1. Introduction
Pulmonary arterial hypertension (PAH) remains a progressive and fatal disorder. Present treatments provide some relief of symptoms, but to date had limited effect upon long-term survival. Studies in animal models and human subjects have identified a number of signalling mechanisms that are dysregulated in the setting of PAH including hyperactive vasoconstriction and loss of vasodilation in general and nitric oxide (NO)-mediated vasodilation in particular., In some cases, a genetic mutation has been linked to human disease. Still, in most instances, the causative events remain unknown and this likely contributes to the delay in therapeutic progress.
The secreted matricellular protein thrombospondin-1 (TSP1) is thought to play a role in vascular health and disease. In the systemic vasculature, TSP1 modulates vascular response and at pathological levels promotes vascular dysfunction., In cells and animal models, overactive TSP1 signalling inhibits vasodilation in part by limiting NO production and signalling., In addition, TSP1 is reported to be involved in arteriosclerosis-associated vascular remodelling. In the lung, TSP1 has been reported to be important in the maintenance of homeostasis, and to inhibit cancer growth., Beyond these functions, a role for TSP1 in promoting pulmonary vasculopathy is now being appreciated. We and others have found that tsp1−/− mice are protected from hypoxia-mediated PAH., We also reported that TSP1 is up-regulated in lungs from PAH patients compared with non-PAH controls.,, However, the molecular mechanisms that regulate TSP1 in the lung are still unknown.
Hypoxia stimulates pulmonary vasoconstriction and, if chronic, causes hypertrophy of the medial layer of pulmonary arteries (PAs). In a feed-forward manner, vascular deterioration due to decreased blood flow through the lungs further exacerbates tissue hypoxia. Most responses to hypoxia are mediated through the induction of a specific gene expression programme regulated by a family of α/β heterodimeric transcription factors known as hypoxia-inducible factors (HIFs). Under normoxic conditions, HIFα subunits are unstable and their integrity is dependent on hydroxylation by oxygen-dependent enzymes and binding to the von Hippel-Lindau (VHL) protein, the substrate recognition component of an E3 ubiquitin ligase complex that targets HIF for proteosomal degradation., Of the three known alpha subunits, HIF-1α and HIF-2α have been the most studied. Although HIF-2α is abundantly expressed in the lung, studies in mutant mice suggest that both HIF-1α and HIF-2α are involved in the hypoxic adaptive process in the lung vasculature.,,, In heterozygous hif-1α+/− mice, hypoxia-induced vascular remodelling is decreased. Likewise, heterozygous hif-2α+/− mice did not develop pulmonary hypertension following prolonged hypoxia. Furthermore, dysregulation of the HIF pathway has been reported to promote pulmonary hypertension both in mouse models and in human patients with HIF-2α mutations., However, the molecular changes triggered by HIF are incompletely understood. It has been shown that hypoxia induces vascular cell expression of TSP1, while in tumour cells hypoxia decreases TSP1 levels by non-transcriptional mechanisms. Nonetheless, it is largely unknown how hypoxia regulates TSP1 in the lung, whether this occurs in an HIF-dependent manner, and if this regulation contributes to pulmonary vascular dysfunction and PAH.
We now report that hypoxia induces TSP1 in murine lungs and in human and murine pulmonary vascular and non-vascular cells. Using a murine model of constitutive hypoxia (induced by deletion of the vhl gene), we found increased levels of pulmonary TSP1. On the other hand, in mice mutated to lack both vhl and hif-2α, and in homozygote hif-2α−/− mice exposed to chronic hypoxia, TSP1 induction in the lung was reverted. In contrast, in mice mutated to lack both vhl and hif-1α, TSP1 induction was maintained. Furthermore, luciferase reporter assays demonstrated transcriptional activity of HIF-2α, but not HIF-1α, when it binds to hypoxia-response elements (HREs) close to the tsp1 promoter. Additionally, under hypoxia, increased levels of TSP1 accelerate fibroblast and pulmonary artery smooth muscle cell (PASMC) migration and destabilize endothelial cell–cell interactions. In functional studies with PAs from wild-type (WT) and tsp1−/− mice, TSP1 promoted endothelial dysfunction under hypoxia, in part by targeting specific voltage-gated channels. Finally, analysis of lungs from individuals with and without end-stage PAH found that both TSP1 and HIF-2α increased in the pulmonary vasculature of diseased lungs. Taken together, these studies provide novel mechanistic insights into the regulation of pulmonary TSP1 and its contribution to PAH.
2. Methods
2.1 Animals
Age-matched male C57BL/6 WT and tsp1−/− mice (stock numbers 000664 and 006141, respectively) were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Vhlfl/fl-UBC-Cre-ERT2, HIF-2αfl/fl-UBC-Cre-ERT2, and HIF-1αfl/fl-UBC-Cre-ERT2 mice were used to generate VHL (vhl−/−), or HIF-2α (hif-2α−/−) knockout mice, respectively, or double VHL/HIF-2α, VHL/HIF-1α knockout mice (vhl−/−/hif-2α−/−, vhl−/−/hif-1α−/−, respectively). The gene deletion procedure employed to generate these animals was previously described., To induce hypoxia in vivo, mice were placed in an airtight chamber with inflow and outflow valves, and infused with a mixture of 10% O2 and 90% N2 (Carburos Metálicos, Madrid, Spain). All mice in this work were sacrificed by first administering inhalation general anaesthesia (isoflurane 1.5%) followed by cervical dislocation. All studies were performed under the supervision of the Head of Animal Welfare and Health with a protocol approved by the Committee for Research and Ethics of the Universidad Autonoma of Madrid in accordance with the Spanish and European guidelines (Directive 2010/63/EU of the European Parliament).
2.2 Antibodies and reagents
The following reagents were employed: mouse anti-TSP1 clone A6.1 (Pierce, Alcobendas, Spain), TSP1 in human samples was detected with monoclonal anti-TSP1 ab1823 (Abcam, Cambridge, UK), HIF-2α was detected with anti-HIF-2α ab199 (mice) or ab73895 (human; Abcam), and HIF-1α was detected with polyclonal anti-HIF-1α C-term (Cayman Chemical Company, Ann Arbor, MI, USA), or monoclonal anti-HIF-1-a (610958, BD Biosciences, human samples) anti-Vinculin hVIN-1 (Sigma-Aldrich, Tres Cantos, Spain), anti-α-Tubulin T6199 (Sigma-Aldrich), anti-β-Actin (Cell Signaling Technology, Danvers, MA, USA), and rabbit anti-ZO-1 (Thermo Fisher Scientific, Alcobendas, Spain). Secondary antibodies were anti-IgG + IgM of mouse and rabbit conjugated with Peroxidase (Pierce), as well as goat anti-rabbit and goat anti-mouse antibodies conjugated with Alexa Fluor 488 (Invitrogen, Alcobendas, Spain). Alexa Fluor 568 phalloidin (Life Technologies, Alcobendas, Spain). TSP1 from human platelets was obtained from (Athens Research and Technology, Athens, GA, USA).
2.3 Cell culture
Primary murine pulmonary artery smooth muscle cells (mPASMCs) were obtained from 8- to 10-week-old male C57BL/6 WT or tsp1−/− mice. Mice were sacrificed as previously described and flushed with sterile PBS to remove blood, and lungs were then extracted under sterile conditions. PAs were carefully dissected and the adventitia was removed under a dissecting microscope. Arteries were then cut into rings (1.8–2 mm length) and explanted in a 35 mm culture dish in DMEM with 20% FBS, penicillin (100 U/mL), streptomycin (100 U/mL), amphotericin B (100 µg/mL), HEPES (200 µg/mL), and Heparin 500× (1/500 mL media). Contaminating fibroblasts were separated from mPASMCs by taking advantage of differential adhesive ability. The cells migrated from the explants within 6–9 days and grew to confluence in ∼2 weeks. When explanted cells grew to confluence, they were plated on a 2% gelatin-coated culture plate, allowed to adhere for 30 min, during which contaminating fibroblasts attached to the plate. Non-attached mPASMCs were separated and re-plated. Cell purity was confirmed by immunostaining with mouse anti-SMA (clone 1A4, Dako, Carpinteria, CA, USA) and rabbit anti-Calponin (CNN1) EP798Y ab46794 (Abcam). Primary pulmonary fibroblasts (mFib) were isolated by enzymatic digestion with collagenase A from Clostridium histolyticum (Sigma-Aldrich). Briefly, mice were sacrificed as above and lungs were perfused with PBS, extracted, cut into small pieces, and then incubated with 3 mL of 2 mg/mL collagenase solution for 30 min. After digestion, cells were washed twice in DMEN with 10% FBS and then cultured in DMEM supplemented with 20% FBS, penicillin (100 U/mL), streptomycin (100 U/mL), and 1% HEPES buffer. Cells were grown for 2 days and then cultured for an additional 3 days in minimum media with 5% FBS to minimize contaminating endothelial or smooth muscle cells. Following this, cells were maintained in media with 20% FBS at 37°C and 5% CO2. Human pulmonary artery endothelial cells (hPAECs) and smooth muscle cells (hPASMCs) from ATCC (ATCC-PCS-100-022 or PCS-100-023, respectively) or Lonza (Allendale, NJ, USA) were cultured following manufacturer's recommended specifications. To induce hypoxia, cells were placed into an in vivo 2400 humidified hypoxia workstation (Ruskinn Technologies, Bridgend, UK) with 5% CO2 and 1% oxygen for the indicated time intervals. The human umbilical vein cell line EA.hy926 (ATCC, CRL-2922) was cultured in DMEN supplemented with 1% HAT (hypoxanthine–aminopterin–thymidine), 10% heat-inactivated FBS, 100 U/mL of penicillin and 100 µg/mL of streptomycin, and maintained in an atmosphere of 5% CO2 and 37°C.
