Introduction
Fatty acid oxidation defects (FAODs) are a group of genetic metabolic disorders which are inherited in an autosomal recessive manner. There is a disruption of the carnitine shuttle or the mitochondrial beta oxidation pathway due to the reduced or absent function of the transporter proteins or enzymes, leading to deficient energy production and accumulation of the intermediate metabolites upstream to the block or defect. The spectrum of clinical presentations ranges from recurrent acute hypoglycemic episodes with hepatic dysfunction in neonates to myopathy and cardiomyopathy in older children and adults. They are potentially fatal, and a high index of suspicion is needed to quickly identify the patients with fatty acid oxidation defects in order to manage the metabolic decompensations promptly.
Epidemiology
Numerous newborn screening (NBS) programs have been conducted amongst different populations through which the incidence and/or prevalence of all fatty acid oxidation defects have been reported. Marsden et al. observed that, in general, the combined incidence of all FAODs ranges from 0.9 to 15.2 per 100,000 (Marsden et al., 2021). The incidence of all FAODs is lower in Asian populations than in non-Asian populations, ranging from 0.9 to 4.9 per 100,000. However, as there are no population screening programs in India, the true incidence of fatty acid oxidation defects in the Indian population is not known at present.
Pathophysiology
The energy requirements of our body are met from metabolism of carbohydrates (40-60%), lipids (30-40%) and to a lesser extent from proteins (10-15%). The primary fuel for the brain is glucose which is derived from carbohydrates. Fatty acids are the primary fuel for the heart, skeletal muscle and liver (McGuinness et al., 2023). They are also the major fuel source during periods of prolonged fasting beyond 6 hours. The hepatic glycogen stores are completely exhausted after 12 to 18 hours of fasting (Bender & Mayes, 2018). The lipolysis of the stored triacylglycerols, the body's main fuel reserve, in the adipose tissue releases free fatty acids which are taken up by the liver and further oxidized by beta oxidation to form acetyl coenzyme A. Acetyl CoA, thus formed, may enter the citric acid cycle to be oxidized to carbon dioxide and water, may enter the cholesterol synthesis pathway or may be used to synthesize ketone bodies. Fatty acids are the main metabolic fuels during inter-prandial periods and during periods of metabolic stress like infections and exercise (Houten & Wanders, 2010).
Fatty acids in our body are derived from diet (predominantly, long chain fatty acids), from the adipose tissue (stored as triacylglycerols), and from lipogenesis (from carbohydrates and amino acids) as shown in Figure 1. Fatty acids can be classified into four types according to the length of their aliphatic carbon chain:
Short chain fatty acids (SCFA) are fatty acids with aliphatic tails of 2-6 carbon atoms.
Medium chain fatty acids (MCFA) are fatty acids with aliphatic tails of 6-12 carbon atoms.
Long chain fatty acids (LCFA) are fatty acids with aliphatic tails of 14-20 carbon atoms.
Very long chain fatty acids (VLCFA) with aliphatic tails of 22 or more carbon atoms.
