeres. This increase can happen longitudinally or in parallel, resulting in longer or wider cardiomyocytes, respectively. Longer cardiomyocytes are observed in hearts with eccentric hypertrophy, while cardiomyocytes with larger cross-sectional area (CSA) are characteristic of a concentric response. Intercalated discs, the cardiac specific junctions, are responsible for intercellular communication related to mechanical and electrical coupling. Those terminal structures consist of adherence junctions, where the myofibrils anchor, desmosomes, for mechanical stabilization and gap junctions, for ion transfer. It is shown that ultrastructural changes affect the assembly of sarcomeres, while, in hypertrophic conditions, intercalated discs appear to have a denser architecture, fact that requires further research., Finally, the sarcolemma consists of many proteins and complexes, associated with mechanotransduction, and in hypertrophic responses appears to be altered indicated by an irregular cell surface, t-tubule dilation, and enhanced Ca2+ efflux, through stretch-sensitive ion channels.,,,
Despite the various pathological stimuli, there are many common features in the hypertrophic response in different cardiac diseases. In addition to increased cardiomyocyte mass, sarcomere rearrangement, and ECM deposition, other common features have recently been appreciated including inflammatory signalling, immune cell activation, and decreased angiogenesis. Numerous cell types are involved in orchestrating this complex pathological response. The heart consists of a heterogeneous population of cells including cardiomyocytes and non-cardiomyocytes, and it is now clear that intercellular signalling and communication between these cell types is critical in the pathophysiology of ventricular hypertrophy and remodelling. Although we will not elaborate on the different intercellular communication pathways contributing to the cardiac response to stress, here below we discuss the main different cells and processes that occur during the development of the disease and that involve other cell types rather than CMs alone.
4. Non-cardiomyocyte events
4.1 Inflammation
Upon a hypertrophic stimulus, a strong inflammatory response is also observed, which is triggered in the first stages of pathologic hypertrophy. Due to haemodynamic overload and subsequent mechanical stress, mostly cardiomyocytes, but other cardiac cell types also, release autocrine and paracrine factors such as hormones, cytokines, growth factors (GFs), and chemokines. These factors, that may be meant to contribute to the adaptation to stress, when sustained, become maladaptive mediators contributing to decompensation to heart failure (reviewed in,).
Initially, the Sympathetic Nervous System (SNS) is activated, as a compensatory mechanism to the hypertrophic trigger, followed by activation of Renin-Angiotensin-Aldosterone-System. Neurohumoral systems are found to interact with the immune system and trigger fibrosis, while chronic activation of those systems is correlated with the maladaptive responses and characteristics of the failing heart., Inflammatory cytokines target many cell types in the hypertrophic heart, such as cardiomyocytes, immune cells or fibroblasts, in a complex process that remain poorly studied. However, they positively correlate to the disease progress and severity. Some studies show evidence that cardiac inflammation is directly initiated by the cardiomyocyte as being a potent source of inflammatory cytokines. Early after transverse aortic constriction (TAC), cardiomyocytes are known to release cytokines such as monocyte chemoattractant protein 1, chemokine (C-X-C-motif) ligand1, interleukin-18, -6, and -32 (IL-18, IL-6, and IL-32), which may all contribute to the pro-inflammatory environment of the stressed heart., Other researchers reported cytokine immunoreactivity also in the vasculature in a TAC model, implicating ECs in the initiation of immune cell infiltration in the heart., Additionally, the natriuretic peptide system appears to affect the inflammatory modulators in a double and contradicting way, having an impact in the following cardiac events. It includes atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP) and C-type natriuretic peptide, as major molecular regulators, each of which acts through different mechanisms during remodelling processes in the failing heart.
4.2 Immune activation and infiltration
It is known that cardiac tissue hosts an important amount of resident immune cells. Macrophages, B cells, T cells, Dendritic Cells (DCs), Natural Killer cells (NK), neutrophils, and mast cells are the immune cellular subpopulations, present in the healthy, as well as the diseased heart. During a hypertrophic reaction, they become active, serving different aspects of a strong inflammatory response and attracting additional immune subpopulations with significant roles in the progress of the disease. In pressure overload induced hypertrophy, they infiltrate the heart in a time-dependent manner, determining the cardioprotective, or cardiodestructive effects. Neutrophils are the first responders of the innate immune system during inflammation. In hypertrophic animal models of pressure overload, their population remained increased during the whole study period, and it was positively correlated to the mass index. They have a pivotal role in recruiting macrophages and DCs, by the secretion of immunomodulating factors. Mast cells are responsible for vascular homeostasis and angiogenesis in heart, but also play a modulator’s role during hypertrophy. With the process of degranulation, they release inflammation mediators and their accumulation has been positively correlated with adverse remodelling of pressure overload models. DCs are antigen-presenting cells, serving as a bridge between innate and adaptive immune system. Evidence show that they play a pathogenic role. Although they are found to increase during the progress human heart failure, no statistical significance was proven. Those subcellular populations are proposed to have detrimental role in cardiac hypertrophy due to pressure overload, while others appear to have time-dependent and controversial roles.
Macrophages and their different subpopulations play key roles in the progress of the inflammatory reaction, upon hypertrophic stimulation. Cardiac-resident macrophages have cardiac pro-repair and angiogenesis regulatory capacity, while a smaller subpopulation of them is capable of activating T cells by antigen presentation, serving as immunomodulators., Furthermore, blood-circulating monocytes are reported to reach the heart, during health and in higher degree in disease conditions, and differentiate in macrophages with a pro-inflammatory character., The recruitment of non-resident macrophages [specifically the C-C chemokine receptor 2+ (CCR2+) population] in the heart is known to have a pivotal role in the adverse cardiac remodelling induced by pressure overload., NK cells are known by their cytolytic and tissue homeostatic function, as well as their immunomodulatory properties of macrophage and T cell activation, in the cardiac tissue. Decrease in number or dysfunction of NK cells was positively correlated with chronic heart failure in human. The adaptive immune system also participates in the inflammatory reaction of a pathologically hypertrophic heart. T cells have the ability to activate or suppress inflammation, according to their subtype, and are known to participate in the pathophysiology of a diseased heart. Activated T cells migrate from lymphoid organs to the heart, where they expand in response to pressure overload. B cells, on the other side, are well known for antibody production. In the diseased heart, they release pro-inflammatory molecules, by which LV function is attenuated, and activate the complement, acting by damaging the cardiomyocytes., Finally, eosinophils, able to control cardiac death and fibroblast activity, as well as a specific subset of T cells, NK T cells, provide evidence of cardioprotective actions.,
4.3 Fibrosis
Cardiac fibrosis, where activated myofibroblasts are the main effector cells, is primarily driven through activation of resident interstitial cell populations. Although, considered a protective mechanism upon tissue or ECM damage occur, aiming at wound healing and repair, complete restoring of the initial tissue functionality is seldomly achieved. Several other cell types, including cardiomyocytes, ECs, pericytes, macrophages, lymphocytes, and mast cells may also contribute to the formation of fibrotic lesions by secreting fibrogenic mediators and matricellular proteins, or just by modifying specific communication axes that will influence the fibroblast’s phenotype. It was suggested that the first stage of a hypertrophic response is accompanied by loss of cell–cell interactions and protective factors secreted by fibroblasts. The mechanisms of induction of fibrosis and the contribution of the various cell types to the process are dependent on the underlying cause of myocardial injury. Activation of neurohumoral pathways stimulates fibroblasts both directly and by affecting certain immune cell populations. Immune cell infiltration, a strong inflammatory response, and increased oxidative stress due to ROS accumulation, are major regulators of the fibrotic response that characterizes the maladaptive hypertrophic heart. During chronic hypertrophic stimuli, the small number of cardiac-resident fibroblasts become activated and acquire the ability to proliferate and differentiate into myofibroblasts. Myofibroblasts, apart from the ability to produce excessive amounts of collagen and other ECM elements, are recognized as cellular types with both fibrotic and contractile properties, playing an important contribution to muscle repair.,, Furthermore, they present high resistance to cell death, as a survival mechanism in the abundant cell death signals microenvironment of a wound, fact that may have negative consequences, as persistent cardiac fibrosis can be detrimental. The extensive fibrosis and ECM deposition are main factors that directly stiffen and impair contractility of the cardiac muscle, both characteristics of the failing heart. Attenuation or inhibition of the fibrotic response, through pharmacological (personalized) interventions, is believed to have a positive effect in heart failure.,,
4.4 Angiogenesis and hypoxia
Vasculature density and angiogenesis in the heart have been positively correlated with physiological hypertrophy that is observed in a growing organism towards adulthood or in cases of athletic or pregnancy hypertrophy, attributed to the increased CM size and consequent oxygen and metabolic product demands. In healthy conditions, CMs have the ability to secrete angiogenic factors and promote angiogenesis, through an important crosstalk with ECs, the most abundant cell type in the cardiac muscle., Although it is also proven that angiogenesis alone is able to stimulate hypertrophic responses in cardiomyocytes, this review mainly focuses on cases of an already existing hypertrophic stimulus, apart from angiogenesis. Miscommunication and disproportional CM growth, compared to the capillary net/vasculature bed leads to hypoxic conditions within the cardiac muscle, contributing to the transition into a pathologic hypertrophied heart., During this stage, capillary rarefaction and endothelial dysfunction cause underperfusion of the heart, leading, apart from hypoxia, to fibrosis, CM death, and triggering of inflammation. Moreover, hypoxia is known to contribute to the metabolic switch in CMs and facilitate further pathological alterations. Tube-like structure formation as well as EC number appears to decrease with the progress of heart failure. The latter may be attributed to molecular signals that either inhibit endothelial proliferation, promote cell death or the Endothelial to Mesenchymal transition., Recently, our own study revealed a new process by which, stressed cardiomyocytes can influence the surrounding endothelium through release of exosomes that are enriched in miR-200c-3p. Once stressed, cardiomyocyte-derived exosomes are taken up by cardiac ECs, these will lose their angiogenic capacity and become dysfunctional contributing to the severity of the cardiac phenotype caused by prolonged increased pressure overload of the LV. All these, indicate that restoring the angiogenetic ability of the failing heart may have a positive impact in delaying or even preventing the progress of the disease.
5. Aortic stenosis
AS is among the most common valvular diseases of the heart and is described as an obstructive disorder of blood flow, during systole, from the LV towards the ascending aorta, due to the narrowing of the aortic valve. This narrowing is usually caused by idiopathic degenerative sclerosis accompanied by increasing calcium deposition in the valve and which, progresses over time; congenital bicuspid valve or rheumatic fever. An important consequence of the disease is the progressive pressure overload that triggers ‘compensatory’ mechanisms in the LV myocardium, resulting in increased LV mass, without the respective, and expected cavity enlargement. This LV wall thickening translates to a concentric hypertrophic response, in which cardiac function is compensated, while Ejection Fraction (EF) is preserved. EF, an haemodynamic parameter, represents the percentage of the blood that is ejected from the LV during systole and, when preserved to normal levels, is indicative of good systolic cardiac function, low EF, in turn, indicates poor efficiency and function of the organ. Moreover, in cases of diastolic dysfunction with other comorbidities implicated, EF can remain relatively high and assessment of other parameters is need for diagnosis. If left untreated, chronic pressure overload in the hypertrophied heart results in progressive impairment of cardiac function involving systolic dysfunction, LV wall thinning and dilation and enlargement of the LV cavity, recognized as an eccentric hypertrophic reaction and characterized by reduced EF.