2.4 HIF reporter in vitro assay
TSP1 HREs (HRE1: GGCGGCTGACGTCCCATCCCGAAGA and HRE2: CCAAGGCTGCGTGGGCGGGC ACCGA) were introduced (three copies in tandem for each HRE) in the luciferase reporter plasmid pGL4.23 vector (Promega, Alcobendas, Spain) between KpnI and HindIII, generating pGL4.23-HRE1 and pGL4.23-HRE2. As an HRE validated control, we used the HIF-responsive firefly luciferase reporter, expressing the luciferase gene under the control of nine copies in tandem of the VEGF HRE (p3EGR.Luc-9xHRE.VEGF). Renilla, pRL-SV40 Vector (Promega), was used as an internal control. In addition, to test HIF functional activity, we used retroviral vectors pRV-GFP encoding HIF-mutated constitutive active forms, HIF-2α (P-A)2 or HIF-1α (P-A)2, or a mutation lacking transcriptional activity HIF-1α(P-A)2Bhlh. Chinese Hamster Ovary cells (CHO.K1) were cultured in p24 plate at the 75% optimum confluence in 1% glucose DMEN, 10% FBS, penicillin (100 U/mL), and streptomycin (100 U/mL). Then, cells were transiently cotransfected with the pRV vectors (0.25 µg), luciferase vectors (0.25 µg), and Renilla (0.05 µg) using 2.5 µg/well of the transfection reagent jetPEI (Polyplus, Illkirth, France). After 24 h, reporter activity was determined using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. Firefly luciferase activity was normalized based on the Renilla luciferase activity and Luciferase activity was measured using the Glomax Multidetection system (Promega).
2.5 Human tissue
Control non-PAH and end-stage PAH lungs were obtained immediately following explantation under ongoing University of Pittsburgh IRB protocols (970946 and PRO14010265). Informed consent was given for the use of human samples and the study conformed to the principles outlined in the Declaration of Helsinki. Under sterile conditions and employing magnification, lung parenchyma and distal fifth-order PAs were dissected for further processing using a minimal ‘touch’ technique to prevent tissue injury.
2.6 Immunofluorescence
Cells were seeded onto fibronectin-coated 13-mm glass coverslips (5 μg/mL fibronectin) and then incubated in normoxia or hypoxia (1% O2) for 24 h. Afterwards, cells were fixed with 4% paraformaldehyde in PBS and permeabilized with 0.5% Triton in PBS with 1% BSA, 100 µg/mL of gamma globulin and 0.05% azide. Cells were blocked for 30 min with 5% BSA in PBS with 100 µg/mL of gamma globulin and 0.05% azide, and stained with the indicated primary antibodies followed by Alexa Fluor 488 or 568 labelled secondary antibodies or Alexa Fluor 568 phalloidin. DAPI (Sigma-Aldrich) to stain cell nuclei was used. Cells were mounted in Prolong Gold (Invitrogen) and imaged with a Leica fluorescence microscope 020-525.024 (Leica, Madrid, Spain). Images were collected using Leica TCS software. Focal adhesion (FA) contacts were quantified with ImageJ following the protocol described by Horzum et al.
2.7 Protein expression by western blot analysis
Lysates of snap-frozen lung tissues (murine and human) and isolated murine pulmonary cells were prepared in RIPA buffer [50 mM TRIS (pH 7.5), 1% NP-40, 1 mM EDTA, 125 mM NaCl, 0.25% sodium deoxycholate, 1 mM sodium orthovanadate, 1 mM sodium fluoride, and 1× phosphatase/protease inhibitors cocktail (Roche Applied Science, Hercules, CA, USA)]. Cell lysates were centrifuged at 17 000 × g for 20 min. A bicinchoninic acid assay (Bio-Rad, Life Sciences Research, Hercules, CA, USA) was used to quantify total protein. Lysates (30 μg/lane) mixed with 1× reducing Laemmli buffer (Bio-Rad) were boiled at 95°C for 5 min, electrophoretically separated on SDS–PAGE gels, and transferred onto nitrocellulose membranes (Bio-Rad). Blots were probed with primary antibody to the respective proteins and afterwards with HRP-conjugated secondary antibodies. Proteins were visualized with HRP substrate (Luminata Forte, Millipore, Madrid, Spain) on ImageQuant LAS 4000 (GE Healthcare Life Sciences, Madrid, Spain). Alternatively, human samples were blocked in Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE, USA), incubated overnight at 4°C with primary fluorescent-labelled antibodies, and visualized on an Odyssey Imaging System (Licor). The intensity of the bands was quantified using ImageQuant 5.2 or ImageJ (NIH, Bethesda, MD, USA).
2.8 RNA quantification by RT–PCR
Analysis of mRNA was performed by RT–PCR with StepOne Plus (Applied Biosystems, Carlsbad, CA, USA). Cells were grown to 90% confluence in 60 mm culture dishes, and total RNA was extracted from frozen lung tissues or cells using Hybrid-R™ (GeneAll Biotechnology, Co., Ltd) following the manufacturer's instructions. RNA (0.5 µg/sample) was reverse-transcribed to cDNA with MultiScribe RT of Gold RNA PCR core kit (Gene Amp, Foster City, CA, USA) and 1 µL of cDNA was amplified by RT–PCR using the StepOne Plus detection system and power SYBR green (Applied Biosystems). The primer pairs used to analyse tsp1 were designed to amplify exon 2 and 3 of the tsp1 sequence, which is missing in tsp1−/− mice (F: GGTGTCCTGTTCCTGTTGCA; R: CCGTTATCTCCCCCAGACTCT). Other primers used were: hprt (F: GTTAAGCAGTACAGCCCCAAA; R: AGGGCATATCCAACAACAA ACTT), phd3 (F: TGGACAACCCCAATGGTGAT; R: GCAGGACCCCTCCATGTAACT), β-actin (F: CGATGCCTGAGGCTCTTT; R: TGGATGCCACAGGATTCCA), Kv1.5 (F: CTGGGTCAGCAAGAGCCATT; R: TCAGGCAGAGTCTCCAAGCA), Human hif-1α (F: AGCCGAGGAAGAACTATGAACATAA; R: GTGGCCTGTGCAGTGCAA) and hif-2α (F: CTCATCCCTGCGACCATGA; R: TTCCCAAAACCAGAGCCATT).
2.9 siRNA-mediated gene silencing
siRNA experiments were carried out with specific pools of siRNAs directed against human TSP1, HIF-1α, or HIF-2α (Santa Cruz, Heidelberg, Germany) or with a non-targeted pool of control siRNAs (scr). Cells were transfected with Lipofectamine 2000 (Invitrogen), according to the manufacturer's instruction. Two days after transfection, cells were subjected to normoxia or hypoxia as indicated in each experiment.
2.10 Cell migration
Cells were serum-starved for 3 h and then allowed to migrate across Transwell filters (6.5 mm diameter, 8 µm pore size, Costar Corning, NY, USA) for 7 h at 37°C and 5% CO2 under normoxia or hypoxia (1% O2). As a chemoattractant DMEM with 20% FBS was added into the lower chamber, while basal media were used as a negative control. Non-migrating cells on the upper surface of the membrane were gently removed with Q-tips, while the migrating cells on the lower surface were fixed, stained with Diff-Quick (International Reagent, Kobe, Japan), and counted under the microscope at a magnification of ×10.
2.11 Transwell permeability assays
Transendothelial flux of FITC-dextran (molecular mass of 70 kDa; Sigma) was used as an index of endothelial paracellular permeability. hPAECs were seeded at passages 5–8 at a density of 2–3 × 104 cells/cm2 on Transwell polycarbonate filters, 6.5 mm diameter, 0.4 μm pore size (Costar Corning). FITC-dextran (10 mg/mL) (Sigma) in cell medium was added to the upper chambers of the Transwell system and the monolayers were then exposed for 6–7 h to normoxia or hypoxia (1% O2). Transfer of FITC-dextran across hPAEC monolayers was quantified after 1 h in 100 μL taken from the lower chamber. As a permeability control, we treated hPAEC monolayers with 1 mM EGTA, which alters intercellular junctions increasing the FITC-dextran flux across the cell monolayer. The fluorescence was measured with a spectrophotometer (FLUOstar Omega, BMG Labtech, Cheswick, PA, USA) using 480 and 515 nm as the excitation and emission wavelengths, respectively.
2.12 Vascular contractility
Murine PAs were carefully dissected free of surrounding tissue, cut into rings (1.8-2 mm length), and maintained for 16 h under normoxic or hypoxic (1% O2) conditions in basal medium [DMEN with penicillin (100 U/mL) and streptomycin (100 U/mL)]. Afterwards, vessel segments were mounted on a wire myograph in the presence of Krebs physiological solution. To maintain normoxic or hypoxic conditions, buffer solutions were continuously aerated with 21% O2, 5% CO2, and 74% N2 (pO2 = 17–19 kPa) or with 95% N2 and 5% CO2 (pO2 = 2.6–3.3 kPa), respectively, and stretched to a transmural pressure equivalent to 30 mmHg. Contractility was recorded by an isometric force transducer and a displacement device coupled with a digitalization and data acquisition system (PowerLab, Paris, France). To confirm smooth muscle viability arteries were first stimulated by raising the K+ concentration of the buffer to 80 mmol/L and then allowed to recover. Arteries were then stimulated with serotonin (5-HT, 10 µM) (Sigma) and treated with a concentration–response curve of the endothelium-dependent and independent vasodilators acetylcholine (Ach, 0.001–10 µmol/L) (Sigma) or sodium nitroprusside (SNP, 0.01–1000 nmol/L) (Sigma). In other experiments, vessels were pretreated with XE991 (0.3 µmol/L) or diphenyl phosphine oxide-1, DPO-1 (1 µmol/L), to inhibit Kv7 and Kv1.5 channels, respectively (Sigma).