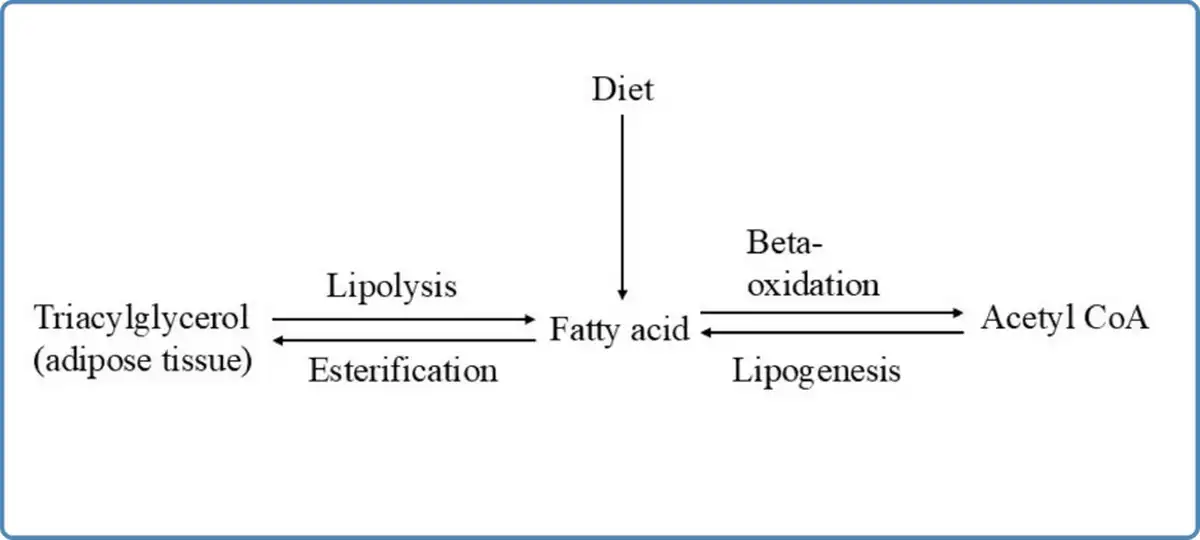
Figure 1
Overview of fatty acid metabolism
The oxidation of fatty acids is a biochemical process through which the fatty acids are broken down into smaller molecules during which energy is generated. There are different types of fatty acid oxidation occurring in different cellular organelles namely, alpha oxidation, beta oxidation and omega oxidation. Alpha oxidation helps in the degradation of phytanic acid, the by-product of green vegetables consumed in the diet. It occurs in the peroxisomes to release carbon dioxide. Beta oxidation of fatty acids occurs both in the mitochondria and the peroxisomes. Beta oxidation of short chain, medium and long chain fatty acids occurs in the mitochondria whereas the beta oxidation of very long chain fatty acids occurs in the peroxisomes of the cells. Omega oxidation is an alternative pathway to beta oxidation occurring in the endoplasmic reticulum, to degrade large fatty acid molecules which would be otherwise toxic to the body in higher concentrations. These water insoluble fatty acids are hydroxylated to water soluble dicarboxylic acids which are then excreted in the urine.
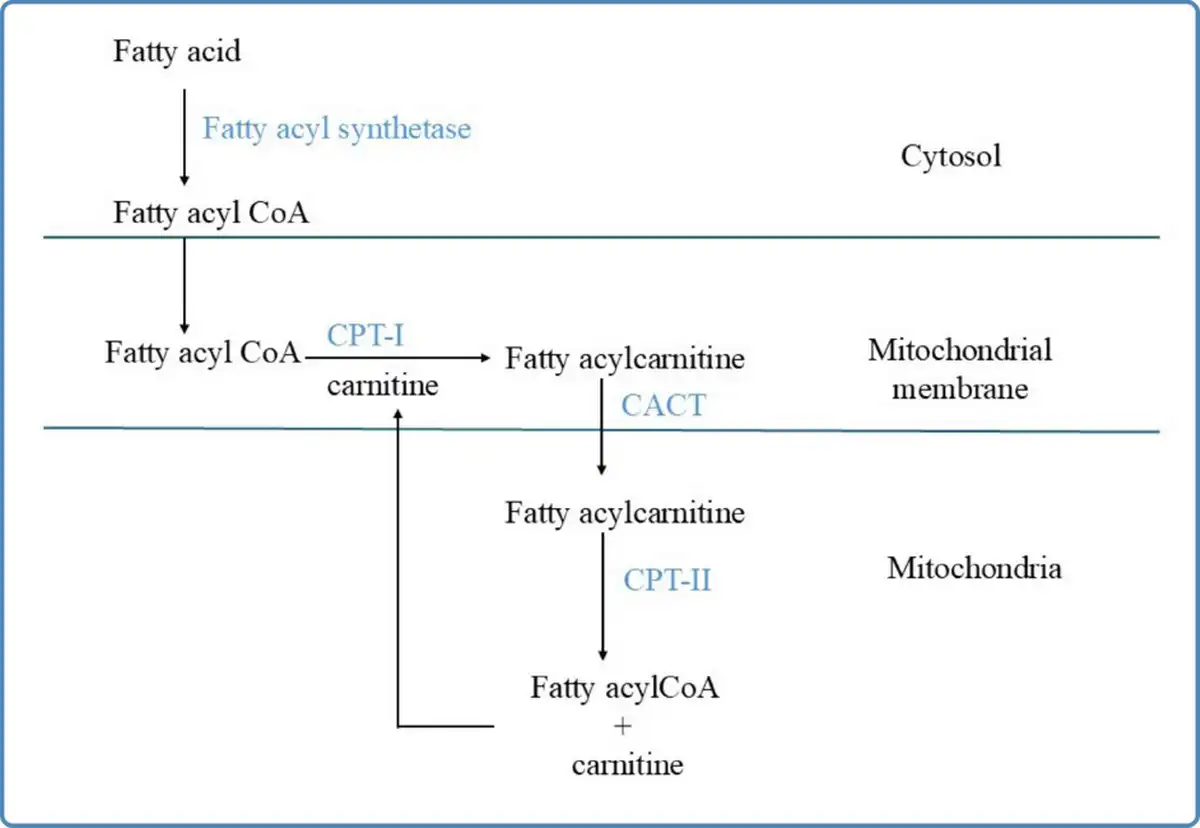
Figure 2
Schematic representation of the carnitine shuttle. CPT-I: Carnitine palmitoyl transferase-I; CPT-II: Carnitine palmitoyl transferase-II;CACT: Carnitine acylcarnitine translocase
Fatty acids are transported in the bloodstream bound to albumin and lipoproteins (Kerner & Hoppel, 2000). The transport of short chain and medium chain fatty acids across the plasma membrane occurs by passive diffusion and long chain fatty acids are transported via specific fatty acid transporter proteins (Guerra et al., 2022). After entry into the cytosol, the long chain fatty acids are activated to their specific acyl CoA esters by fatty acyl CoA synthetase, before their uptake into the mitochondrial matrix. The short and medium chain fatty acids diffuse passively across the mitochondrial membrane and are activated inside the mitochondrial matrix. The mitochondrial membrane is not permeable to long chain fatty acids. Hence, the