Diagnosis of AS is confirmed by echocardiography, aiming at quantifying the degree of stenosis by jet velocity, transvalvular systolic pressure gradient, and aortic valve area. AS can be classified in mild, moderate, severe, and very severe, considering the valve anatomy, the severity of valve dysfunction, and the respective response of the ventricle and pulmonary circulation. Clinical symptoms of the disease may include syncope, angina, and exertional dyspnea and often heart failure and arrhythmias. AS without symptoms usually requires no treatment, unless accompanied by LV dysfunction or conditions that require cardiac surgery. Severe symptomatic AS can be treated either by a transcatheter repair device or by surgical valve replacement, based primarily on symptoms or reduced ventricular systolic function. Upon onset of the disease, progression velocity is associated with the severity and the only available intervention, as an effort to prevent this progress, is symptomatic treatment. However, by the time that severe symptoms that require a surgical intervention develop, the systolic dysfunction of a dilated or failing heart is usually irreversible. The increasing number of patients with AS and the deficiency of early treatment options, emerges the investigation of novel possible therapeutic approaches that may involve the prevention or reverse of the transition to heart failure and the restoration of cardiac function.
Due to the complexity of the disease, and the many different cellular components involved towards the development of a pathological cardiac phenotype, it is important to develop and characterize animal models that best represent the human condition regarding the morphological and physiological changes that in the stressed heart. Not only different approaches to cause chronic pressure overload to the heart have been and are being developed, also different animals have been used to study different aspects of the disease. In the next section of this review, we focus on the different animal species that have been experimentally tested in the context of LVH, the experimental outcomes, their advantages and disadvantages as models of the human condition.
6. Animal models of LVH, induced by pressure overload
Animal experimentation has been practiced since ancient years and was used as one of the first tools to explore the field of anatomy of living organisms. Possible therapies and treatment testing in animals has also been performed through the history of medicine. However, published scientific articles are only available since the last decades, describing more advanced models and procedures. Lately, new advances and technologies, as well as new ethical restrictions, have significantly and progressively altered the perception of animal use, and new principles have been applied according to the 3Rs. The 3Rs refer to the Reduce of animals in the research field, the Refinement of the experiments to eliminate animal suffering and the Replacement of animals when feasible. Despite the rapid progress in science, complete replacement of animals cannot be achieved with the current knowledge and technology, as there is lack of adequately representative in vitro models, and the use of human samples is very restricted. Within the upcoming years, progress in the in vitro and computational models, as well as developing minimally invasive techniques to acquire human samples, are expected to substitute a great percentage of animal models. As for now, animal experimentation still plays a pivotal role in many aspects of the medical field. The most common animal models used for heart research are mice and rats, which, while accommodating many scientific purposes, yet, due to size limitations and differences with human in physiology parameters and life span, often fail to provide accurately results representative to human conditions. Rodents provide an ‘easy’ to handle, low-cost tool that may elucidate cellular and molecular mechanisms, but fail in mimicking the human architecture and heart rate, O2 consumption, contractility, protein expression, and cellular microenvironment. This limitation leads to the need of larger animal models that can surmount the important issue of organism representativeness.
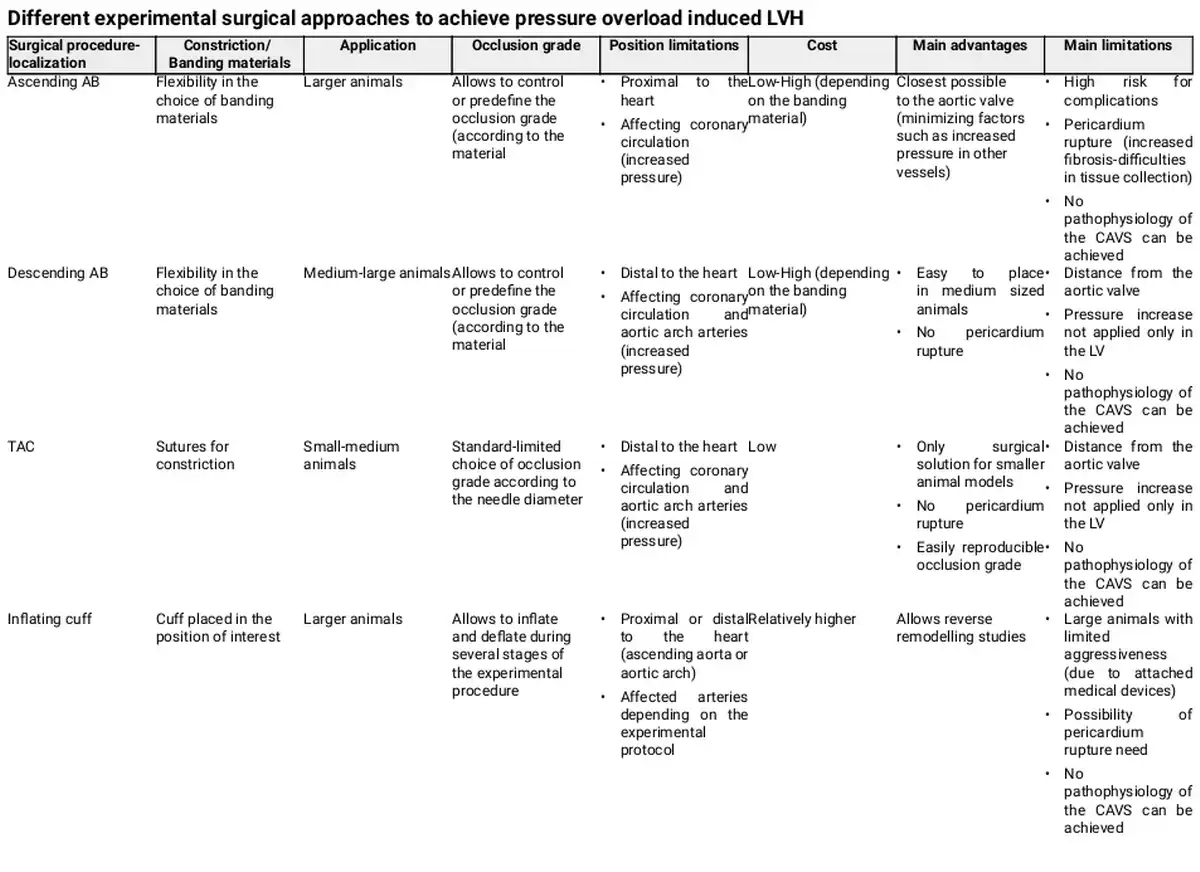
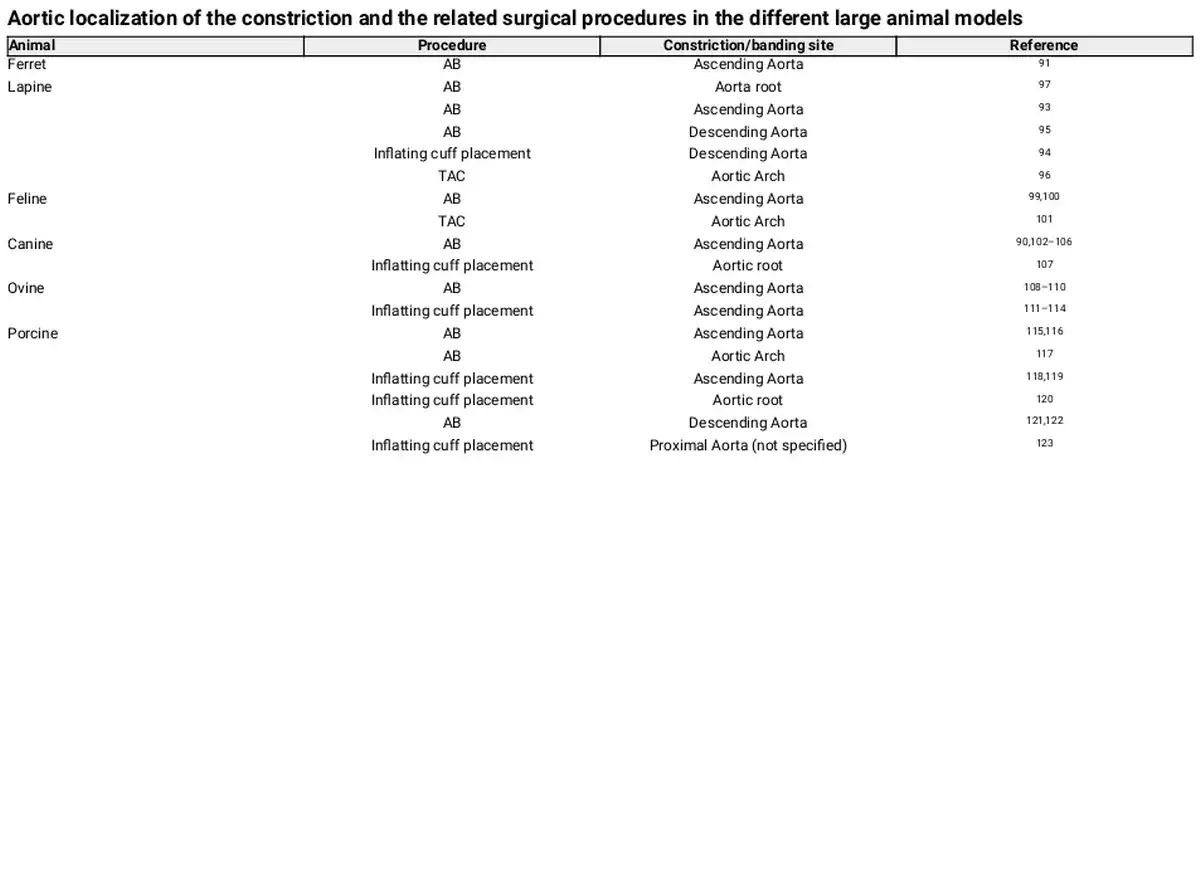
In the field of cardiology, many, surgical or not, large animal models are or have been used to elucidate physiological but also disease mechanisms, drug effects, and toxicity. Among commonly used animal models are the ferrets, dogs, cats, rabbits, sheep, pigs, horses, cows, and others. Due to differences in physiology, anatomy, cellular, or molecular mechanisms, for each experiment, the animal model has to be carefully selected to avoid as much potential confounders as possible. Furthermore, essential for the selection of the suitable animal and model is the principal scientific question behind the study. Especially for LVH, caused in human by AS, a surgical model that involves the constriction of aorta in order to cause an increase in blood pressure inside the LV, is found to be the most suitable method to investigate the physiological and pathological hypertrophic mechanisms. This model, aims, by mimicking the AS-induced cardiac phenotype, at triggering mechanisms that are responsible for the consequent effect of cardiomyocyte hypertrophy. The constriction or ‘banding’ of the aorta can be performed in the ascending or descending aorta or in some cases in the aortic arch, resulting in a local partial occlusion and a controlled decrease of its inside diameter. Table 1 describes the most commonly used surgical approaches to achieve pressure overload in the LV, mimicking human AS, and specifies their important advantages and disadvantages. Table 2, in turn, specifies the surgical models and respective constriction localization in the aorta, in the animal species included in this review. Although LVH can also be initiated by constriction of the abdominal or suprarenal part of the aorta, this is not covered in the present review as it is more representative of a model of hypertension than AS, and may conceal important differences. Additionally, cardiomyocyte hypertrophy achieved by non-surgical methods, fails to cover many of the disease aspects, involves metabolic disorders or is sometimes not feasible in large animal models and will be excluded. Figure 2 summarizes the most notorious observations for each animal model described in this review.