2.13 Live cell calcium measurement
Calcium was measured in mPASMC 10 days after harvest from age-matched male WT and tsp1−/− mice. Cells were seeded on Lab-Tek 4 wells (Chambered Coverglass, Thermo Scientific). When cells reached 70% confluence, they were incubated for 16 h in normoxic or hypoxic (1% O2) conditions. They were then washed with 4.5 g/L of glucose supplemented Hank's Balanced Salt Solution (HBSS) and incubated at 37°C, 5% CO2 for 30 min with the cytosolic calcium probe Fluo-4 AM (Thermo Fisher) at 1 µmol/L in HBSS glucose. After incubation, the wells were washed and incubated for additional 30 min. Next, cells were time lapse photographed with an Leica SP5 Confocal Microscope every 7 s using a dry ×20 objective and illuminated with a 488 nm Ar laser using spectral filtering and hybrid (HyD) detectors. To interrogate the role of potassium Kv1.5 channels in this process, cells were treated with DPO-1 (2 μmol/L). Images were collected using Leica TCS software, and the increase in the fluorescence peak was quantified using ImageJ (NIH, Bethesda, MD, USA). As a control of calcium measurement, we quantified the fluorescence of cells treated with Ionomycin (1 µmol/L; Sigma-Aldrich).
2.14 Statistical analysis
The data are presented as the mean ± S.E.M. for all the studies. ANOVA followed by Bonferroni's post hoc test was used when comparing three or more groups and two-tailed Student's t-test was used to compare two groups, according with the conditions of normality and homoscedasticity. Shapiro–Wilk normality test and Brown–Forsythe test were used to analyse these conditions. In case the assumptions of normality and homoscedasticity were not accomplished, we used non-parametrical statistical tests; Mann–Whitney test to compare two groups and Kruskal–Wallis followed by Dunn's post hoc test to compare three or more groups. A P-value of <0.05 was considered significant. Additionally, we used a trend test in those experiments where we wanted to show changes along the time, P-trend <0.05 was considered significant.
3. Results
3.1 Hypoxia-mediated induction of pulmonary TSP1 parallels stabilization of HIF-2α
Previously we found that chronic hypoxia increases TSP1 expression in the lung. Extending these results, we found that WT mice subjected to hypoxia (10% O2) experience an increase in pulmonary TSP1 mRNA levels concurrent with induction of phd3, a known hypoxia-sensitive gene (Figure 1A). Analysis of TSP1 mRNA levels in tsp1−/− mice found no detectable levels of TSP1 message but demonstrated significant induction of phd3 under hypoxia (Figure 1A). To investigate the kinetics of this in vivo response, we performed a time course study. Analysis of protein levels in WT mice under hypoxia demonstrated a rapid time-dependent increase in TSP1 that was matched by parallel increase in the levels of HIF-2α (Figure 1B). As expected, tsp1−/− mice lungs showed no evidence of protein post-hypoxia (Figure 1B).
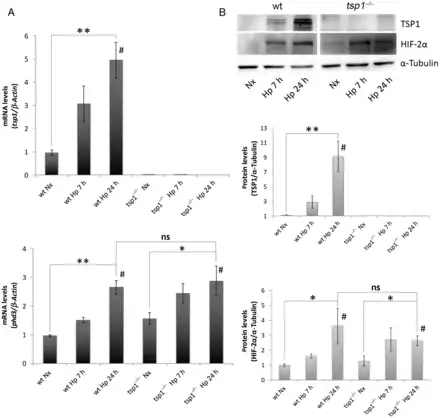
Figure 1
Hypoxia-mediated induction of pulmonary TSP1 parallels stabilization of HIF-2α. (A) Quantitative RT–PCR analysis was performed to determine TSP1 mRNA expression levels in lung samples from WT and tsp1−/− mice exposed to normoxia (Nx) or hypoxia (Hp) (10% O2) for the indicated time points. mRNA levels are expressed as fold change over WT in normoxic conditions and controlled with β-Actin as the housekeeping gene. The hypoxia reporter gene phd3 was analysed in the same samples as a control of hypoxic stress. Statistical analysis between different conditions was made using a murine type stratified one-way ANOVA test followed by Bonferroni's post hoc test, *P < 0.05, **P < 0.01; #P-trend < 0.05. Student's t-test was made between WT Hp 24 h and tsp1−/− Hp 24 h. Results are expressed as means ± S.E.M. (n = 4), ns (not significant). (B) Protein levels in lung samples from WT and tsp1−/− mice challenged with normoxia (Nx) or hypoxia (Hp) were detected via western blot probed against TSP1, HIF-2α, and α-Tubulin as a loading control. Quantification of TSP1 and HIF-2α bands was done by densitometry and controlled with α-Tubulin. Protein levels are expressed as fold change over WT in normoxic conditions. Statistical comparisons between different conditions were made using a murine type stratified Kruskal–Wallis followed by Dunn's post hoc test, *P < 0.05. **P < 0.01. #P-trend <0.05. Mann–Whitney test was made between WT Hp 24 h and tsp1−/− Hp 24 h. Results are expressed as means ± S.E.M. (n = 4), ns (not significant).
3.2 Hypoxia up-regulates TSP1 in pulmonary vascular and non-vascular cells
TSP1 is produced and secreted by systemic arterial VSMCs, fibroblasts, and endothelial cells. However, it is not known if mPASMC and endothelial cells, and pulmonary fibroblasts, produce TSP1 and whether this process is modulated by changes in oxygen tension. To test this, we isolated pulmonary fibroblasts (mFib) and mPASMC harvested from murine (WT and tsp1−/−) PAs and confirmed their linage by staining with specific markers for SMC, like α-SMA and CNN-1 (see Supplementary material online, Figure S1). Then, we challenged them with hypoxia (1% O2) for 24 h. Our results demonstrated that hypoxia significantly increased TSP1 mRNA levels in mFib and hPAECs, and stimulated a modest but not significant increase in mPASMC (Figure 2A). Accordingly, TSP1 protein levels were significantly increased in all murine and human (hPAEC and hPASMC) cell types after a hypoxic challenge (Figure 2B). To more precisely define the hypoxic induction of pulmonary TSP1, we employed fluorescent imaging of normoxic and hypoxic cells. IF imaging confirmed increased TSP1 expression in hypoxic mPASMC, mFib, and hPAEC that was localized to the perinuclear cytoplasm (Figure 2C).
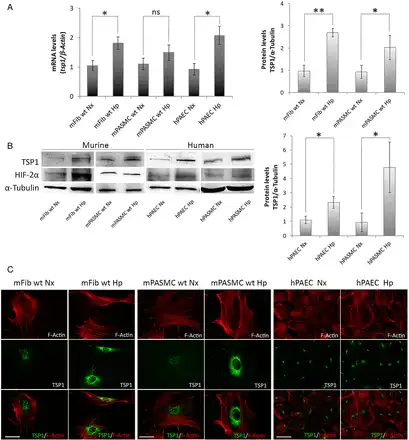
Figure 2
Hypoxia up-regulates TSP1 in pulmonary vascular and non-vascular cells. (A) Pulmonary murine fibroblasts (mFib) and murine PASMC (mPASMC) from WT, and human PAECs (hPAECs) were exposed to normoxia (Nx) or hypoxia (Hp) (1% O2) for 24 h and changes in mRNA levels determined by RT–PCR. TSP1 mRNA levels are expressed as fold change over normoxic conditions and controlled with β-Actin as the housekeeping gene. Average ± S.E.M. of n = 3 performed is shown. *P < 0.05, **P< 0.01. Student's t-test, ns (not significant). (B) Protein levels from mFib, mPASMC, hPAEC, and hPASMC exposed to normoxia (Nx) or hypoxia (Hp) (1% O2) for 24 h were detected by western blot probed against TSP1, HIF-2α, and α-Tubulin as a loading control. Densitometric analysis was performed to quantify TSP1 bands and levels were controlled with α-Tubulin and expressed as fold change over Nx. Average ± S.E.M. of n = 3 performed is shown. *P < 0.05. Student's t-test, ns (not significant). (C) Visualization of TSP1 (green) and F-Actin (red) in mFib, mPASMC, and hPAEC grown on fibronectin (5 μg/mL)-coated coverslips. Images shown are representative of three experiments. Bars, 50 μm.