carnitine shuttle is an obligatory pre-requisite for their uptake into the mitochondrial matrix (Houten & Wanders, 2010;Kerner & Hoppel, 2000). The carnitine shuttle needs proper functioning of three distinct membrane-bound proteins namely carnitine palmitoyl transferase-I (CPT-I), carnitine acylcarnitine translocase (CACT), and carnitine palmitoyl transferase- II (CPT-II). Carnitine palmitoyl transferase I (CPT-I) is located at the outer mitochondrial membrane and catalyzes the formation of acylcarnitine by addition of L-carnitine to the fatty acyl CoA esters. This is the rate-limiting step of beta oxidation and is regulated by the energy status of the cell. Malonyl CoA, formed during lipogenesis, is the inhibitor of this enzyme. The acylcarnitine, thus formed, is transported across the inner mitochondrial membrane in exchange for carnitine by CACT. Inside the mitochondrial matrix, CPT-II is located bound to the inner mitochondrial membrane and converts the acylcarnitine to fatty acyl CoA ester by releasing the carnitine, as shown in Figure 2. This carnitine is again recycled by its addition into the carnitine pool. The transport of carnitine across the plasma membrane into the cytosol is by organic cation/carnitine transporter 2 (OCTN2) (Tamai et al., 1998).
Inside the mitochondrial matrix, the activated acyl CoA esters are degraded into acetyl CoA, a two-carbon intermediate metabolite, through a cyclic process known as beta oxidation. Each cycle of beta oxidation comprises of four sequential steps through which one unit of acetyl CoA is formed, thus shortening the length of the fatty acyl CoA chain by two carbons (Houten & Wanders, 2010). The even chain fatty acids are completely broken down to form acetyl CoA molecules. The odd chain fatty acids are broken down to finally form propionyl CoA (three-carbon molecule) which is an intermediate product in the tricarboxylic acid cycle. The acetyl CoA formed in the skeletal muscle and the cardiac muscle enters the citric acid cycle for adenosine triphosphate (ATP) production, and that formed in the liver tissue is also supplied for the synthesis of ketone bodies.