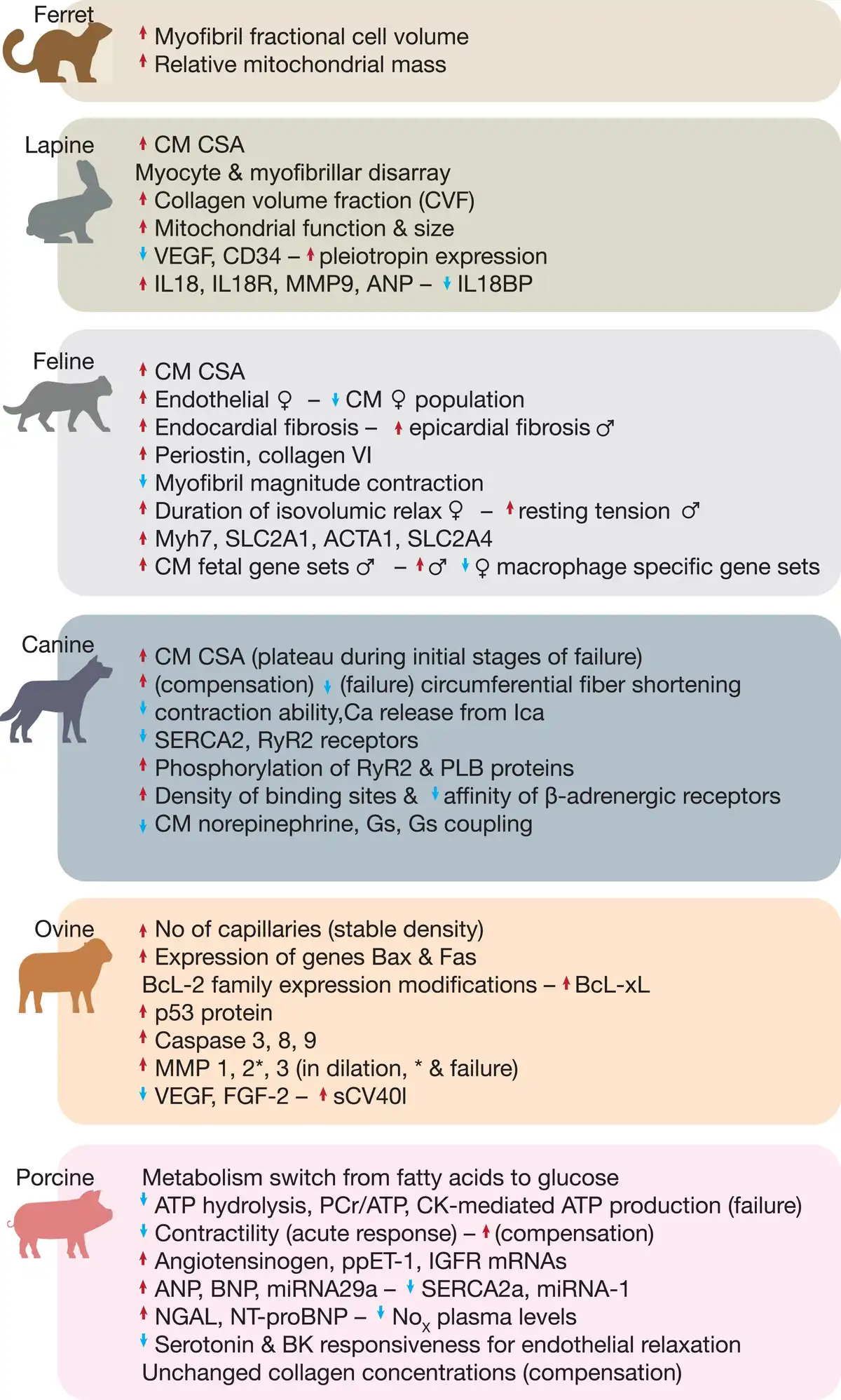
Figure 2
Large animal models of left ventricular hypertrophy induced by cardiac pressure overload. Phenotypical characteristics of different animal models after inducing hypertrophy by pressure overload, and respective important scientific achievements and discoveries per animal.
6.1 Ferret
Among the animals used to study LVH induced by pressure overload is the ferret (Mustello purtorius furo). In a study conducted in 1980, adult male animals were submitted to Banding of Ascending Aorta (AAB), by a circular wire clip, covered with polyethylene. The constriction occluded 70% of the aorta, achieving an overall LV mass increase of 45% of LV, already after two weeks and determined by the Left Ventricle/Body Weight (LV/BW) ratio. In parallel, hypertrophic growth regression was studied in a different group of animals, that were allowed to recover from the banding period of two weeks, for six more weeks after debanding. The results showed that LV mass was able to regress and adjust to similar values in comparison to the control animals. Haemodynamic data indicated an immediate increase in Left Ventricular Systolic pressure (LVSP), upon banding, but no significant differences in the Left Ventricular End Diastolic pressure (LVEDP). Heart rates, immediately upon banding, were slightly increased in both banding and regression groups, but their mean rates indicated no significant difference compared to controls. In a tissue/cellular level, it was shown that both the banded and regression animals had greater fractional cell volume of myofibrils in endocardial regions compared to the anterior papillary muscle, a difference that was not observed between different heart regions of control animals. Subcellularly, the morphology and cell fraction volume were examined, showing no significant differences rather than the mean relative mass of mitochondria to be greater in banded animals compared to controls, similarly to the mean relative mass of the myofibrils. This fact was attributed to the increased need of energy by the producing and consuming structures, i.e. mitochondria and myofibrils, as a response to the hypertrophic stimulus of pressure overload.
6.2 Lapine
New Zealand White Rabbit (Oryctolagus cunigulus) has, lately, become very common as an experimental animal model to study CVDs. It can combine the benefits of smaller animal models, while rabbits are phylogenetically closer to human, with more physiological and genetic resemblance. Experiments conducted in adult as well as neonatal/infant animals achieved a pathologic hypertrophy formation and were able to correlate with the human pathologic hypertrophic condition, caused by AS, by presenting comparable results. Nevertheless, the size and life span differences to human, deprive lapine models of high representativeness necessary to model cardiac diseases.
Haemodynamically, it has been shown that AAB, resulting in 75% of reduction of the aortic CSA, in adult rabbits, can cause significantly elevated LVSP, while preserving the animal viability. Pressure overload for the whole period of the constriction (up to 24 weeks) was confirmed by weekly echocardiography, while decreased to normal upon removal of the constriction. Until a period of two weeks after the banding, Fraction Shortening (FS) increased, starting to diminish afterwards. In contrast, Left Ventricle Systolic and Diastolic Dimensions appeared to decrease until week 2, but both showed an extravagant increase between weeks 4 and 24. During the first four weeks after banding, LV and Interventricular Septum (IVS) thickness were progressively increasing, while right after appeared to attenuate, in accordance with the diastolic wall thickness to LV cavity radius (h/r), marking the passage from a concentric compensated hypertrophic response to an eccentric decompensation state. No statistical significance was shown by the slight increase of heart rate in the banding animals and the total LV Weight to Body Weight ratio (LVW/BW), was found to be at the peak of 92,3% at week 8. In a different, ‘non-surgical’, attempt to induce LV hypertrophy in adult rabbits, by the placement of a partially deployed stent in descending aorta by catheterization, after three months an increase of 10% in Heart weight to Body weight ratio (HW/BW) was achieved, indicating the possibility of a more chronic model of the disease. Experiments in neonatal (10 days old) rabbits indicated a compensated state, four weeks after the banding, by a preserved FS and highly increased LV mass to volume ratio, which both appear to be reduced at week 6, representing the decompensated phase; during those time points the HW/BW appeared to be equal and higher than the control animals. Recently, it was proven that severe TAC (75%) can establish heart failure in a period of 6 weeks in neonatal rabbits, while a moderate constriction (50%) failed to do so. While the pressure after ligation was increased 26 times in six weeks the LV wall thickness also appeared to increase with time, being two times higher than in the control animals. Moreover, the progress from a concentric compensated to an eccentric decompensated heart, in these animals, was found to happen between week 4 and 6, indicated by alterations in LV dimension, EF, and FS.,
Histology results indicate that AB in rabbits can cause myocyte and myofibrillar disarray, accompanied by interstitial fibrosis. Collagen Volume Fraction indicates two to three times increase in collagen, which does not normalize to control levels upon removal of the banding, in contrast to other parameters determined., Myocytes showed increased thickness, with an increased CSA by 38.4% after three months in moderate model of chronic hypertrophy and almost 200% in a severe neonatal model, after six weeks., An old study, focusing on the mitochondria of CMs, indicated an increase of their function following pressure overload which, gradually decreases in time. Mitochondrial alterations can already be observed 2 days after banding and include larger size, decreased number of cristae or occasionally emptiness. At day 7, completely destroyed or swollen mitochondria are present with significantly augmented mass. At day 45, a relative normalization in size and structure of the organelles is observed, in contrast to the observations at eight months where the cell’s mitochondrial fraction appears to be smaller, with many elongated incompletely divided complexes. Moreover, experimental models of rabbits have evidenced a decrease in expression of Vascular Endothelial Growth Factor (VEGF) and CD34, and increased Pleiotrophin, suggesting impaired angiogenesis and an imbalance between the microcapillary network and the cellular needs to cope with hypertrophic growth and remodelling. Finally, increased pro-inflammatory factors such as cytokine IL-18, IL-18 Receptor, Matrix metallopeptidase 9 (MMP9) and ANP, and decreased antagonist IL-18 Binding Protein, during all the stages of remodelling induced by pressure overload, are believed to play a role in endothelial cell death and other pathological mechanisms of heart failure.
6.3 Feline
Cats (Felis catus) have been also used as large animal models in variable conditions. In CVDs, and especially cardiac hypertrophy due to pressure overload, young cats were used to reproduce a mild state of the disease that increases in severity as the animal grows. The slowly evolving constriction mimics the progressive nature of the chronic human compensated hypertrophy towards a decompensated state. The majority of the experiments were towards generating a model of Heart Failure with preserved Ejection Fraction (HFpEF). Feline appear to be suitable animal models, as their physiological properties can be comparable to human with translational power, even though their small size may be a limitation for the use of human equipment and measurements. Nevertheless, metabolic differences, such as concentration of high density lipoproteins, may affect responses to drugs or the development of a disease in a way that is not common in humans, confounding experimental results.