3.3 HIF-2α regulates TSP1 levels in the lung
From the above studies, it was apparent that hypoxia rapidly up-regulated TSP1 transcript in the lung. Nonetheless, it remained unknown at what level HIF controlled TSP1 expression. Since both HIF-1α and HIF-2α null mice are resistant to pulmonary hypertension, as well as the tsp1−/− mice,, we wondered whether HIF-α activation was sufficient to produce TSP1 induction in the lung. To this aim, we generated mice deficient in the VHL protein. Mice lacking the VHL gene (vhl−/−) no longer process HIF-α protein for degradation under normoxia,, and therefore phenocopy hypoxic WT mice. Pulmonary TSP1 mRNA and protein levels in normoxic vhl−/− mice were significantly increased compared with the levels in normoxic WT mice (Figure 3A and B). To assess whether HIF-1α or HIF-2α was required for hypoxia-mediated induction of pulmonary TSP1, we generated mice in which both VHL and HIF-2α or VHL and HIF-1α were simultaneously inactivated (vhl−/−/hif-2α−/− or vhl−/−/hif-1α−/−, respectively) and analysed pulmonary TSP1 levels. In contrast to the induction observed in vhl−/− mice, pulmonary TSP1 mRNA and protein levels in vhl−/−/hif-2α−/− double knockout mice were decreased (Figure 3A and B), whereas in vhl−/−/hif-1α−/− TSP1 mRNA levels remained induced (Figure 3A). To confirm the role of HIF-2α in regulating pulmonary TSP1, we exposed hif-2α−/− mice to chronic hypoxia. As in double knockout mice, hypoxia did not up-regulate pulmonary TSP1 in hif-2α−/− mice (Figure 3B). To further extend these results to human lung, we analysed TSP1 levels in hPAEC and hPASMC following HIF-1α or HIF-2α interference. Interestingly, hypoxia-mediated induction of TSP1 mRNA was down-regulated in hPAEC treated with the HIF-2α siRNA (Figure 3C). When protein levels were analysed, we observed in hypoxia either no increase in TSP1 (hPAEC) or a decrease in TSP1 (hPASMC) following HIF-2α suppression (Figure 3D). Consistently, HIF-2α protein levels proved difficult to detect by western blot in hPASMC.
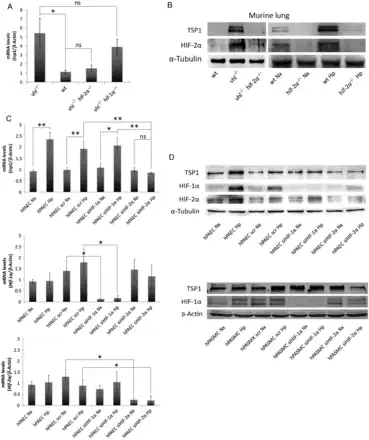
Figure 3
HIF-2α regulates TSP1 levels in the lung. (A) Quantitative RT–PCR analysis was performed to determine TSP1 mRNA expression levels in lung samples from WT, VHL deficient (vhl−/−), VHL/HIF-2α (vhl−/−/hif-2α−/−), and VHL/HIF-1α (vhl−/−/hif-1α−/−) double-deficient mice. mRNA levels are expressed as fold change over WT and controlled with β-Actin as the housekeeping gene. Average ± S.E.M. of n = 4 performed is shown. Statistical comparisons between different conditions were made using Kruskal–Wallis followed by a Dunn's post hoc test, *P < 0.05, ns (not significant). (B) Protein levels in lung samples from WT, vhl−/−, vhl−/−/hif-2α−/−, and hif-2α−/− mice under normoxia (Nx) or hypoxia (Hp) (10% O2) for 3 days were analysed by western blot. Representative images of three experiments are shown. (C) TSP1, HIF-1α, and HIF-2α mRNA expression levels were analysed in hPAEC untreated or transfected with scrambled (scr), HIF-1α or HIF-2α siRNA and then exposed to Nx or Hp (1% O2) for 24 h. Statistical comparisons between different conditions were made using a cell type stratified Mann–Whitney's test, *P < 0.05, **P < 0.01. Mann–Whitney's test was made between different cell types in Hp, *P < 0.05 was considered significant, n = 4. (D) Western blot of hPAEC and hPASMC, untreated or transfected with scrambled (scr), HIF-1α or HIF-2α siRNA and then exposed to Nx or Hp (1% O2) for 24 h, probed against TSP1, HIF-1α, HIF-2α, and α-Tubulin or β-Actin as a loading control. Representative images of five experiments are shown.
Extending these cell and animal studies, we performed western blot analysis of lung parenchyma and fifth-order PAs from individuals with (n = 15) and without PAH (n = 8). Paralleling results obtained in mice, we found up-regulation of pulmonary TSP1 protein in both parenchymal and PA samples from PAH compared with non-PAH individuals (Figure 4A and B). Interestingly though, HIF-2α as well as HIF-1α protein expression was increased in PA samples from PAH lungs, but decreased in parenchymal samples from the same (Figure 4A and B).
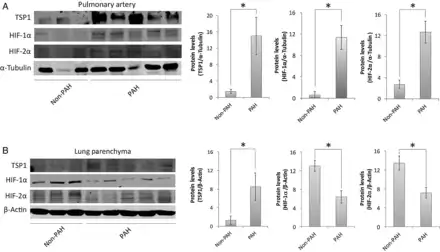
Figure 4
Lung samples of PAH patients. Western blot analysis of lysates of fifth-order PAs (A) and lung parenchyma (B) from non-PAH and PAH human lungs was performed against TSP1, HIF-1α, HIF-2α, and α-Tubulin or β-Actin as a loading control. Representative blots and densitometry (average ± S.E.M.) are presented as the mean ratio of target protein to α-Tubulin or β-Actin, respectively (n = 8 normal and 15 PAH samples); Mann–Whitney test corrected by Bonferroni's post hoc test was performed, *P < 0.05.
3.4 The proximal promoter region of TSP1 contains functional HREs
We analysed the proximal promoter region of TSP1 and identified two putative HREs between positions −1120 to −1196 (site 1) and −249 to −225 (site 2) relative to the transcription starting site. These sites contained the core RCGTG/C sequence and were selected based on the highest score corresponding to potential HRE sites published in the literature. Furthermore, these sites corresponded with open chromatin and transcription factors binding site clusters that are highly conserved among different mammalian species (Figure 5A). To establish a possible direct interaction of HIF with these putative TSP1 HRE regulatory sites, we performed luciferase reporter assays. We inserted TSP1 HRE site 1 or HRE site 2 with three copies in tandem in the luciferase reporter plasmid pGL4.23. CHO.K1 cells were cotransfected with these HREs and the constitutive active forms of HIF-1α or HIF-2α [HIF-1α (P-A)2, or HIF-2α (P-A)2, respectively]. To validate these vectors, we employed as a control the luciferase vector p3EGR.Luc bearing a well-known functional HRE of VEGF (see Supplementary material online, Figure S2). Reporter activity demonstrated an HIF-2α-mediated significant induction (4- and 18-fold in HRE site 1 and HRE site 2, respectively; Figure 5B). These results clearly indicate that HIF-2α interacts with both HRE, and on the other hand, this interaction reports functional activity. It is worth mentioning that HIF-2α displayed a higher reporter activity on TSP1 HRE site 2 compared with HRE site 1.
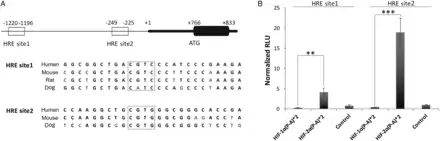
Figure 5
HIF-2α functionally binds to HREs of the tsp1 proximal promoter sequence. (A) Blast sequence alignment of the tsp1 proximal promoter sequence of different mammalian species containing conserved HRE sites (sites 1 and 2), and boxes mark core sequences of HRE sites. (B) Luciferase reporter activity assay of TSP1 HREs. Equal numbers of CHO.K1 cells were plated in 24-well plate (5 × 105 cells per well) and cotransfected with PGL4.23-3xHRE1 (HRE site 1) or PGL4.23-3xHRE2 (HRE site 2) and with pRV-GFP-HIF-1α(P-A)2 (HIF-1α(P-A)*2) or pRV-GFP-HIF-2α(P-A)2 (HIF-2α(P-A)*2) or pRV-GFP empty vector as a control. pRL-SV40 (Renilla) was included in all transfections as a luciferase internal control. Twenty-four h after cotransfection cells were lysed and analysed for luciferase activity. Results are expressed as means ± S.E.M. of relative light units (RLU) normalized to control. Statistical comparisons between different conditions were made using Kruskal–Wallis, followed by a Dunn's post hoc test, **P < 0.01, ***P < 0.001, n = 8.
3.5 TSP1 stimulates de-adhesion to promote hypoxia-mediated migration of pulmonary fibroblasts and PASMCs
In pulmonary vasculature activation, subsequent migration of fibroblasts and myofibroblasts into the medial layer of vessels have been suggested to contribute to vessel remodelling., Although TSP1 is known to promote migration of cells under normoxia, it is unknown if TSP1 controls the migratory activity of mFib and mPASMC under either normoxia or hypoxia. To assess this, we tested in vitro cell migration with the classic transwell assay. mFib and mPASMC harvested from WT and tsp1−/− mice were incubated under normoxia or hypoxia (1% O2) for 7 h and migration determined. In response to normoxia, both WT and tsp1−/− mPASMC and mFib displayed similar migratory capacity (Figure 6A and B). In contrast, under hypoxic conditions, WT mFib displayed significantly greater migratory response compared with tsp1−/− cells (Figure 6A). Similarly, hypoxia increased migration in mPASMC from WT, but not from tsp1−/−, mice (Figure 6B).