There are four substrate-specific acyl CoA dehydrogenase enzymes specific to the chain length of fatty acyl CoA on which they act (Sim et al., 2002). However, there is some overlap between the substrates of each enzyme. The short chain acyl CoA dehydrogenase (SCAD) acts on substrates of chain length between C4 and C6, the medium chain acyl CoA dehydrogenase (MCAD) acts on substrates of chain length between C6 and C12, and the very long chain acyl CoA dehydrogenase (VLCAD) acts on substrates of chain length between C14 and C24. After the first step of dehydrogenation, hydrogenation of trans-2-enoyl CoA is carried out by the action of 2-enoyl CoA hydratase, which produces 3-hydroxyacyl CoA. 3-hydroxyacyl CoA is dehydrogenated by the action of 3-hydroxyacyl CoA dehydrogenase. The medium and short chain 3-hydroxyacyl CoA (chain length between C4 to C10) are dehydrogenated by medium/short chain 3-hydroxyacyl CoA dehydrogenase (M/SCHAD). The long chain 3-hydroxyacyl CoA dehydrogenase (LCHAD), which is present at the alpha subunit of the mitochondrial trifunctional protein (MTP) reduces the long chain hydroxy acyl CoA substrates (chain length between C12 to C16) (Guerra et al., 2022). The fourth and final step is the cleavage of 3-hydroxyacyl CoA to yield a molecule of acetyl CoA, catalyzed by 3-ketoacyl CoA thiolase. The medium chain 3-ketoacyl CoA thiolase (MCKT) acts on substrates of length between C4 and C12 whereas the long chain 3-ketoacyl CoA thiolase (LCKT) acts on long chain substrates. The long chain enoyl CoA hydratase and long chain 3-hydroxy acyl CoA dehydrogenase and the long chain 3-ketoacyl CoA thiolase are a part of the mitochondrial trifunctional protein.
Classification
Fatty acid oxidation defects can be broadly classified as carnitine shuttle defects, beta oxidation defects, and defects of electron transfer.
Carnitine shuttle defects are subclassified, based on the defect in the transporter protein involved, as:
Carnitine transporter deficiency (CTD)/ Systemic primary carnitine deficiency
Carnitine palmitoyl transferase 1A (CPT1A) deficiency
Carnitine-acylcarnitine translocase (CACT) deficiency
Carnitine palmitoyl transferase II (CPT II) deficiency
Beta-oxidation defects can be subclassified, on the basis of the defect in the oxidation of specific acyl CoA chain, as:
Disorders of long chain fatty beta oxidation -
Very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency
Long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD) deficiency
Mitochondrial trifunctional protein deficiency (MTPD1 & MTPD2)
Disorders of medium chain fatty acid beta oxidation -
Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency
Disorders of short chain fatty acid beta oxidation -
Short-chain acyl-CoA dehydrogenase (SCAD) deficiency
3-hydroxyacyl-CoA dehydrogenase (HADH) deficiency
Defects of electron transfer includes:
Multiple acyl- CoA dehydrogenase (MAD) deficiency (also known as Glutaric aciduria II)
The pathological features seen in this group may be due to the intracellular accumulation of the fatty acids and/or their metabolites upstream to the defect/ block in the cycle and also due to metabolic decompensation secondary to deficient ATP production. Lack of energy supply disrupts the pathways of gluconeogenesis, ketogenesis and urea cycle leading the manifestations of the same in the affected individual.