In adult mice, banding of the aortic arch with a nylon thread, causing an average pressure gradient of 64 mmHg, after six weeks of banding, resulted in a significant decrease of Left Ventricular Inner Dimension in systole, accompanied by an increase in LV wall thickness, EF and FS, compared to the baseline measurements. Left Ventricular Inner Dimension in diastole, Cardiac Output and Stroke Volume remained unchanged. Altogether, these data suggest a concentric hypertrophic response, that can be classified as level 1 of the New York Heart Association. Young cats subjected to AAB with a clip of a standard diameter of 2,8–3 mm, presented signs of hypertrophy after one week of banding, including increased IVS wall thickening and Left Atria (LA) size, while LV diameter and FS remained comparable to sham animals, in a period of two to three months of banding. Animals were categorized, one week after the banding, according to the constriction degree with a cut-off of 85%; animals with a constriction greater than 85% presented a steadily increased pressure gradient, around the banding, which was almost doubled compared to sham after two to three months. Animals with a constriction of less than 85%, also presented increased LA size and pressure gradient in the banding site, compared to sham, but still at a lower degree than the ones with more severe constriction. Another study aiming at comparing sex-related differences in HFpEF and where loose constrictions were performed in the aorta of male and female young cats, resulted in similar phenotype in both sexes without any disturbances in the physiological growth of the animals. In detail, four months post-surgery the pressure gradient remained the same in all the banded animals and HW/BW and LV wall thickness were increased, compared to sham animals. EF indicated a small but significant decrease only in male animals. LVEDP was also higher in males but this increase did not reach the significance cut-off. In these studies, while there was a sex-dependent increase in CM CSA compared to sham, in female a decrease in CMs was observed, accompanied by increased endothelial cell number. Moreover, sex-independent significantly high endocardial fibrosis developed, while increased epicardial fibrosis was only significant in male animals with many of the ECM proteins, such as periostin and collagen VI, to be upregulated. Regarding the contractility properties, a depression in magnitude contraction of the myofibrils was observed, with a tendency to increase proportionally to the constriction degree. In females, the isovolumic relaxation was found to be prolonged compared to sham and males, while resting tension was significantly increased only in males. Furthermore, the credibility of hypertrophic response in this animal model was proven by RNA sequencing for hypertrophic markers including Myosin Heavy Chain 7, Solute Carrier Family 2 Member 2, Actin Alpha 1, and Solute Carrier Family 2 Member 4, which appeared to be elevated in the banded animals compared to sham. Finally, there are indications of fetal gene program activation, and many altered CM gene sets, which seem mainly to occur in male. Similarly, macrophage-associated activation of heart growth, tissue regeneration, and leucocyte differentiation signalling were found to be upregulated in male, but depressed in female. In contrast, fibroblasts appeared to have a common response mechanism, in both sexes, in terms of gene activation. The sex-related activation of specific gene sets may elucidate the reasons of the different prevalence of disease observed in human male and female.
6.4 Canine
Dogs (Canis lupus familiaris) used to be one of the most common animal models in the field of biomedical sciences during the past years. Due to their abundancy in the human environment, they are easy to access and approach, but also facilitate the understanding of pain or discomfort expressions, necessary for animal welfare. Furthermore, canine organ physiology, and in particular the cardiovascular system, presents many similarities to human physiology, allowing the utilization of same medical instruments, such as human catheter and echocardiography instruments. Being phylogenetically closer to human when compared to smaller animals, they are more likely to share common cellular and molecular mechanisms both in healthy and disease conditions. Lately, due to the increase of ethical concerns and restrictions, this model tends to be less used and is being substituted by other methods. Importantly and similarly to feline models, dogs do not develop common comorbidities that are observed in human CVDs, such as atherosclerosis, and therefore, may not be suitable for the study of particular cardiac conditions.
Nevertheless, large studies in dogs have been essential to achieve a better disease representation, while avoiding mortality factors, such as aortic rupture, congestive heart failure and others. A 3-year-long large study, apart from establishing a progressive aortic constriction model in adult animals, concluded that animals presenting high baseline wall stress are more prone to heart dysfunction. Comparisons between chronic hypertrophy, during normal growth of young animals, and hypertrophic stimuli during adulthood, indicate a similar hypertrophic degree in all banded animals, increase in coronary perfusion pressure proximal to the banding site and attenuated minimum coronary resistance. The myocardial blood flow was, however, only significantly increased in the young models compared to their respective controls. Furthermore, regression experiments in banded puppies with normal FS indicated that a significant increase of more than 100% in LV mass and three times in pressure gradient, after 10 months, could be reversed with tendency to normalize, upon removal of the band, presenting an important decrease in both pressure gradient and LV mass. When a fixed-diameter band was placed in young animals for 12 months, two different types of cardiac hypertrophy developed, varying between 40 and 100% of increase in LV mass. One presented compensated heart failure with an LV/BW ratio 7.7 g/kg, LVEDP of 8 mmHg and mean systolic pressure of 202 g/cm2, while the other, decompensated, presented values of 9.8 g/kg, 25 mmHg, and 313 g/cm2, respectively, in addition to a decrease in subendocardial blood flow.
Cardiomyocytes of banded animals under the same conditions, that presented compensated or decompensated hypertrophic response after 12 months, appeared to reach a plateau in their CSA between six and nine months, with the CMs from failing hearts to grow first in size, followed by those from compensated. In the same study, a decrease in circumferential fibre shortening was observed in failing hearts and an increase in compensated hearts, compared to control, after 12 months. Regarding the contraction abilities of CMs from puppies subjected to banding for 15 months, it appears to be impaired at baseline but also upon Ca2+ trigger, in accordance to a decreased ability of L-type Ca2+ channels to release Ca2+. The number of receptors of SERCA2a and RyR2 was also assessed, showing a significant decrease of 68 and 35%, respectively, and proteins such as RyR2 and phospholamban appeared hyperphosphorylated. Studies involving a chronic model of fully compensated hypertrophic hearts, revealed β-adrenergic receptors with higher density of binding sites but with lower affinity and decreased myocyte norepinephrine, indicating the implication of SNS. Later, in another study, a decrease of 59% in guanil nucleotide binding protein (Gs) was demonstrated, justifying the observed deficiency in Gs coupling. The decreased adrenergic responsiveness and defects in the adenylate cyclase system were attributed to the Gs coupling deficiency, in the sarcolemma of a hypertrophied heart.
6.5 Ovine
Lambs and sheep (Ovis aries) have also been subjected in aortic banding procedures in order to reproduce models of LVH related to AS. These animals are manageable and easily operated, with nutritional and housing tolerance, go through wound healing without complications and display decreased risk of arrhythmogenic incidences during open-heart surgeries. Interestingly, it was observed that intraspecies interaction acts in a motivational way regarding animal recovery after surgery, limiting the factor of space availability. The large size and anatomy relatively similar, to human allow for chronic and translational instrumentation of the animals. Also, compared to other experimental animals, such as dogs and pigs, and despite similar degree of anatomical differences, sheep seem to develop more consistent degrees of LVH and have a moderate body mass increase wile ageing, which facilitates surgical procedures.
A large experiment was performed to evaluate AAB in juvenile sheep as a model of LVH induced by AS. The model resulted in a condition of progressive AS with low probability of complications during the surgical procedures. When subjecting both lambs to six weeks of banding, both groups developed a similar increase in LV/BW, as wells as similar pressure gradients between the LV and aorta, similar LVEDP and heart rates. Despite the similar conditions, the sheep group showed impaired cardiac function, while the lamb group remained similar to the control (sham) group, revealing age-related differences in response to cardiac stress. These observations could be attributed to the hyperplastic capability of the juvenile heart, to the different degree of neovascularization or to altered calcium handling properties between the two groups. Nonetheless, those results indicate the existence of a possible confounder, related to maturity of the myocardium, which in many, if not most, studies were not taken into account. A series of studies in adult animals, where an inflating device was used to partially and gradually constrict the aorta, demonstrated a reproducible model in which the LVH, evaluated by Left Ventricular mass index, is established three to four weeks after the constriction, with stable cardiac function and progressive LV dilation up to seven to eight weeks, were the failure stage initiates. The failure stage was reflected by decreased FS, the animals’ inability to move, unwillingness to eat and tachypnea. Ultimately, a more chronic model, using the same device, was achieved, with the peak of LVH at approximately 32 weeks after the constriction, LV dilation starting at 103 and decreased EF at 117 weeks. Notably, removal of the hypertrophic stimulus by deflation of the device resulted in restoration of EF to control levels.
Assessment of the vasculature bed capacity, during hypertrophic stimulus, in young sheep, revealed an increase of 32% in the number of capillaries in LV, while capillary density, coronary flow reserve, and minimal coronary resistance remained stable, in comparison with control animals. In the same study, when protamine, a protein that correlates to neovasculature impairment during hypertrophy, was administered, the subendocardial capillary density was reduced and the minimal coronary resistance was increased, with the LVH degree remaining the same compared to the control LVH animals. As the experiment involved young animals, with increased capacity of neovascularization, further investigation of the mechanisms involved in the process may unveil new therapeutic targets and approaches. Adult animals, in turn, revealed a significant increased expression of the pro-apoptotic genes Bax and Fas, upon the development of hypertrophy, while the anti-apoptotic gene Bcl-2 were not altered. This increased Bax to Bcl-2 ratio observed during pathologic hypertrophy, positively correlates with the expression of p53, a direct regulator of the anti-apoptotic family of Bcl-2. Furthermore, Bcl-xL, another member of the Bcl-2 family, was found to progressively increase during the process and was associated with LV dilation and the failing stage, suggesting its role in the transition to failure. Apoptosis was assessed, by investigating the Expression levels of caspases 3, 8, and 9, supported further implication of mitochondrial- and death receptor-mediated pathways. Caspase 3 was progressively increased to almost eight times in all stages of the disease and, in particular, in the failing stage; caspase 8 was found almost two times overexpressed in the failing stage and caspase 9 progressively increased its expression up to approximately six times through all the stages of the disease. The role of matrixins, GFs and inflammatory mediators, in the ECM changes during the progress of the disease was also investigated. MMP-1 and MMP-3 were significantly increased during cardiac dilation and MMP-2 was increased in the failing stage, but were decrease again upon removal of the hypertrophic stimulus. In turn, VEGF and Fibroblast growth factors-2 were found to significantly decrease towards failing, while VEGF increased again at the recovery stage. Regarding inflammation, Interferon alpha-2 was decreased along with cardiac hypertrophic growth, in contrast to the soluble ligand sCD40l that increased during the dilation stage.
Sheep and lamb models are currently being used and constantly being improved to study different aspects of cardiac disease, and as they exhibit important cardiac pathology differences, similar to other animal species, the choice of model should always be based on the specific scientific question.