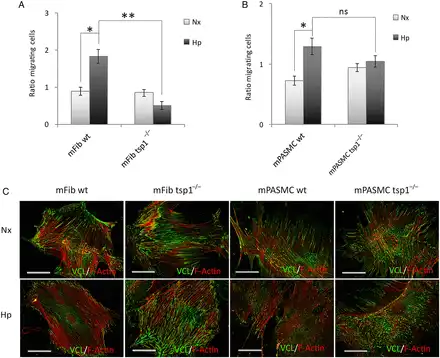
Figure 6
TSP1 stimulates de-adhesion to promote hypoxia-mediated migration of mFib and mPASMC. Migration of mFib (A) and mPASMC (B) from WT and tsp1−/− mice was assessed in transwell assays. Cells (12 × 103 cells/well) were serum-starved for 3 h and then allowed to migrate for 7 h at 37°C and 5% CO2 under normoxia (Nx) or hypoxia (Hp) (1% O2). As chemoattractant, DMEM with 20% FBS was added into the lower chamber, and basal media was used as a negative control. The number of cells migrated are represented as fold ± S.E.M. over WT under normoxic conditions. Statistical comparisons between different conditions were made using a murine stratified Student's t-test, *P < 0.05 **P < 0.01. Student's t-test was made between WT Hp 24 h and tsp1−/− Hp 24 h, *P < 0.05 was considered significant, n = 4, ns (not significant). (C) Visualization of cell adhesion plaques. mFib and mPASMC grown on fibronectin (5 μg/ml)-coated coverslips were cultured under Nx or Hp (1% O2) for 24 h, then fixed, permeabilized, and incubated with mAb to Vinculin (VCL), visualized with Alexa 488 (green), together with Alexa Fluor 568 phalloidin (red) to stain actin filaments (F-Actin). Images shown are representative of three experiments and at least 30 cells per condition. Bars, 50 μm.
FA disassembly promotes cell motility. Interestingly TSP1 is an intermediate of cell adhesion and stimulates disassembly of FA contacts. Consistent with this, hypoxic WT mFib and mPASMC exhibited reduced adhesion to fibronectin substrate, which correlated with a decrease in FA contacts, compared with cells from tsp1−/− mice (Figure 6C). Quantification of the percentage of total FA contacts per cell area, the average area of an individual FA contacts, and the number of FA contacts per cell area, all demonstrated increased adhesiveness of tsp1−/− mFib and mPASMC under hypoxic conditions, consistent with their demonstrated decreased migratory capacity (Table 1).

3.6 Hypoxia-mediated increase in TSP1 destabilizes pulmonary artery endothelial cell junctions and increases paracellular permeability
The above results indicated that TSP1 induces FA disassembly in mFib and mPASMC. Related to this, prior reports have shown that TSP1 also regulates intercellular junctions in PAEC. Therefore, we aimed to determine whether the hypoxia-mediated induction of TSP1 could also influence endothelial cell function. To this aim, we transfected hPAEC with a control or siRNA against TSP1 and then cultured them in normoxia or hypoxia and conducted permeability studies in the absence or presence of exogenous TSP1. As predicted, hPAEC treated with the TSP1-targeting siRNA demonstrated significantly less TSP1 protein following hypoxia compared with cells treated with the scrambled control siRNA or untreated cells (Figure 7A). As a permeability control, we treated hPAEC monolayers with 1 mM EGTA (see Supplementary material online, Figure S2). Interestingly, hypoxia and exogenous TSP1 (20 µg/mL) both increased cell permeability (Figure 7B). Conversely, knockdown of TSP1 blocked the hypoxic-mediated increase in cell permeability (Figure 7B). To inquire whether this was due to changes in intercellular junctions, we performed immunofluorescent staining of an essential constituent of tight junctions, the protein Zonulin-1 (ZO-1) in these cells. Interestingly, hPAEC transfected with the control siRNA under hypoxia displayed an irregular immunofluorescent staining pattern of ZO-1 distribution. However, treating the cells with a TSP1 siRNA ameliorated the hypoxia-mediated dysregulation of ZO-1 (Figure 7C).
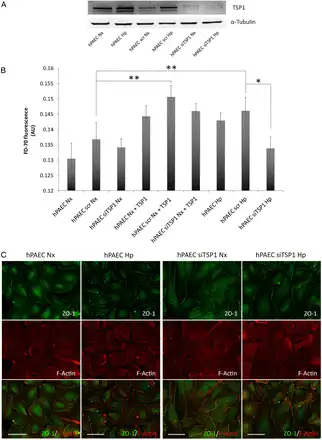
Figure 7
Hypoxia-mediated increase in TSP1 destabilizes hPAEC junctions and increases paracellular permeability. hPAECs were untreated or transfected with scrambled (scr) or specific TSP1 siRNA (siTSP1) and 24 h after transfection cells were exposed to normoxia (Nx) or hypoxia (Hp) (1% O2). (A) Analysis of TSP1 and α-Tubulin protein levels by western blot, and images shown are representative of four experiments. (B) hPAEC monolayers were exposed to normoxia (Nx) or hypoxia (Hp) (1% O2) or treated with TSP1 exogenous (20 µg/mL) for 7 h, and flux of FITC-dextran 70 kDa (FD-70) across hPAEC monolayers for 1 h. Fluorescence was quantified with a spectrophotometer at 515 nm. Average ± S.E.M. of n = 6 performed is shown. Statistical comparisons between different conditions were made using a cell type stratified one-way ANOVA test followed by Bonferroni's post hoc test, *P < 0.05, **P < 0.01. (C) IF of hPAEC with ZO-1-Alexa 488 (green) and Alexa Fluor 568 phalloidin (red) to visualize actin filaments (F-Actin). Two days after transfection cells were grown on fibronectin (2 μg/mL)-coated coverslips and cultured under Nx or Hp (1% O2) for 24 h. Images shown are representative of three experiments. AU, arbitrary units.
3.7 TSP1 limits hypoxia-mediated vascular responses in PAs
Vascular contraction and dilation are oxygen-sensitive processes that deteriorate under hypoxic conditions.In vivo, hypoxia promotes vasodilation of the systemic circulation and increased tissue perfusion, whereas in the pulmonary circulation acute hypoxia promotes vasoconstriction to limit perfusion of parenchyma that is less ventilated. We have reported that, under normoxia, systemic arterial blood flow in skeletal muscles and perfusion of skin flaps is limited by TSP1 basally, and in response to ischaemia–reperfusion injury.,,, However, it was not clear if endogenous TSP1 limited pulmonary arterial function under hypoxia. To investigate this, we challenged PA from male WT and tsp1−/− mice to SNP, a pro-drug metabolized by smooth muscle cells to NO, or Ach, an endothelial cell activator and stimulator of endogenous NO production, and assessed vasodilation under both normoxia and hypoxia (1% O2). Vasodilation induced by SNP under normoxic or hypoxic conditions was similar among WT and tsp1−/− PAs (see Supplementary material online, Figure S3). Similarly, under normoxia, WT and tsp1−/− PAs displayed comparable sensitivity to Ach (Figure 8A and B). However, endothelium-dependent vasodilation induced with Ach was significantly reduced in hypoxic WT PA. In contrast, hypoxic tsp1−/− PA maintained Ach sensitivity comparable to normoxic vessels (Figure 8A and B).
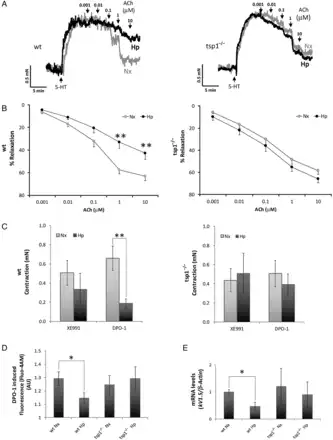
Figure 8
TSP1 limits hypoxia-mediated vascular responses in PAs. Vascular responses were analysed in endothelium-intact PAs from WT or tsp1−/− mice previously incubated for 16 h under normoxic (Nx) or hypoxic (Hp) (1% O2) conditions. Representative traces (A) and average values (B) of the ACh-induced relaxation (ACh) in serotonin (5-HT)-stimulated PAs. (C) Average values of the contraction induced by XE991 (0.3 µmol/L) and DPO-1 (1 µmol/L), Kv7 and Kv1.5 channels inhibitors, respectively. (D) Life cell calcium measurement with cytosolic calcium probe Fluo-4 AM. Average values of DPO-1-induced fluorescence (2 µmol/L) in mPASMC from WT or tsp1−/− mice previously incubated for 16 h under normoxic (Nx) or hypoxic (Hp) (1% O2) conditions. Statistical comparisons in (A–D) were made using two-way ANOVA, followed by Bonferroni's post hoc test; *P < 0.05, **P < 0.01. Results are expressed as means ± S.E.M. (n = 10). AU, arbitrary units. (E) Quantitative RT–PCR analysis was performed to determine Kv1.5 mRNA expression levels in mPASMC from WT or tsp1−/− mice under normoxia (Nx) or hypoxia (Hp) (1% O2) for 24 h. mRNA levels are expressed as fold change over WT in normoxic conditions and controlled with β-Actin as the housekeeping gene. Results are expressed as means ± S.E.M. Statistical comparisons between different conditions were made using a mice type stratified Student's t-test, *P < 0.05, n = 3.