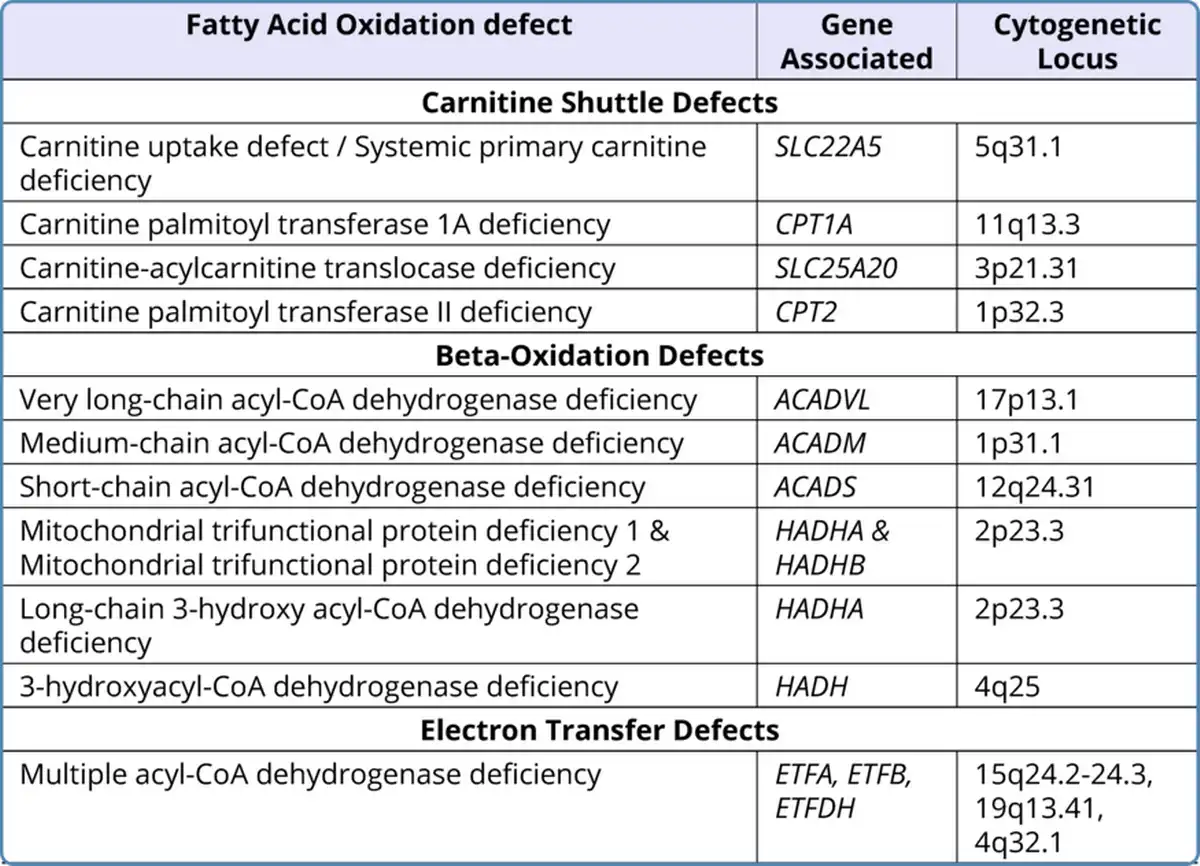
Table 1
Genetic Basis of Fatty Acid Oxidation Defects
Genetic Basis
All the defects in fatty acid oxidation were identified to be inherited in an autosomal recessive pattern and the presence of bi-allelic pathogenic variants (homozygous or compound heterozygous) in the associated gene is required for the manifestation of the disease in an individual. Heterozygous carriers are usually unaffected clinically. However, there are some exceptions to this. Heterozygous mothers carrying affected fetuses of long-chain 3-hydroxyacyl CoA dehydrogenase (LCHAD)/trifunctional protein (TFP)/CPT-1A deficiency have been observed to be at risk of developing acute fatty liver of pregnancy (AFLP) and HELLP syndrome (hemolysis, elevated liver enzymes and low platelets) (Spiekerkoetter et al., 2003;Karall et al., 2015;Innes et al., 2000). Table 1 sums up the genetic basis of the FAODs.
Clinical Manifestations
During fetal life, the main source of energy is from the continuous supply of glucose to the growing fetus from the maternal circulation. After birth, the neonate derives energy from the beta oxidation of fatty acids. These fatty acids are obtained from breast milk which predominantly contains long chain fatty acids. Hence, individuals with long-chain fatty acid oxidation defects present in the neonatal period. After weaning, during infancy, most of the fatty acids in the diet are medium-chain fatty acids. Hence, the symptoms of medium-chain fatty acid oxidation defects develop in late infancy after dietary transition from breastfeeding. Metabolic decompensations are precipitated by prolonged fasting, exercise, intercurrent illnesses and the patient may appear relatively well in the periods between these episodes (Morris & Spiekerkoetter, 2016).
Affected individuals may have broadly three main clinical presentations - hepatic manifestations, cardiac manifestations, and myopathy. Hepatic manifestations are due to hepatic dysfunction leading to Reye-like symptoms with acute hypoglycemic episodes with dullness of activity, lethargy, vomiting, seizures, encephalopathy and coma. This hypoglycemia is not associated with ketone body production and is of hypoketotic type. During the acute episode, the patient may have hepatomegaly with elevated transaminases and hyperammonemia. Liver biopsy, if done at this time, has been reported to show microvesicular lipid deposition, similar to Reye syndrome. In some patients, chronic liver dysfunction has also been seen.