6.6 Porcine
Porcine (Sus scrofa) is a very common and highly efficient model that can predict and resemble human conditions and responses. Pigs display similar heart physiology, vasculature and function, disease progress, gene sequence, and chromosome analogy to human, while availability of tissue samples never being an issue.,, Comorbidities related to CVDs in human, are also observed in pigs, indicating similar reactions and underlying mechanisms, as well as a highly resembling immune system., Furthermore, due to their size, the animals can be subjected to surgical practices with human equipment and conditions, mimicking procedures that are performed in patients. The downside is that research in pigs takes longer, requires expensive surgical equipment, specialized facilities, and expertise, with a very limited number of institutions being able to accommodate such procedures.,
Studies describing surgical protocols of aortic constriction have demonstrated hypertrophic and fibrotic responses in animals 12 weeks after the intervention, with transition to a decompensated heart happening between weeks 4 and 6 and cardiac dilation establishing at the 8th week., Furthermore, disappearance of muscle layer in the mitral valve may contribute to the onset of cardiac dysfunction. Immediately after banding, increased LVESD and myocardial strain were detected but again normalized to baseline 12 h later, similar to the increased LVEDD that was normalized 3 h later. Systolic wall thickness of the LV, declined during the first 6 h, but was significantly increased after 72 h, suggesting an acute cardiac adaptation to pressure overload. High adaptability of the cardiac muscle was also indicated by the active compensating remodelling that could initially prevent the increase in systolic pressure, accompanied by elevated active tension; leading though to a consequent decrease in diastolic stress but increase in diastolic pressure, during a period of approximately one month. Diastolic stress of the LV was attributed to cardiac stiffness and not fibrosis. Placement of an inflating cuff in the aorta of young animals, for 30 days, causing a 45% increase in LV/BW, indicated a concurrent increase of 16% weight of the right ventricle (RV) and decreased myocardial blood flow of the LV, during exercise, in comparison to control animals. Furthermore, minimal coronary vascular resistance, specifically in the endomyocardium, was significantly increased in the hypertrophic hearts compared to their respective controls. Transplantation of mesenchymal stem cells (MSCs) overexpressing VEGF, through the venous coronary system, in banded animals resulted in a significantly higher myocardial blood flow compared to banded, with normal MSCs or without transplantation, and sham animals, indicating the important role of VEGF in regulating myocardial blood flow during pathologic hypertrophy. A more chronic model of five months, suggested that a progressive stenosis is responsible for mean circumferential wall thickness and isovolumic relaxation constant (tau) increase, LV and RV mass increase and fibrosis, as well as a secondary pulmonary artery pressure increase.
Investigation of the metabolic energetics of hypertrophied pig CMs, by magnetic resonance imaging, of animals with concentric hypertrophy and HFpEF, demonstrated the characteristic metabolic switch from fatty acids to glucose substrate. Furthermore, severe cardiac dysfunction was correlated with reduced ATP hydrolysis, decreased myocardial phosphocreatine to ATP ratio (phosphocreatine/ATP) and Creatine Kinase-mediated ATP production. Data indicated that ATP hydrolysis rates, which were proven to increase linearly with cardiac workload, dynamically change during the different stages of the disease. Immediate responses of CMs implicated a decreased contractility during the first 6 h, that was later normalized and progressively increased 4 and 7 days later, and a 13-fold angiotensinogen, 112-fold ppET-1 and 37-fold Insulin-like Growth Factor Receptor peak mRNA overexpression, reported at 2, 3, and 24 h, respectively. Taken the findings together, it was suggested that GFs provide an initial support in the CM contractility as acute response to the hypertrophic stimulus. In later stages of chronic models, high expression of ANP, BNP, and miR-29a and decreased miR-1 and SERCA2a were demonstrated, accompanied by increased circulating neutrophil gelatinase-associated lipocalin levels, indicative of fibrosis, and NT-pro-BNP levels, five months after the banding. Endothelial dysfunction in this model has been correlated with Gi and Gq protein-mediated relaxation of the coronary arteries, and a decrease in endothelial relaxation in response to serotonin and bradykinin (Bk) was also observed. In addition, no significant decreases were detected in the endothelium-derived hyperpolarizing factors pathway, in contrary with the plasma NO levels that appeared declined, indicating a contribution to the progression of heart failure, as NO is known for its antihypertrophic effect. Concerning the ECM, a study reported unchanged collagen concentrations in banded animals with a degree of 98% increase in HW/BW, four to six weeks later, compared to controls.
Despite all the scientific advantages of using pig models to study cardiac disease, logistics remains and will remain an issue for many institutions. Furthermore, experiments in these animals are very costly not only due to the costs of breeding and maintaining the animals but any pharmacological study will require higher amounts of drugs, oligonucleotides, and reagents which also makes it often impossible to perform well-designed studies with significant statistical power.
7. Conclusions
Cardiomyocytes, upon a hypertrophic stimulus, suffer significant alterations at multiple levels, often involving the return to fetal gene programmes or the activation of molecular mechanisms that can be fatal to them. The alterations at the cellular level, with their accumulation and the progression of time, have an impact at the organ level, resulting in clinical symptoms that progress in severity and, if left untreated, to death. It is shown that if CM pathologic alterations are inhibited before a critical point, that most probably corresponds to the transition from a compensated state to a decompensated one, a reverse in cardiac dysfunction to a functional heart can occur. The fact that those subcellular alterations precede the organ functionality implications, provides an exploitable opportunity in terms of therapeutic approaches. Due to the inadequacy of human tissues to be provided in research and the still non-advanced and inefficient in vitro tools, that reproduce low complexity structures, the only existing solution for results with high reliability and translatability, are experimental animal models. For CVDs, in particular, large animal models are required, in order to overcome the many limitations of the smaller models, regarding the more delicate and robust outcomes. The contribution of small animals in many discoveries is highly recognized; nevertheless, with the advances in research nowadays more precision to the representation of a human condition is required. Large animal models are already being used in pre-clinical trials and represent a step in translational research that can no longer be omitted. A recently and so far, successful example, is the work by Thum and co-workers, where pre-clinical results from studies in mice were validate in a pig model of myocardial infarction, and later on in a clinical phase 1B study in ischaemic heart failure patients. Although such advances were not yet achieved for AS, much cardiovascular research is currently being performed in larger animals, mostly pigs, in order to translate results from mice studies to a more human-like situation.
The mentioned animal models used to reproduce LVH, by induction of pressure overload, representing a condition of the LV comparable to the hypertrophy caused by human AS, have elucidated many unknown or not well-understood cellular and molecular mechanisms that are crucial to better understand the process of cardiac hypertrophy, and to better design strategies to diagnose the disease as well as to therapeutically target the disease process and/or the intervening cellular/molecular players. Ethical issues and model-specific limitations are driving science towards the use of stem cells and organoid technology. But will these ever replace animal models? Organoids, 3D and ex-vivo models are, to some extent, able to recapitulate human physiology by using human cells, are relatively quickly established and robust. They also allow for genetic modification and patient-specific applications. However, most of them still lack a (functional) vascular system, are mostly cultured under static conditions, and as cells grow into a 3D structure, not all will be able to receive nutrients while exchange of waste also becomes an issue. Furthermore, the systemic component is also missing. More and more, we realize how cells, tissues, and organs need to communicate with each other in order to maintain a healthy status of the organism, in a complex network that will be hard to achieve with these non-animal alternative models. Both have their advantages and limitations and combining them is currently, and by far, the best strategy to generate new knowledge and to find and test new drug targets for different diseases.
Acknowledgements
L.D.W. acknowledges support from ERA-CVD JCT2016 EXPERT, the Dutch Heart Foundation, Dutch Federation of University Medical Centers, ZonMW and the Royal Netherlands Academy of Sciences. L.D.W. was further supported by grant 311549 from the European Research Council (ERC), the VICI award 918-156-47 from The Netherlands Organization for Scientific Research (NWO) and the Marie Sklodowska-Curie grant agreement no. 813716 (TRAIN-HEART). P.C.M. is supported by a Dutch CardioVascular Alliance (DCVA) awarded to the Phaedra consortium as well as the Impulse Grant 2018 awarded to the Phaedra IMPACT consortium (CVON-2018-29) and is an Established Investigator of the Dutch Heart Foundation (NHS2015T066). P.C.M. was further supported by the H2020 Twinning project RESETageing (GA 952266) and the TKI-LSH project MEDIATOR (LSHM21016) from Health-Holland.
Abbreviations
AAB: Ascending Aorta/Aortic Banding
ACTA1: Actin Alpha 1
AHA: American Heart Association
ANP: Atrial Natriuretic Peptide
AS: Aortic Stenosis
ATP: Adenosine triphosphate
Bk: Bradykinin
BMP2: Bone morphogenetic protein 2
BNP: Brain Natriuretic Peptide
CCR2: C-C chemokine receptor 2
CK: Creatine Kinase
CMs: cardiomyocytes
CNP: C-type natriuretic peptide
CO: Cardiac Output
CSA: Cross Sectional Area
CVDs: Cardiovascular diseases
CVF: Collagen Volume Fraction
CXCL1: chemokine (C-X-C-motif) ligand 1
DCs: Dendritic Cells
ECM: Extracellular Matrix
ECs: Endothelial Cells
EDHF: Endothelium Derived Hyperpolarizing Factors (
EF: Ejection Fraction
EMT: Endothelial to Mesenchymal transition
FGF: Fibroblast growth factors
ppET-1: preproEndothelin-1
FS: Fraction Shortening
GFs: growth factors
Gs: guanil nucleotide binding protein
H/r: diastolic wall thickness to LV cavity radius ratio
HFpEF: Heart failure with preserved ejection fraction
HW/BW: Heart weight to Body weight ratio
ICa: L-type Ca2+ channels
IFN-a-2: Interferon alpha-2
IGFR: Insulin-like Growth Factor Receptor
IL6: interleukin 6
IL32: interleukin 32
IL18: interleukin 18
IL-18BP: Interleukin-18 Binding Protein
IL-18R: Interleukin-18 Receptor
IVS: Interventricular Septum
LA: Left Atria
LV/BW: Left Ventricle/Body Weight ratio
LV: Left Ventricle/Left Ventricular
LVDD: Left ventricle Diastolic dimensions
LVEDP: Left Ventricular End Diastolic pressure
LVH: Left Ventricular Hypertrophy
LVIDd: Left Ventricular Inner Dimension in diastole
LVIDs: Left Ventricular Inner Dimension in systole
LVMI: Left Ventricular mass index
LVSD: Left ventricle Systolic dimensions
LVSP: Left Ventricular Systolic pressure
MCP1: monocyte chemoattractant protein 1
MMP: Matrix metallopeptidase
MSCs: Mesenchymal Stem Cells
MYH7: Myosin Heavy Chain 7
NGAL: Neutrophil gelatinase-associated lipocalin
NK: Natural Killer cells
NO: Nitric Oxide
NT-proBNP: N-terminal pro-hormone Brain Natriuretic Peptide
NYHA: New York Heart Association
PCr/ATP: phosphocreatine to ATP ratio
PLB: Phospholamban
PTN: Pleiotrophin
RAAS: Renin-Angiotensin-Aldosterone-System
ROS: Reactive Oxygen Species
RV: Right Ventricle
RyR2: Ryanodine Receptor 2
SERCA2a: Sarco/Endoplasmic Reticulum Ca2+-ATPase
SLC2A: Solute Carrier Family 2 Member 4
SNS: Sympathetic Nervous System
SV: Stroke Volume
TAC: Transverse Aortic Constriction
tau: isovolumic relaxation constant
UPS: ubiquitin-proteasome system
VEGF: Vascular Endothelial Growth Factor
WHO: World Health Organization
References
- 1. World_Health_Organization. W.H.O. Cardiovascular Diseases: Health Topics; 2023. https://www.who.int (January 2023, date last accessed).
- 2. Vigil-Garcia M, Demkes CJ, Eding JEC, Versteeg D, de Ruiter H, Perini I, Kooijman L, Gladka MM, Asselbergs FW, Vink A, Harakalova M, Bossu A, van Veen TAB, Boogerd CJ, van Rooij E. Gene expression profiling of hypertrophic cardiomyocytes identifies new players in pathological remodelling. Cardiovasc Res 2021;117:1532–1545.