3.8 TSP1 affects hypoxia-mediated down-regulation of Kv1.5
It is reported that hypoxia inhibits the function and expression of several Kv channels in mPASMC which results in membrane depolarization and leads to an increase in intracellular Ca2+ concentrations. Therefore, we tested the role of TSP1 on the modulatory effect of hypoxia on Kv channels and whether this affected intracellular calcium concentration. We analysed the contractile responses induced by Kv1.5 or Kv7 channel inhibitors in PA from WT or tsp1−/− mice exposed to normoxia or hypoxia. We found that the Kv7 channel inhibitor XE991 produced a similar degree of contraction under normoxia and hypoxia in both, WT and tsp1−/− PA (Figure 8C). Conversely, contraction mediated by the Kv1.5 channel inhibitor DPO-1 was markedly diminished in hypoxic, when compared with normoxic, WT PA (Figure 8C). Remarkably, hypoxic tsp1−/− PA treated with DPO-1 had no loss of vasoconstriction (Figure 8C). Next, we analysed changes in intracellular Ca2+, using cells treated with ionomycin, a calcium ionophore, as controls (see Supplementary material online, Figure S4). In agreement with the data in Figure 8C, the increase in intracellular Ca2+ induced by DPO-1 was attenuated in hypoxic when compared with normoxic WT mPASMC. Of note, this difference was not observed in tsp1−/− mPASMC (Figure 8D). Furthermore, hypoxia significantly decreased Kv1.5 mRNA levels in mPASMC harvested from lungs of WT mice, while Kv1.5 mRNA levels in cells from tsp1−/− mice were not altered by hypoxia (Figure 8E).
4. Discussion
Oxygen is essential for mammalian life and cells are well designed to rapidly alter gene expression profiles in response to changes in the partial pressure of oxygen. Hypoxia activates cellular sensing mechanisms focused on restoring oxygen to the hypoxic regions to maintain cell viability. Previous studies from human and animal models point to the family of HIF-1α and HIF-2α as important regulators in pulmonary vascular responses to both acute and chronic hypoxia.,,,, However, results from human subjects and mice models highlight the relevance of HIF-2α in PAH. Individuals with Chuvash polycythaemia, which is caused by a mutation in VHL, are found to develop PAH. In addition, pulmonary HIF-2α activity was found to be increased in a murine model of Chuvash polycythaemia, whereas loss of one copy of the HIF-2α gene was associated with less pulmonary hypertension in these animals. Not unexpectedly, HIF-2α gain-of-function mutations are associated with PAH.,, Moreover, we have seen that the arterial remodelling phenotype of vhl−/− was partially decreased in the vhl−/−/hif-2α−/− double null mice (unpublished data). Furthermore, erythropoietin (EPO), a downstream target of HIF-2α, is increased in the peripheral blood and endothelial cells from explanted lungs of end-stage PAH patients., In hPASMC hypoxia, via HIF-2α, increases expression of the transcription factor forkhead box M1 (FoxM1) to promote cell proliferation, and the downstream gene target octamer-binding transcription factor (Oct4).
We now find that TSP1 induction in vhl−/− mice, that have constitutive HIF activity and mimic chronic hypoxia, was only reverted in the absence of HIF-2α, but not when HIF-1α was eliminated, suggesting that HIF-2α is necessary for hypoxic-mediated pulmonary induction of TSP1. In human pulmonary vascular cells, hypoxia increased TSP1 mRNA and protein levels that reverted when HIF-2α was silenced. In addition, our in vitro luciferase reporter assays proved that a transcriptional mechanism mediated by HIF-2α-binding to HREs close to the tsp1 promoter was involved. Altogether, these results clearly indicate that HIF-2α induces TSP1 levels in hypoxic murine and human lungs.
Curiously, in mPASMC, hypoxic challenge resulted in a modest and non-significant increase in TSP1 transcript. Although the reasons for this remain to be determined, it is possible that cell viability was adversely altered under the serum-restricted conditions of the experiment. However, we cannot exclude that other HIF-independent events could also affect TSP1 protein levels in hypoxic pulmonary vascular cells. Finally, preliminary results in a cohort of human PAH and non-PAH samples suggest that both HIF-1α and HIF-2α might be involved in the regulation of TSP1 in human lungs, predominantly in the pulmonary vasculature. A more extensive investigation in human PAH will be required to confirm these initial findings.
Migration of PA fibroblasts and PASMC contributes to pulmonary arterial remodelling and luminal narrowing in PAH. Previous studies from our group found tsp1−/− systemic arterial smooth muscle cells deficient in PDGF-driven migration compared with WT cells. Consistent with this, we observed that, under hypoxia, mFib and mPASMC from tsp1−/− mice had decreased migration compared with cells from WT mice. These in vitro data predict an effect in vessel remodelling in hypoxic lungs, in keeping with our previous results demonstrating decreased pulmonary arterial remodelling in tsp1−/− mice following chronic hypoxia. Hypoxia-mediated increases in pulmonary TSP1 likely stimulated mFib and mPASMC migration, in part, through limiting adhesion in WT cells. In contrast, hypoxic tsp1−/− mFib and mPASMC demonstrated increased expression of FA contacts along with decreased migration. In addition, the pathogenesis of PAH also involves endothelial cell dysfunction that plays an integral role in mediating the structural changes in the pulmonary vasculature. New findings herein demonstrate that hypoxia-mediated induction of TSP1 levels contribute to increase endothelial permeability, mediated in part by changes in cell–cell adhesion. This, in fact, may facilitate PASMC migration through the endothelial barrier contributing to vessel remodelling in PAH.
Taken together, these data provide genetic evidence that TSP1 drives hypoxic pulmonary vascular remodelling. These findings also provide possible mechanistic insights into the previously reported finding that HIFs stimulate pulmonary fibroblast migration that, based on results presented, is mediated to some degree through the induction of TSP1.
At a functional level, we found that endothelial-dependent vasodilation elicited by ACh was impaired in hypoxic PA from WT mice. In contrast, hypoxic tsp1−/− PA retained sensitivity to Ach. In addition, the contraction and the increase in intracellular calcium induced by the Kv1.5 channel inhibitor DPO-1 were markedly decreased in hypoxic vs. normoxic WT PA, whereas tsp1−/− PAs were resistant to these DPO-1 effects. The reduced contraction to DPO-1 in hypoxic WT PAs is consistent with the down-regulation of Kv1.5 mRNA levels and with the decreased accumulation of intracellular calcium observed in mPASMC. These results are in agreement with studies showing reduced Kv1.5 channel activity and expression in cultured mPASMC,, and intact animals, exposed to chronic hypoxia. Down-regulation of Kv1.5 and other oxygen-sensitive Kv channels is associated with loss of acute hypoxia-mediated pulmonary vasoconstriction., As Kv1.5 channel expression is preserved in tsp1−/− mice, it is expected that hypoxic pulmonary vasoconstriction would be preserved rather than lost when these mice are exposed to chronic hypoxia. However, the mechanisms underlying the preservation of Kv1.5 channel activity in isolated PA from tsp1−/− mice remain unknown. Of some possible importance in this regard, we previously reported that endothelin receptor protein levels were down-regulated in lungs from tsp1−/− compared with WT mice. As endothelin-1 is known to inhibit Kv1.5 channels,, this raises the possibility that the TSP1 effects on Kv channel-mediated contraction in hypoxic PA could be mediated through endothelin-1. Alternatively, the resistance of tsp1−/− PA to hypoxia may be due to effects on other signalling moieties, including reactive oxygen species (ROS), that are increased in hypoxia and promote vasoconstriction. We have reported that TSP1 can directly activate NADPH oxidases in aortic vascular smooth muscle cells and renal tubular epithelial cells to increase superoxide production. It remains to be seen if TSP1 directly stimulates enzymatic ROS production in the pulmonary vasculature.
The present results show that hypoxia, in a HIF-2α-dependent manner, elicits an increase on TSP1 levels in tissues and pulmonary artery cells mediating structural changes in the pulmonary vasculature. Taken together, these new findings suggest multiple mechanisms through which TSP1 may promote PAH. As TSP1 has been found to be increased in several pulmonary diseases,,, our present findings likely have implications beyond PAH.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by the Instituto de Salud Carlos III (grant PI13/01866 and PIE 13/00041) and Red Cardiovascular RD12/0042/0065 (M.J.C.), and by NIH (grants P01 HL103455, R01 HL-108954, and 1R01HL112914-01A1) and American Heart Association grant 11BGIA7210001 (J.S.I.). This work was also supported by the Institute for Transfusion Medicine, the Hemophilia Center of Western Pennsylvania, and the Heart, Lung, Blood and Vascular Medicine Institute of the University of Pittsburgh (J.S.I.). Funding to pay the Open Access publication charges for this article was provided by the Instituto de Salud Carlos III (grant PI13/01866 from M.J.C).
Acknowledgements
We acknowledge Miguel Vicente-Manzanares, Luis Del Peso, and Hortensia de la Fuente Flores for their technical assistance and providing reagents. We also thank Lorena Vega Piris from the Methodology Unit (Instituto de Investigación Sanitaria Princesa) for her assistance with the statistical analysis and Alba Juanes García, Carlos García Briz, and Javier Sevilla Montero for experimental support and suggestions to this work.
Conflict of interest: J.S.I. is chair of the Scientific Advisory Boards of Vasculox, Inc. (St Louis, MO, USA) and Radiation Control Technologies, Inc. (Jersey City, NJ, USA) and holds equity interest in the same.
References
- 1. Zamanian RT, Kudelko KT, Sung YK, de Jesus Perez V, Liu J, Spiekerkoetter E. Current clinical management of pulmonary arterial hypertension. Circ Res 2014;115:131–147.
- 2. Grunig E, Dehnert C, Mereles D, Koehler R, Olschewski H, Bartsch P, Janssen B. Enhanced hypoxic pulmonary vasoconstriction in families of adults or children with idiopathic pulmonary arterial hypertension. Chest 2005;128:630S–633S.
- 3. Crosswhite P, Sun Z. Nitric oxide, oxidative stress and inflammation in pulmonary arterial hypertension. J Hypertens 2010;28:201–212.