Cardiac manifestations present with symptoms related to hypertrophic cardiomyopathy which may gradually progress to heart failure. The individual may also develop pericardial effusion, arrythmias and sudden death may occur due to the arrhythmias. Cardiac manifestations are predominantly seen with accumulation of long chain fatty acids in the cardiac muscle as they interfere with the ion channels and cause arrythmia.
Some individuals may present with muscle weakness, fatigue and muscle pain (myalgia). There may also be episodes of rhabdomyolysis with variable severity characterized by elevated serum creatine phosphokinase (CPK), myoglobinuria and acute renal failure (Morris & Spiekerkoetter, 2016).
A proportion of infant deaths certified as sudden infant death syndrome (SIDS) have been later attributed to fatty acid oxidation disorders (identified either postmortem or retrospectively after the diagnosis of an affected sibling). Majority of the cases have been linked to long chain fatty acid oxidation defects like MTP deficiency and LCHAD deficiency (Boles et al., 1998;Mathur et al., 1999).
Clinical manifestations of specific fatty acid oxidation defects
Carnitine Transporter Deficiency (CTD)
CTD, also known as 'Carnitine Uptake Defect' or 'Systemic Primary Carnitine Deficiency', occurs due to the abnormality in the transporter protein of carnitine in the cell membrane, 'organic cationic transporter 2 (OCTN2)' which is encoded by SLC22A5 gene on 5q (Wang et al., 2001). There is increased renal loss of carnitine leading to low plasma concentrations of carnitine. The most common presentation is with cardiac failure (due to progressive cardiomyopathy) although there may be accompanying myopathy manifesting as proximal muscle weakness. (Stanley et al., 2006). The median age of onset of cardiac symptoms is three years. Routine 2D echocardiography done at the time of diagnosis may detect asymptomatic cardiac findings. Majority of individuals identified to have primary carnitine deficiency by newborn screening remain asymptomatic even without treatment (Crefcoeur et al., 2023).
Carnitine palmitoyl transferase I deficiency
Three isoforms of CPT-1 have been identified (Morris & Spiekerkoetter, 2016). CPT-1A is present in the liver and kidney. CPT-1B is present in the cardiac and skeletal muscles. CPT-1C is present in the brain. Only deficiency of CPT-1A has been described in literature, to date (Jones et al., 2020). As CPT-1A is present only in the liver and kidneys, the long-chain fatty acid uptake into the mitochondrial matrix of the hepatocytes is affected and these individuals present with hepatic manifestations of non-ketotic hypoglycemia and coma. The usual age of manifestations is between 6- 12 months of life. In rare instances, CPT- 1A deficiency has been observed to cause renal tubular acidosis along with hepatic dysfunction. There is no cardiac or skeletal muscle involvement seen in this defect.
Carnitine-acylcarnitine translocase (CACT) deficiency
This is a rare defect occurring due to bi-allelic variants in the SLC25A20 gene. It is associated with high mortality. The age of presentation is during the neonatal period, typically in the first days of life. The newborn presents with hypoketotic hypoglycemia, hyperammonemia, encephalopathy features, hepatopathy and myopathy with poor head control. A notable feature is the presence of cardiac arrhythmias which may contribute to sudden death in these patients.