- 3. Hein S, Arnon E, Kostin S, Scho[Combining Diaeresis]nburg M, Elsa[Combining Diaeresis]sser A, Polyakova V, Bauer EP, Klo[Combining Diaeresis]vekorn W-P, Schaper J. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart. Circulation 2003;107:984–991.
- 4. Tsao CW, Aday AW, Almarzooq ZI, Anderson CA, Arora P, Avery CL, Baker-Smith CM, Beaton AZ, Boehme AK, Buxton AE, Commodore-Mensah Y. Heart disease and stroke statistics-2023 update: a report from the American Heart Association. Circulation 2023;147:e93–e621.
- 5. Xie Y, Gao Y, Gao R, Yang W, Dong Z, Moses RE, Sun A, Li X, Ge J. The proteasome activator REGγ accelerates cardiac hypertrophy by declining PP2Acα–SOD2 pathway. Cell Death Differ 2020;27:2952–2972.
- 6. McGonigle P, Ruggeri B. Animal models of human disease: challenges in enabling translation. Biochem Pharmacol 2014;87:162–171.
- 7. Savoji H, Mohammadi MH, Rafatian N, Toroghi MK, Wang EY, Zhao Y, Korolj A, Ahadian S, Radisic M. Cardiovascular disease models: a game changing paradigm in drug discovery and screening. Biomaterials 2019;198:3–26.
- 8. Fliegner D, Gerdes C, Meding J, Stasch J-P. Translational in vivo models for cardiovascular diseases. Handb Exp Pharmacol 2016;232:223–234.
- 9. Tan CMJ, Lewandowski AJ. The transitional heart: from early embryonic and fetal development to neonatal life. Fetal Diagn Ther 2020;47:373–386.
- 10. Ford LE. Heart size. Circ Res 1976;39:297–303.
- 11. Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol 2016;97:245–262.
- 12. Dorn GW II, Robbins J, Sugden PH. Phenotyping hypertrophy. Circ Res 2003;92:1171–1175.
- 13. Heallen TR, Kadow ZA, Wang J, Martin JF. Determinants of cardiac growth and size. Cold Spring Harb Perspect Biol 2020;12:a037150.
- 14. Rohini A, Agrawal N, Koyani CN, Singh R. Molecular targets and regulators of cardiac hypertrophy. Pharmacol Res 2010;61:269–280.
- 15. Oldfield CJ, Duhamel TA, Dhalla NS. Mechanisms for the transition from physiological to pathological cardiac hypertrophy. Can J Physiol Pharmacol 2020;98:74–84.
- 16. Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez-Sarano M. Burden of valvular heart diseases: a population-based study. Lancet 2006;368:1005–1011.
- 17. Komuro I. Molecular mechanism of cardiac hypertrophy and development. Jpn Circ J 2001;65:353–358.
- 18. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol 2018;15:387–407.
- 19. Feher J (ed.). 5.12—Integration of cardiac output and venous return. In: Quantitative Human Physiology. 2nd ed. Boston: Academic Press; 2012. p599–607.
- 20. Litviňuková M, Talavera-López C, Maatz H, Reichart D, Worth CL, Lindberg EL, Kanda M, Polanski K, Heinig M, Lee M, Nadelmann ER, Roberts K, Tuck L, Fasouli ES, DeLaughter DM, McDonough B, Wakimoto H, Gorham JM, Samari S, Mahbubani KT, Saeb-Parsy K, Patone G, Boyle JJ, Zhang H, Zhang H, Viveiros A, Oudit GY, Bayraktar OA, Seidman JG, Seidman CE, Noseda M, Hubner N, Teichmann SA. Cells of the adult human heart. Nature 2020;588:466–472.
- 21. Lim GB. Complexity and plasticity of cardiac cellular composition. Nat Rev Cardiol 2020;17:759.
- 22. Mollova M, Bersell K, Walsh S, Savla J, Das LT, Park S-Y, Silberstein LE, dos Remedios CG, Graham D, Colan S, Kühn B. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc Natl Acad Sci U S A 2013;110:1446–1451.
- 23. Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe[Combining Acute Accent]-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frise[Combining Acute Accent]n J. Evidence for cardiomyocyte renewal in humans. Science 2009;324:98–102.
- 24. Yutzey KE. Cardiomyocyte proliferation. Circ Res 2017;120:627–629.
- 25. Derks W, Bergmann O. Polyploidy in cardiomyocytes. Circ Res 2020;126:552–565.
- 26. Vliegen HW, Eulderink F, Bruschke AV, van Der Laarse A, Cornelisse CJ. Polyploidy of myocyte nuclei in pressure overloaded human hearts: a flow cytometric study in left and right ventricular myocardium. Am J Cardiovasc Pathol 1995;5:27–31.
- 27. Wang ZV, Ferdous A, Hill JA. Cardiomyocyte autophagy: metabolic profit and loss. Heart Fail Rev 2013;18:585–594.
- 28. Sambandam N, Lopaschuk GD, Brownsey RW, Allard MF. Energy metabolism in the hypertrophied heart. Heart Fail Rev 2002;7:161–173.
- 29. Ramachandra CJA, Chua J, Cong S, Kp MMJ, Shim W, Wu JC, Hausenloy DJ. Human-induced pluripotent stem cells for modelling metabolic perturbations and impaired bioenergetics underlying cardiomyopathies. Cardiovasc Res 2021;117:694–711.
- 30. Ritterhoff J, Young S, Villet O, Shao D, Neto FC, Bettcher LF, Hsu Y-WA, Kolwicz SC, Raftery D, Tian R. Metabolic remodeling promotes cardiac hypertrophy by directing glucose to aspartate biosynthesis. Circ Res 2020;126:182–196.
- 31. Mirtschink P, Krek W. Hypoxia-driven glycolytic and fructolytic metabolic programs: pivotal to hypertrophic heart disease. Biochim Biophys Acta 2016;1863:1822–1828.
- 32. Mirtschink P, Krishnan J, Grimm F, Sarre A, Hörl M, Kayikci M, Fankhauser N, Christinat Y, Cortijo C, Feehan O, Vukolic A, Sossalla S, Stehr SN, Ule J, Zamboni N, Pedrazzini T, Krek W. HIF-driven SF3B1 induces KHK-C to enforce fructolysis and heart disease. Nature 2015;522:444–449.
- 33. Yang D, Liu H-Q, Liu F-Y, Guo Z, An P, Wang M-Y, Yang Z, Fan D, Tang Q-Z. Mitochondria in pathological cardiac hypertrophy research and therapy. Front Cardiovasc Med 2022;8:822969.
- 34. Xu M, Xue R-Q, Lu Y, Yong S-Y, Wu Q, Cui Y-L, Zuo X-T, Yu X-J, Zhao M, Zang W-J. Choline ameliorates cardiac hypertrophy by regulating metabolic remodelling and UPRmt through SIRT3-AMPK pathway. Cardiovasc Res 2019;115:530–545.
- 35. Gao S, Li G, Shao Y, Wei Z, Huang S, Qi F, Jiao Y, Li Y, Zhang C, Du J. FABP5 deficiency impairs mitochondrial function and aggravates pathological cardiac remodeling and dysfunction. Cardiovasc Toxicol 2021;21:619–629.
- 36. Facundo HdTF, Brainard RE, Caldas FRdL, Lucas AMB. Advances in experimental medicine and biology. Adv Exp Med Biol 2017;982:203–226.
- 37. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol 2011;301:H2181–H2190.
- 38. Li L, Xu J, He L, Peng L, Zhong Q, Chen L, Jiang Z. The role of autophagy in cardiac hypertrophy. Acta Biochim Biophys Sin (Shanghai) 2016;48:491–500.
- 39. Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 2007;13:619–624.
- 40. Oyabu J, Yamaguchi O, Hikoso S, Takeda T, Oka T, Murakawa T, Yasui H, Ueda H, Nakayama H, Taneike M, Omiya S, Shah AM, Nishida K, Otsu K. Autophagy-mediated degradation is necessary for regression of cardiac hypertrophy during ventricular unloading. Biochem Biophys Res Commun 2013;441:787–792.
- 41. Hariharan N, Ikeda Y, Hong C, Alcendor RR, Usui S, Gao S, Maejima Y, Sadoshima J. Autophagy plays an essential role in mediating regression of hypertrophy during unloading of the heart. PLoS One 2013;8:e51632.
- 42. Forte M, Frati G, Sciarretta S. Regulation of calcium handling by autophagy: a novel mechanism limiting cardiac hypertrophy and dysfunction? Cardiovasc Res 2022;118:1377–1379.
- 43. Ljubojević-Holzer S, Kraler S, Djalinac N, Abdellatif M, Voglhuber J, Schipke J, Schmidt M, Kling K-M, Franke GT, Herbst V, Zirlik A, von Lewinski D, Scherr D, Rainer PP, Kohlhaas M, Nickel A, Mühlfeld C, Maack C, Sedej S. Loss of autophagy protein ATG5 impairs cardiac capacity in mice and humans through diminishing mitochondrial abundance and disrupting Ca2+ cycling. Cardiovasc Res 2022;118:1492–1505.
- 44. Brundel BJJM. The role of proteostasis derailment in cardiac diseases. Cells 2020;9:2317.
- 45. Gilda JE, Gomes AV. Proteasome dysfunction in cardiomyopathies. J Physiol 2017;595:4051–4071.
- 46. Greenberg HZE, Zhao G, Shah AM, Zhang M. Role of oxidative stress in calcific aortic valve disease and its therapeutic implications. Cardiovasc Res 2022;118:1433–1451.
- 47. Fortun[Combining Tilde]o MA, Ravassa S, Fortun[Combining Tilde]o A, Zalba G, Di[Combining Acute Accent]ez J. Cardiomyocyte apoptotic cell death in arterial hypertension. Hypertension 2001;38:1406–1412.
- 48. Yamamoto S, Sawada K-i, Shimomura H, Kawamura K, James TN. On the nature of cell death during remodeling of hypertrophied human myocardium. J Mol Cell Cardiol 2000;32:161–175.
- 49. van Empel VP, De Windt LJ. Myocyte hypertrophy and apoptosis: a balancing act. Cardiovasc Res 2004;63:487–499.
- 50. Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Myocyte death, growth, and regeneration in cardiac hypertrophy and failure. Circ Res 2003;92:139–150.
- 51. Buyandelger B, Mansfield C, Knoll R. Mechano-signaling in heart failure. Pflugers Arch 2014;466:1093–1099.
- 52. Lyon RC, Zanella F, Omens JH, Sheikh F. Mechanotransduction in cardiac hypertrophy and failure. Circ Res 2015;116:1462–1476.
- 53. Russell B, Motlagh D, Ashley WW. Form follows function: how muscle shape is regulated by work. J Appl Physiol 2000;88:1127–1132.
- 54. Ehler E, Horowits R, Zuppinger C, Price RL, Perriard E, Leu M, Caroni P, Sussman M, Eppenberger HM, Perriard J-C. Alterations at the intercalated disk associated with the absence of muscle LIM protein. J Cell Biol 2001;153:763–772.
- 55. Perriard JC, Hirschy A, Ehler E. Dilated cardiomyopathy: a disease of the intercalated disc? Trends Cardiovasc Med 2003;13:30–38.
- 56. Ferrans VJ, Buja LM, Maron BJ. Sarcolemmal alterations in cardiac hypertrophy and degeneration. Recent Adv Stud Cardiac Struct Metab 1976;9:395–419.