- 4. Lakshminrusimha S, Wiseman D, Black SM, Russell JA, Gugino SF, Oishi P, Steinhorn RH, Fineman JR. The role of nitric oxide synthase-derived reactive oxygen species in the altered relaxation of pulmonary arteries from lambs with increased pulmonary blood flow. Am J Physiol Heart Circ Physiol 2007;293:H1491–H1497.
- 5. Best DH, Austin ED, Chung WK, Elliott CG. Genetics of pulmonary hypertension. Curr Opin Cardiol 2014;29:520–527.
- 6. Isenberg JS, Hyodo F, Pappan LK, Abu-Asab M, Tsokos M, Krishna MC, Frazier WA, Roberts DD. Blocking thrombospondin-1/CD47 signaling alleviates deleterious effects of aging on tissue responses to ischemia. Arterioscler Thromb Vasc Biol 2007;27:2582–2588.
- 7. Stenina OI, Krukovets I, Wang K, Zhou Z, Forudi F, Penn MS, Topol EJ, Plow EF. Increased expression of thrombospondin-1 in vessel wall of diabetic Zucker rat. Circulation 2003;107:3209–3215.
- 8. Isenberg JS, Hyodo F, Matsumoto K, Romeo MJ, Abu-Asab M, Tsokos M, Kuppusamy P, Wink DA, Krishna MC, Roberts DD. Thrombospondin-1 limits ischemic tissue survival by inhibiting nitric oxide-mediated vascular smooth muscle relaxation. Blood 2007;109:1945–1952.
- 9. Isenberg JS, Qin Y, Maxhimer JB, Sipes JM, Despres D, Schnermann J, Frazier WA, Roberts DD. Thrombospondin-1 and CD47 regulate blood pressure and cardiac responses to vasoactive stress. Matrix Biol 2009;28:110–119.
- 10. Roth JJ, Gahtan V, Brown JL, Gerhard C, Swami VK, Rothman VL, Tulenko TN, Tuszynski GP. Thrombospondin-1 is elevated with both intimal hyperplasia and hypercholesterolemia. J Surg Res 1998;74:11–16.
- 11. Lawler J, Sunday M, Thibert V, Duquette M, George EL, Rayburn H, Hynes RO. Thrombospondin-1 is required for normal murine pulmonary homeostasis and its absence causes pneumonia. J Clin Invest 1998;101:982–992.
- 12. Chen Y, Wang X, Weng D, Tao S, Lv L, Chen J. A TSP-1 functional fragment inhibits activation of latent transforming growth factor-beta1 derived from rat alveolar macrophage after bleomycin treatment. Exp Toxicol Pathol 2009;61:67–73.
- 13. Yamaguchi M, Sugio K, Ondo K, Yano T, Sugimachi K. Reduced expression of thrombospondin-1 correlates with a poor prognosis in patients with non-small cell lung cancer. Lung Cancer 2002;36:143–150.
- 14. Lee YJ, Koch M, Karl D, Torres-Collado AX, Fernando NT, Rothrock C, Kuruppu D, Ryeom S, Iruela-Arispe ML, Yoon SS. Variable inhibition of thrombospondin 1 against liver and lung metastases through differential activation of metalloproteinase ADAMTS1. Cancer Res 2010;70:948–956.
- 15. Bauer PM, Bauer EM, Rogers NM, Yao M, Feijoo-Cuaresma M, Pilewski JM, Champion HC, Zuckerbraun BS, Calzada MJ, Isenberg JS. Activated CD47 promotes pulmonary arterial hypertension through targeting caveolin-1. Cardiovasc Res 2012;93:682–693.
- 16. Ochoa CD, Yu L, Al-Ansari E, Hales CA, Quinn DA. Thrombospondin-1 null mice are resistant to hypoxia-induced pulmonary hypertension. J Cardiothorac Surg 2010;5:32.
- 17. Malenfant S, Neyron AS, Paulin R, Potus F, Meloche J, Provencher S, Bonnet S. Signal transduction in the development of pulmonary arterial hypertension. Pulm Circ 2013;3:278–293.
- 18. Antezana AM, Antezana G, Aparicio O, Noriega I, Velarde FL, Richalet JP. Pulmonary hypertension in high-altitude chronic hypoxia: response to nifedipine. Eur Respir J 1998;12:1181–1185.
- 19. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001;292:464–468.
- 20. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001;292:468–472.
- 21. Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, Beck H, Plaisance S, Dor Y, Keshet E, Lupu F, Nemery B, Dewerchin M, Van Veldhoven P, Plate K, Moons L, Collen D, Carmeliet P. Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med 2002;8:702–710.
- 22. Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT, Semenza GL. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest 1999;103:691–696.
- 23. Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, Carmeliet P. Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest 2003;111:1519–1527.
- 24. Tan Q, Kerestes H, Percy MJ, Pietrofesa R, Chen L, Khurana TS, Christofidou-Solomidou M, Lappin TR, Lee FS. Erythrocytosis and pulmonary hypertension in a mouse model of human HIF2A gain of function mutation. J Biol Chem 2013;288:17134–17144.
- 25. Shan F, Li J, Huang QY. HIF-1 alpha-induced up-regulation of miR-9 contributes to phenotypic modulation in pulmonary artery smooth muscle cells during hypoxia. J Cell Physiol 2014;229:1511–1520.
- 26. Gale DP, Harten SK, Reid CD, Tuddenham EG, Maxwell PH. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 alpha mutation. Blood 2008;112:919–921.
- 27. Hickey MM, Richardson T, Wang T, Mosqueira M, Arguiri E, Yu H, Yu QC, Solomides CC, Morrisey EE, Khurana TS, Christofidou-Solomidou M, Simon MC. The von Hippel-Lindau Chuvash mutation promotes pulmonary hypertension and fibrosis in mice. J Clin Invest 2010;120:827–839.
- 28. Phelan MW, Forman LW, Perrine SP, Faller DV. Hypoxia increases thrombospondin-1 transcript and protein in cultured endothelial cells. J Lab Clin Med 1998;132:519–529.
- 29. Bienes-Martinez R, Ordonez A, Feijoo-Cuaresma M, Corral-Escariz M, Mateo G, Stenina O, Jimenez B, Calzada MJ. Autocrine stimulation of clear-cell renal carcinoma cell migration in hypoxia via HIF-independent suppression of thrombospondin-1. Sci Rep 2012;2:788.
- 30. Elorza A, Soro-Arnaiz I, Melendez-Rodriguez F, Rodriguez-Vaello V, Marsboom G, de Carcer G, Acosta-Iborra B, Albacete-Albacete L, Ordonez A, Serrano-Oviedo L, Gimenez-Bachs JM, Vara-Vega A, Salinas A, Sanchez-Prieto R, Martin del Rio R, Sanchez-Madrid F, Malumbres M, Landazuri MO, Aragones J. HIF2alpha acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol Cell 2012;48:681–691.
- 31. Miro-Murillo M, Elorza A, Soro-Arnaiz I, Albacete-Albacete L, Ordonez A, Balsa E, Vara-Vega A, Vazquez S, Fuertes E, Fernandez-Criado C, Landazuri MO, Aragones J. Acute Vhl gene inactivation induces cardiac HIF-dependent erythropoietin gene expression. PLoS ONE 2011;6:e22589.
- 32. Aragones J, Jones DR, Martin S, San Juan MA, Alfranca A, Vidal F, Vara A, Merida I, Landazuri MO. Evidence for the involvement of diacylglycerol kinase in the activation of hypoxia-inducible transcription factor 1 by low oxygen tension. J Biol Chem 2001;276:10548–10555.
- 33. Kondo K, Kim WY, Lechpammer M, Kaelin WG Jr. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol 2003;1:E83.
- 34. Horzum U, Ozdil B, Pesen-Okvur D. Step-by-step quantitative analysis of focal adhesions. MethodsX 2014;1:56–59.
- 35. Marxsen JH, Stengel P, Doege K, Heikkinen P, Jokilehto T, Wagner T, Jelkmann W, Jaakkola P, Metzen E. Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-alpha-prolyl-4-hydroxylases. Biochem J 2004;381:761–767.
- 36. Yao M, Roberts DD, Isenberg JS. Thrombospondin-1 inhibition of vascular smooth muscle cell responses occurs via modulation of both cAMP and cGMP. Pharmacol Res 2011;63:13–22.
- 37. Mumby SM, Abbott-Brown D, Raugi GJ, Bornstein P. Regulation of thrombospondin secretion by cells in culture. J Cell Physiol 1984;120:280–288.
- 38. Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999;399:271–275.
- 39. Ortiz-Barahona A, Villar D, Pescador N, Amigo J, del Peso L. Genome-wide identification of hypoxia-inducible factor binding sites and target genes by a probabilistic model integrating transcription-profiling data and in silico binding site prediction. Nucleic Acids Res 2010;38:2332–2345.
- 40. Sartore S, Chiavegato A, Faggin E, Franch R, Puato M, Ausoni S, Pauletto P. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: from innocent bystander to active participant. Circ Res 2001;89:1111–1121.
- 41. Stenmark KR, Bouchey D, Nemenoff R, Dempsey EC, Das M. Hypoxia-induced pulmonary vascular remodeling: contribution of the adventitial fibroblasts. Physiol Res 2000;49:503–517.
- 42. Chandrasekaran L, He CZ, Al-Barazi H, Krutzsch HC, Iruela-Arispe ML, Roberts DD. Cell contact-dependent activation of alpha3beta1 integrin modulates endothelial cell responses to thrombospondin-1. Mol Biol Cell 2000;11:2885–2900.