- 57. Diaz ME, Graham HK, Trafford AW. Enhanced sarcolemmal Ca2+ efflux reduces sarcoplasmic reticulum Ca2+ content and systolic Ca2+ in cardiac hypertrophy. Cardiovasc Res 2004;62:538–547.
- 58. Frieler RA, Mortensen RM. Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation 2015;131:1019–1030.
- 59. Bazgir F, Nau J, Nakhaei-Rad S, Amin E, Wolf MJ, Saucerman JJ, Lorenz K, Ahmadian MR. The microenvironment of the pathogenesis of cardiac hypertrophy. Cells 2023;12:1780.
- 60. Polyakova EA, Mikhaylov EN, Sonin DL, Cheburkin YV, Galagudza MM. Neurohumoral, cardiac and inflammatory markers in the evaluation of heart failure severity and progression. J Geriatr Cardiol 2021;18:47–66.
- 61. De Angelis E, Pecoraro M, Rusciano M, Ciccarelli M, Popolo A. Cross-talk between neurohormonal pathways and the immune system in heart failure: a review of the literature. Int J Mol Sci 2019;20:1698.
- 62. Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S, Brown JH. Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by Ca2+/calmodulin-dependent protein kinase II δ signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation 2018;138:2530–2544.
- 63. Willeford A, Suetomi T, Nickle A, Hoffman HM, Miyamoto S, Heller Brown J. CaMKIIδ-mediated inflammatory gene expression and inflammasome activation in cardiomyocytes initiate inflammation and induce fibrosis. JCI Insight 2018;3:e97054.
- 64. Chen WY, Hong J, Gannon J, Kakkar R, Lee RT. Myocardial pressure overload induces systemic inflammation through endothelial cell IL-33. Proc Natl Acad Sci U S A 2015;112:2749–2754.
- 65. Ghigo A, Franco I, Morello F, Hirsch E. Myocyte signalling in leucocyte recruitment to the heart. Cardiovasc Res 2014;102:270–280.
- 66. Li H, Chen C, Wang DW. Inflammatory cytokines, immune cells, and organ interactions in heart failure. Front Physiol 2021;12:695047.
- 67. Martini E, Kunderfranco P, Peano C, Carullo P, Cremonesi M, Schorn T, Carriero R, Termanini A, Colombo FS, Jachetti E, Panico C, Faggian G, Fumero A, Torracca L, Molgora M, Cibella J, Pagiatakis C, Brummelman J, Alvisi G, Mazza EMC, Colombo MP, Lugli E, Condorelli G, Kallikourdis M. Single-cell sequencing of mouse heart immune infiltrate in pressure overload-driven heart failure reveals extent of immune activation. Circulation 2019;140:2089–2107.
- 68. Liu X, Shi GP, Guo J. Innate immune cells in pressure overload-induced cardiac hypertrophy and remodeling. Front Cell Dev Biol 2021;9:659666.
- 69. Afşin A, Asoğlu R, Kurtoğlu E, Kaya H. Neutrophil to lymphocyte ratio as a predictor of left ventricular hypertrophy in patients with newly diagnosed hypertension. J Hypertens Manag 2019;5:42.
- 70. Brower GL, Janicki JS. Pharmacologic inhibition of mast cell degranulation prevents left ventricular remodeling induced by chronic volume overload in rats. J Card Fail 2005;11:548–556.
- 71. Athanassopoulos P, Balk AHMM, Vaessen LMB, Caliskan K, Takkenberg JJM, Weimar W, Bogers AJJC. Blood dendritic cell levels and phenotypic characteristics in relation to etiology of end-stage heart failure: implications for dilated cardiomyopathy. Int J Cardiol 2009;131:246–256.
- 72. Molawi K, Wolf Y, Kandalla PK, Favret J, Hagemeyer N, Frenzel K, Pinto AR, Klapproth K, Henri S, Malissen B, Rodewald H-R, Rosenthal NA, Bajenoff M, Prinz M, Jung S, Sieweke MH. Progressive replacement of embryo-derived cardiac macrophages with age. J Exp Med 2014;211:2151–2158.
- 73. Sadoshima J. Myocardin-related transcription factor A in macrophages mediates pathological hypertrophy. Cardiovasc Res 2022;118:662–663.
- 74. Liu L, Zhao Q, Kong M, Mao L, Yang Y, Xu Y. Myocardin-related transcription factor A regulates integrin beta 2 transcription to promote macrophage infiltration and cardiac hypertrophy in mice. Cardiovasc Res 2022;118:844–858.
- 75. Vredevoe DL, Widawski M, Fonarow GC, Hamilton M, Martínez-Maza O, Gage JR. Interleukin-6 (IL-6) expression and natural killer (NK) cell dysfunction and anergy in heart failure. Am J Cardiol 2004;93:1007–1011.
- 76. García-Rivas G, Castillo EC, Gonzalez-Gil AM, Maravillas-Montero JL, Brunck M, Torres-Quintanilla A, Elizondo-Montemayor L, Torre-Amione G. The role of B cells in heart failure and implications for future immunomodulatory treatment strategies. ESC Heart Fail 2020;7:1387–1399.
- 77. Hinz B. Myofibroblasts. Exp Eye Res 2016;142:56–70.
- 78. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci 2014;71:549–574.
- 79. Ren Z, Yu P, Li D, Li Z, Liao Y, Wang Y, Zhou B, Wang L. Single-cell reconstruction of progression trajectory reveals intervention principles in pathological cardiac hypertrophy. Circulation 2020;141:1704–1719.
- 80. Frangogiannis NG. Cardiac fibrosis: cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med 2019;65:70–99.
- 81. Gibb AA, Lazaropoulos MP, Elrod JW. Myofibroblasts and fibrosis. Circ Res 2020;127:427–447.
- 82. Kurose H. Cardiac fibrosis and fibroblasts. Cells 2021;10:1716.
- 83. Davis J, Molkentin JD. Myofibroblasts: trust your heart and let fate decide. J Mol Cell Cardiol 2014;70:9–18.
- 84. Díez J, de Boer RA. Management of cardiac fibrosis is the largest unmet medical need in heart failure. Cardiovasc Res 2022;118:e20–e22.
- 85. Oka T, Akazawa H, Naito AT, Komuro I. Angiogenesis and cardiac hypertrophy. Circ Res 2014;114:565–571.
- 86. Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, Tallquist MD. Revisiting cardiac cellular composition. Circ Res 2016;118:400–409.
- 87. Segers VFM, Brutsaert DL, De Keulenaer GW. Cardiac remodeling: endothelial cells have more to say than just NO. Front Physiol 2018;9:382.
- 88. Gogiraju R, Bochenek ML, Schafer K. Angiogenic endothelial cell signaling in cardiac hypertrophy and heart failure. Front Cardiovasc Med 2019;6:20.
- 89. Ottaviani L, Juni RP, de Abreu RC, Sansonetti M, Sampaio-Pinto V, Halkein J, Hegenbarth JC, Ring N, Knoops K, Kocken JMM, Jesus Cd, Ernault AC, el Azzouzi H, Rühle F, Olieslagers S, Fernandes H, Ferreira L, Braga L, Stoll M, Nascimento DS, de Windt LJ, da Costa Martins PA. Intercellular transfer of miR-200c-3p impairs the angiogenic capacity of cardiac endothelial cells. Mol Ther 2022;30:2257–2273.
- 90. Aluru JS, Barsouk A, Saginala K, Rawla P, Barsouk A. Valvular heart disease epidemiology. Med Sci (Basel) 2022;10:32.
- 91. Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction. J Am Coll Cardiol 2013;62:263–271.
- 92. Otto CM, Nishimura RA, Bonow RO, Carabello BA, Erwin III JP, Gentile F, Jneid H, Krieger EV, Mack M, McLeod C, O’Gara PT. ACC/AHA guideline for the management of patients with valvular heart disease: executive summary: a report of the American College of Cardiology/American Heart Association Joint Committee on clinical practice guidelines. Circulation 2021;143:e35–e71.
- 93. European_Commission. European 3Rs Centres. EU Science Hub; 2023. https://joint-research-centre.ec.europa.eu/eu-reference-laboratory-alternatives-animal-testing-eurl-ecvam (March 2023, date last accessed).
- 94. Hickman DL, Johnson J, Vemulapalli TH, Crisler JR, Shepherd R. Commonly used animal models. In: Principles of animal research for graduate and undergraduate students. Cambridge, Massachusetts, USA: Academic Press; 2017. p117–175.
- 95. Greenberg MJ, Daily NJ, Wang A, Conway MK, Wakatsuki T. Genetic and tissue engineering approaches to modeling the mechanics of human heart failure for drug discovery. Front Cardiovasc Med 2018;5:120.
- 96. Vuorenpää H, Björninen M, Välimäki H, Ahola A, Kroon M, Honkamäki L, Koivumäki JT, Pekkanen-Mattila M. Building blocks of microphysiological system to model physiology and pathophysiology of human heart. Front Physiol 2023;14:1213959.
- 97. Riehle C, Bauersachs J. Small animal models of heart failure. Cardiovasc Res 2019;115:1838–1849.
- 98. Geens J, Trenson S, Rega FR, Verbeken EK, Meyns BP. Ovine models for chronic heart failure. Int J Artif Organs 2009;32:496–506.
- 99. Breisch EA, Bove AA, Phillips SJ. Myocardial morphometrics in pressure overload left ventricular hypertrophy and regression. Cardiovasc Res 1980;14:161–168.
- 100. Dutta S, Sengupta P. Rabbits and men: relating their ages. J Basic Clin Physiol Pharmacol 2018;29:427–435.
- 101. Ishihara K, Zile MR, Tomita M, Tanaka R, Kanazawa S, Carabello BA. Left ventricular hypertrophy in a canine model of reversible pressure overload. Cardiovasc Res 1992;26:580–585.
- 102. Zhao Z, Chen L, Xiao Y-B, Hao J, Tang C-Z, Zheng D-Z. A rabbit model to study regression of ventricular hypertrophy. Heart Lung Circ 2013;22:373–382.
- 103. Yoshida T, Friehs I, Mummidi S, del Nido PJ, Addulnour-Nakhoul S, Delafontaine P, Valente AJ, Chandrasekar B. Pressure overload induces IL-18 and IL-18R expression, but markedly suppresses IL-18BP expression in a rabbit model. IL-18 potentiates TNF-α-induced cardiomyocyte death. J Mol Cell Cardiol 2014;75:141–151.
- 104. Zhang X, He X, Jing H, Luo K, Shi B, Zhu Z, Zheng J. Neonatal rabbit model for pressure-overloaded heart failure and preliminary exploration of mechanism. Ann Thorac Surg 2021;112:1537–1545.
- 105. Meerson FZ, Zaletayeva TA, Lagutchev SS, Pshennikova MG. Structure and mass of mitochondria in the process of compensatory hyperfunction and hypertrophy of the heart. Exp Cell Res 1964;36:568–578.
- 106. Freeman LM, Rush JE, Stern JA, Huggins GS, Maron MS. Feline hypertrophic cardiomyopathy: a spontaneous large animal model of human HCM. Cardiol Res 2017;8:139–142.