- 43. Higuchi M, Kihara R, Okazaki T, Aoki I, Suetsugu S, Gotoh Y. Akt1 promotes focal adhesion disassembly and cell motility through phosphorylation of FAK in growth factor-stimulated cells. J Cell Sci 2013;126:745–755.
- 44. Murphy-Ullrich JE. The de-adhesive activity of matricellular proteins: is intermediate cell adhesion an adaptive state? J Clin Invest 2001;107:785–790.
- 45. Goldblum SE, Young BA, Wang P, Murphy-Ullrich JE. Thrombospondin-1 induces tyrosine phosphorylation of adherens junction proteins and regulates an endothelial paracellular pathway. Mol Biol Cell 1999;10:1537–1551.
- 46. Leach RM, Robertson TP, Twort CH, Ward JP. Hypoxic vasoconstriction in rat pulmonary and mesenteric arteries. Am J Physiol 1994;266:L223–L231.
- 47. Casey DP, Joyner MJ. Compensatory vasodilatation during hypoxic exercise: mechanisms responsible for matching oxygen supply to demand. J Physiol 2012;590:6321–6326.
- 48. Naeije R, Dedobbeleer C. Pulmonary hypertension and the right ventricle in hypoxia. Exp Physiol 2013;98:1247–1256.
- 49. Bauer EM, Qin Y, Miller TW, Bandle RW, Csanyi G, Pagano PJ, Bauer PM, Schnermann J, Roberts DD, Isenberg JS. Thrombospondin-1 supports blood pressure by limiting eNOS activation and endothelial-dependent vasorelaxation. Cardiovasc Res 2010;88:471–481.
- 50. Isenberg JS, Romeo MJ, Maxhimer JB, Smedley J, Frazier WA, Roberts DD. Gene silencing of CD47 and antibody ligation of thrombospondin-1 enhance ischemic tissue survival in a porcine model: implications for human disease. Ann Surg 2008;247:860–868.
- 51. Park WS, Firth AL, Han J, Ko EA. Patho-, physiological roles of voltage-dependent K+ channels in pulmonary arterial smooth muscle cells. J Smooth Muscle Res 2010;46:89–105.
- 52. Yao L, Nie X, Shi S, Song S, Hao X, Li S, Zhu D. Reciprocal regulation of HIF-1alpha and 15-LO/15-HETE promotes anti-apoptosis process in pulmonary artery smooth muscle cells during hypoxia. Prostaglandins Other Lipid Mediat 2012;99:96–106.
- 53. Smith TG, Brooks JT, Balanos GM, Lappin TR, Layton DM, Leedham DL, Liu C, Maxwell PH, McMullin MF, McNamara CJ, Percy MJ, Pugh CW, Ratcliffe PJ, Talbot NP, Treacy M, Robbins PA. Mutation of von Hippel-Lindau tumour suppressor and human cardiopulmonary physiology. PLoS Med 2006;3:e290.
- 54. Formenti F, Beer PA, Croft QP, Dorrington KL, Gale DP, Lappin TR, Lucas GS, Maher ER, Maxwell PH, McMullin MF, O'Connor DF, Percy MJ, Pugh CW, Ratcliffe PJ, Smith TG, Talbot NP, Robbins PA. Cardiopulmonary function in two human disorders of the hypoxia-inducible factor (HIF) pathway: von Hippel-Lindau disease and HIF-2alpha gain-of-function mutation. FASEB J 2011;25:2001–2011.
- 55. Rankin EB, Biju MP, Liu Q, Unger TL, Rha J, Johnson RS, Simon MC, Keith B, Haase VH. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest 2007;117:1068–1077.
- 56. Farha S, Asosingh K, Xu W, Sharp J, George D, Comhair S, Park M, Tang WH, Loyd JE, Theil K, Tubbs R, Hsi E, Lichtin A, Erzurum SC. Hypoxia-inducible factors in human pulmonary arterial hypertension: a link to the intrinsic myeloid abnormalities. Blood 2011;117:3485–3493.
- 57. Fijalkowska I, Xu W, Comhair SA, Janocha AJ, Mavrakis LA, Krishnamachary B, Zhen L, Mao T, Richter A, Erzurum SC, Tuder RM. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol 2010;176:1130–1138.
- 58. Raghavan A, Zhou G, Zhou Q, Ibe JC, Ramchandran R, Yang Q, Racherla H, Raychaudhuri P, Raj JU. Hypoxia-induced pulmonary arterial smooth muscle cell proliferation is controlled by forkhead box M1. Am J Respir Cell Mol Biol 2012;46:431–436.
- 59. Firth AL, Yao W, Remillard CV, Ogawa A, Yuan JX. Upregulation of Oct-4 isoforms in pulmonary artery smooth muscle cells from patients with pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 2010;298:L548–L557.
- 60. Barlassina C, Lanzani C, Manunta P, Bianchi G. Genetics of essential hypertension: from families to genes. J Am Soc Nephrol 2002;13(Suppl 3):S155–S164.
- 61. Isenberg JS, Calzada MJ, Zhou L, Guo N, Lawler J, Wang XQ, Frazier WA, Roberts DD. Endogenous thrombospondin-1 is not necessary for proliferation but is permissive for vascular smooth muscle cell responses to platelet-derived growth factor. Matrix Biol 2005;24:110–123.
- 62. Eul B, Rose F, Krick S, Savai R, Goyal P, Klepetko W, Grimminger F, Weissmann N, Seeger W, Hanze J. Impact of HIF-1alpha and HIF-2alpha on proliferation and migration of human pulmonary artery fibroblasts in hypoxia. FASEB J 2006;20:163–165.
- 63. Platoshyn O, Yu Y, Golovina VA, McDaniel SS, Krick S, Li L, Wang JY, Rubin LJ, Yuan JX. Chronic hypoxia decreases K(V) channel expression and function in pulmonary artery myocytes. Am J Physiol Lung Cell Mol Physiol 2001;280:L801–L812.
- 64. Wang J, Juhaszova M, Rubin LJ, Yuan XJ. Hypoxia inhibits gene expression of voltage-gated K+ channel alpha subunits in pulmonary artery smooth muscle cells. J Clin Invest 1997;100:2347–2353.
- 65. Wang J, Weigand L, Wang W, Sylvester JT, Shimoda LA. Chronic hypoxia inhibits Kv channel gene expression in rat distal pulmonary artery. Am J Physiol Lung Cell Mol Physiol 2005;288:L1049–L1058.
- 66. Hong Z, Weir EK, Nelson DP, Olschewski A. Subacute hypoxia decreases voltage-activated potassium channel expression and function in pulmonary artery myocytes. Am J Respir Cell Mol Biol 2004;31:337–343.
- 67. Pozeg ZI, Michelakis ED, McMurtry MS, Thebaud B, Wu XC, Dyck JR, Hashimoto K, Wang S, Moudgil R, Harry G, Sultanian R, Koshal A, Archer SL. In vivo gene transfer of the O2-sensitive potassium channel Kv1.5 reduces pulmonary hypertension and restores hypoxic pulmonary vasoconstriction in chronically hypoxic rats. Circulation 2003;107:2037–2044.
- 68. Reeve HL, Michelakis E, Nelson DP, Weir EK, Archer SL. Alterations in a redox oxygen sensing mechanism in chronic hypoxia. J Appl Physiol (1985) 2001;90:2249–2256.
- 69. Rainbow RD, Norman RI, Everitt DE, Brignell JL, Davies NW, Standen NB. Endothelin-I and angiotensin II inhibit arterial voltage-gated K+ channels through different protein kinase C isoenzymes. Cardiovasc Res 2009;83:493–500.
- 70. Whitman EM, Pisarcik S, Luke T, Fallon M, Wang J, Sylvester JT, Semenza GL, Shimoda LA. Endothelin-1 mediates hypoxia-induced inhibition of voltage-gated K+ channel expression in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 2008;294:L309–L318.
- 71. Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol 2006;91:807–819.
- 72. Csanyi G, Yao M, Rodriguez AI, Al Ghouleh I, Sharifi-Sanjani M, Frazziano G, Huang X, Kelley EE, Isenberg JS, Pagano PJ. Thrombospondin-1 regulates blood flow via CD47 receptor-mediated activation of NADPH oxidase 1. Arterioscler Thromb Vasc Biol 2012;32:2966–2973.
- 73. Yao M, Rogers NM, Csanyi G, Rodriguez AI, Ross MA, St Croix C, Knupp H, Novelli EM, Thomson AW, Pagano PJ, Isenberg JS. Thrombospondin-1 activation of signal-regulatory protein-alpha stimulates reactive oxygen species production and promotes renal ischemia reperfusion injury. J Am Soc Nephrol 2014;25:1171–1186.
- 74. Agarwal AR, Mih J, George SC. Expression of matrix proteins in an in vitro model of airway remodeling in asthma. Allergy Asthma Proc 2003;24:35–42.
- 75. Ide M, Ishii H, Mukae H, Iwata A, Sakamoto N, Kadota J, Kohno S. High serum levels of thrombospondin-1 in patients with idiopathic interstitial pneumonia. Respir Med 2008;102:1625–1630.
- 76. Smadja DM, Nunes H, Juvin K, Bertil S, Valeyre D, Gaussem P, Israel-Biet D. Increase in both angiogenic and angiostatic mediators in patients with idiopathic pulmonary fibrosis. Pathol Biol (Paris) 2014;62:391–394.