- 107. Tsigkas G, Katsanos K, Apostolakis E, Papadimitriou E, Koutsioumpa M, Kagadis GC, Koumoundourou D, Hahalis G, Alexopoulos D. A minimally invasive endovascular rabbit model for experimental induction of progressive myocardial hypertrophy. Hypertens Res 2016;39:840–847.
- 108. Ishikawa Y, Uechi M, Hori Y, Takashi E, Yamano S, Enomoto Y, Ugai J. Effects of enalapril in cats with pressure overload-induced left ventricular hypertrophy. J Feline Med Surg 2007;9:29–35.
- 109. Pollack PS, Bailey BA, Budjak R, Fernandez E, Houser SR. Progressive feline pressure-overload: noninvasive assessment correlates with abnormalities in single cells. Am J Physiol 1993;264(4 pt. 2):H1307–H1314.
- 110. Song L-S, Pi Y, Kim S-J, Yatani A, Guatimosim S, Kudej RK, Zhang Q, Cheng H, Hittinger L, Ghaleh B, Vatner DE, Lederer WJ, Vatner SF. Paradoxical cellular ca2+ signaling in severe but compensated canine left ventricular hypertrophy. Circ Res 2005;97:457–464.
- 111. Eaton DM, Berretta RM, Lynch JE, Travers JG, Pfeiffer RD, Hulke ML, Zhao H, Hobby ARH, Schena G, Johnson JP, Wallner M, Lau E, Lam MPY, Woulfe KC, Tucker NR, McKinsey TA, Wolfson MR, Houser SR. Sex-specific responses to slow progressive pressure overload in a large animal model of HFpEF. Am J Physiol Heart Circ Physiol 2022;323:H797–H817.
- 112. Gaasch WH, Zile MR, Hoshino PK, Apstein CS, Blaustein AS. Stress-shortening relations and myocardial blood flow in compensated and failing canine hearts with pressure-overload hypertrophy. Circulation 1989;79:872–883.
- 113. Keech GB, Smith AC, Swindle MM, Koide M, Carabello BA, DeFreyte G. An adult canine model of progressive left ventricular pressure overload. J Invest Surg 1997;10:295–304.
- 114. Bache RJ, Alyono D, Sublett E, Dai XZ. Myocardial blood flow in left ventricular hypertrophy developing in young and adult dogs. Am J Physiol 1986;251(5 pt. 2):H949–H956.
- 115. Walters EM, Prather RS. Advancing swine models for human health and diseases. Mo Med 2013;110:212–215.
- 116. Bikou O, Miyashita S, Ishikawa K. Methods in molecular biology. Methods Mol Biol 2018;1816:337–342.
- 117. Dixon JA, Spinale FG. Large animal models of heart failure: a critical link in the translation of basic science to clinical practice. Circ Heart Fail 2009;2:262–271.
- 118. Walters EM, Wells KD, Bryda EC, Schommer S, Prather RS. Swine models, genomic tools and services to enhance our understanding of human health and diseases. Lab Anim (NY) 2017;46:167–172.
- 119. Li X, Tan W, Li X, Zheng S, Zhang X, Chen H, Pan Z, Zhu C, Yang FH. A surgical model of heart failure with preserved ejection fraction in Tibetan minipigs. J Vis Exp 2022;180:e63526. doi:10.3791/63526.
- 120. Zaragoza C, Gomez-Guerrero C, Martin-Ventura JL, Blanco-Colio L, Lavin B, Mallavia B, Tarin C, Mas S, Ortiz A, Egido J. Animal models of cardiovascular diseases. J Biomed Biotechnol 2011;2011:497841.
- 121. Flanagan MF, Fujii AM, Colan SD, Flanagan RG, Lock JE. Myocardial angiogenesis and coronary perfusion in left ventricular pressure-overload hypertrophy in the young lamb. Evidence for inhibition with chronic protamine administration. Circ Res 1991;68:1458–1470.
- 122. Pabst R. The pig as a model for immunology research. Cell Tissue Res 2020;380:287–304.
- 123. Lunney JK. Advances in swine biomedical model genomics. Int J Biol Sci 2007;3:179–184.
- 124. Vatner DE, Homcy CJ, Sit SP, Manders WT, Vatner SF. Effects of pressure overload, left ventricular hypertrophy on beta-adrenergic receptors, and responsiveness to catecholamines. J Clin Invest 1984;73:1473–1482.
- 125. Longabaugh JP, Vatner DE, Vatner SF, Homcy CJ. Decreased stimulatory guanosine triphosphate binding protein in dogs with pressure-overload left ventricular failure. J Clin Invest 1988;81:420–424.
- 126. Walther T, Volkmar Falk C. Experimental aortic stenosis and corresponding left ventricular hypertrophy in sheep. J Invest Surg 2000;13:327–331.
- 127. Aoyagi T, Mirsky I, Flanagan MF, Currier JJ, Colan SD, Fujii AM. Myocardial function in immature and mature sheep with pressure-overload hypertrophy. Am J Physiol 1992;262(4 pt. 2):H1036–H1048.
- 128. Moorjani N. A pressure overload model to track the molecular biology of heart failure. Eur J Cardiothorac Surg 2003;24:920–925.
- 129. Moorjani N, Catarino P, Trabzuni D, Saleh S, Moorji A, Dzimiri N, Al-Mohanna F, Westaby S, Ahmad M. Upregulation of Bcl-2 proteins during the transition to pressure overload-induced heart failure. Int J Cardiol 2007;116:27–33.
- 130. Moorjani N, Ahmad M, Catarino P, Brittin R, Trabzuni D, Al-Mohanna F, Narula N, Narula J, Westaby S. Activation of apoptotic caspase cascade during the transition to pressure overload-induced heart failure. J Am Coll Cardiol 2006;48:1451–1458.
- 131. Quttainah M, Al-Hejailan R, Saleh S, Parhar R, Conca W, Bulwer B, Moorjani N, Catarino P, Elsayed R, Shoukri M, AlJufan M, AlShahid M, Ouban A, Al-Halees Z, Westaby S, Collison K, Al-Mohanna F. Progression of matrixin and cardiokine expression patterns in an ovine model of heart failure and recovery. Int J Cardiol 2015;186:77–89.
- 132. Casellas J, Vidal O, Pena RN, Gallardo D, Manunza A, Quintanilla R, Amills M. Genetics of serum and muscle lipids in pigs. Anim Genet 2013;44:609–619.
- 133. Modesti PA, Vanni S, Bertolozzi I, Cecioni I, Polidori G, Paniccia R, Bandinelli B, Perna A, Liguori P, Boddi M, Galanti G. Early sequence of cardiac adaptations and growth factor formation in pressure- and volume-overload hypertrophy. Am J Physiol Heart Circ Physiol 2000;279:H976–H985.
- 134. Zheng Y, Chan WX, Charles CJ, Richards AM, Lee LC, Leo HL, Yap CH. Morphological, functional, and biomechanical progression of LV remodelling in a porcine model of HFpEF. J Biomech 2022;144:111348.
- 135. Breisch EA, White FC, Nimmo LE, Bloor CM. Cardiac vasculature and flow during pressure-overload hypertrophy. Am J Physiol 1986;251(5 pt. 2):H1031–H1037.
- 136. Wang X, Hu Q, Mansoor A, Lee J, Wang Z, Lee T, From AHL, Zhang J. Bioenergetic and functional consequences of stem cell-based VEGF delivery in pressure-overloaded swine hearts. Am J Physiol Heart Circ Physiol 2006;290:H1393–H1405.
- 137. Gyöngyösi M, Pavo N, Lukovic D, Zlabinger K, Spannbauer A, Traxler D, Goliasch G, Mandic L, Bergler-Klein J, Gugerell A, Jakab A, Szankai Z, Toth L, Garamvölgyi R, Maurer G, Jaisser F, Zannad F, Thum T, Bátkai S, Winkler J. Porcine model of progressive cardiac hypertrophy and fibrosis with secondary postcapillary pulmonary hypertension. J Transl Med 2017;15:202.
- 138. Charles CJ, Lee P, Li RR, Yeung T, Ibraham Mazlan SM, Tay ZW, Abdurrachim D, Teo XQ, Wang WH, de Kleijn DP, Cozzone PJ. A porcine model of heart failure with preserved ejection fraction: magnetic resonance imaging and metabolic energetics. ESC Heart Fail 2020;7:92–102.
- 139. Xiong Q, Zhang P, Guo J, Swingen C, Jang A, Zhang J. Myocardial ATP hydrolysis rates in vivo: a porcine model of pressure overload-induced hypertrophy. Am J Physiol Heart Circ Physiol 2015;309:H450–H458.
- 140. Malo O, Carrier M, Shi YF, Tardif J-C, Tanguay J-F, Perrault LP. Specific alterations of endothelial signal transduction pathways of porcine epicardial coronary arteries in left ventricular hypertrophy. J Cardiovasc Pharmacol 2003;42:275–286.
- 141. Harper J, Harper E, Covell JW. Collagen characterization in volume-overload- and pressure-overload-induced cardiac hypertrophy in minipigs. Am J Physiol 1993;265(2 pt. 2):H434–H438.
- 142. van der Velden J, Asselbergs FW, Bakkers J, Batkai S, Bertrand L, Bezzina CR, Bot I, Brundel BJJM, Carrier L, Chamuleau S, Ciccarelli M, Dawson D, Davidson SM, Dendorfer A, Duncker DJ, Eschenhagen T, Fabritz L, Falcão-Pires I, Ferdinandy P, Giacca M, Girao H, Gollmann-Tepeköylü C, Gyongyosi M, Guzik TJ, Hamdani N, Heymans S, Hilfiker A, Hilfiker-Kleiner D, Hoekstra AG, Hulot J-S, Kuster DWD, van Laake LW, Lecour S, Leiner T, Linke WA, Lumens J, Lutgens E, Madonna R, Maegdefessel L, Mayr M, van der Meer P, Passier R, Perbellini F, Perrino C, Pesce M, Priori S, Remme CA, Rosenhahn B, Schotten U, Schulz R, Sipido KR, Sluijter JPG, van Steenbeek F, Steffens S, Terracciano CM, Tocchetti CG, Vlasman P, Yeung KK, Zacchigna S, Zwaagman D, Thum T. Animal models and animal-free innovations for cardiovascular research: current status and routes to be explored. Consensus document of the ESC working group on myocardial function and the ESC working group on cellular biology of the heart. Cardiovasc Res 2022;118:3016–3051.
- 143. Foinquinos A, Batkai S, Genschel C, Viereck J, Rump S, Gyöngyösi M, Traxler D, Riesenhuber M, Spannbauer A, Lukovic D, Weber N, Zlabinger K, Hašimbegović E, Winkler J, Fiedler J, Dangwal S, Fischer M, de la Roche J, Wojciechowski D, Kraft T, Garamvölgyi R, Neitzel S, Chatterjee S, Yin X, Bär C, Mayr M, Xiao K, Thum T. Preclinical development of a miR-132 inhibitor for heart failure treatment. Nat Commun 2020;11:633.
- 144. Täubel J, Hauke W, Rump S, Viereck J, Batkai S, Poetzsch J, Rode L, Weigt H, Genschel C, Lorch U, Theek C, Levin AA, Bauersachs J, Solomon SD, Thum T. Novel antisense therapy targeting microRNA-132 in patients with heart failure: results of a first-in-human phase 1b randomized, double-blind, placebo-controlled study. Eur Heart J 2021;42:178.