Carnitine palmitoyl transferase II (CPT-II) deficiency
There are three phenotypic forms of CPT-II deficiency depending on the level of residual enzyme activity in the tissues, i.e., severe neonatal form, intermediate form and the milder adult form (Nyhan et al., 2020). The neonatal form presents as life-threatening coma, cardiomyopathy and hypotonia. This is associated with high mortality in the newborn. It occurs due to CPT2 variations that result in almost negligible enzyme activity. Affected individuals may have associated congenital brain and renal malformations which may have been detected in the prenatal scans. In the antenatal period, there may be oligohydramnios with polycystic kidneys in the prenatal scan. After birth, the neonate may have palpable, enlarged kidneys with associated renal failure and hyperkalemia. The brain may have dysplastic and cystic changes which may be detected in neurosonography. In some neonates, non-specific dysmorphic features in the form of microcephaly, high sloping forehead, bulbous nose, flat occiput and low-set ears, long tapering fingers, widely spaced nipples and contractures of knees, elbows and small joints of the hands have been described. The intermediate form presents during infancy with fasting hypoketotic hypoglycemia. The adult-onset form is milder and presents in the second or third decade of life with low exercise tolerance/ rhabdomyolysis which is triggered by prolonged fasting or exercise. It presents as myopathy with proximal muscle weakness and fatigue. The adult-onset myopathic form is the most common phenotype of CPT2 deficiency (Wieser et al., 2019). The intermediate and adult-onset forms usually occur due to missense variations in both alleles of the CPT2 gene that result in residual or partial enzyme activity.
Acyl-CoA dehydrogenase deficiency
Medium-chain acyl-CoA dehydrogenase deficiency (MCADD) is the most common type of FAOD and is also the least severe form. About 30 - 50% of affected individuals may remain asymptomatic, which has become evident after the advent of newborn screening. Early identification through newborn screening would help prevent metabolic decompensations. MCADD has an exclusively hepatic type of presentation with episodic illness with normal intervals between the episodes. Hepatomegaly may be present during the acute episode. Onset of symptoms is around one to two years of age, when the nocturnal feeds are stopped, and the hypoglycemic episodes occur in the mornings after prolonged overnight fasting. Fasting tolerance of the individual improves with increasing body mass;hence, the frequency of attacks decreases with age.
Short-chain acyl-CoA dehydrogenase deficiency (SCADD) is diagnosed in a vast majority of asymptomatic individuals through newborn screening or family members of affected probands. In previous studies, the clinical findings reported were developmental delay, hypotonia, seizures and failure to thrive (Pederson et al., 2008). However, asymptomatic patients have also been reported. The clinical features do not correlate with the SCAD enzyme activity. Hence, SCAD deficiency is now considered as a biochemical phenotype.
3-hydroxyacyl-CoA dehydrogenase deficiency
Long-chain 3-hydroxyacyl CoA dehydrogenase (LCHAD) deficiency is the more common form. LCHAD enzyme is a component of a mitochondrial trifunctional protein, which has an octameric structure with 4 alpha and 4 beta subunits. The 4 alpha subunits are encoded by HADHA gene, and the 4 beta subunits are encoded by HADHB gene. Patients with LCHAD deficiency may have an isolated LCHAD deficiency or deficient activity of all three component enzymes. Isolated LCHAD deficiency is due to specific variation in HADHA gene. Phenotypes of affected individuals may be variable, ranging from a mild form (resembling MCAD deficiency) to severe disease (resembling VLCAD deficiency). The age of onset of symptoms is usually in infancy. Along with the usual presentations of FAODs, there may be additional manifestations in these individuals in the form of pigmentary retinopathy (Lawlor & Kalina, 1997) and peripheral neuropathy (Grunert et al., 2021). Pigmentary retinopathy may occur in about 70% of patients with LCHAD deficiency presenting with decreased color vision, nyctalopia and field defect in the center of field of view. Peripheral neuropathy is more common in deficiency of MTPD than LCHAD deficiency, with prevalence of 70% and 50% respectively (Grunert et al., 2021). It occurs later in life with onset during adolescence, manifesting as slow, progressive sensorimotor neuropathy with absent deep tendon reflexes. It may be associated with limb-girdle myopathy with recurrent episodes of myoglobinuria. Heterozygous mothers carrying a homozygous affected fetus with LCHAD deficiency have been found to have an increased risk of developing acute fatty liver of pregnancy (AFLP) and hemolysis, elevated liver enzymes, and low platelet count (HELLP) syndrome. About 20% of at-risk pregnancies may have one of these complications.
Short-chain 3-hydroxyacyl CoA dehydrogenase (SCHAD) deficiency, also known as 3-hydroxyacyl-CoA dehydrogenase (HADH) deficiency, is a very rare condition. In SCHAD deficiency, unlike the other subtypes of FAODs, hypoglycemia is associated with hyperinsulinism.
Multiple Acyl-CoA Dehydrogenase (MAD) Deficiency
This is also known as Glutaric aciduria type II. It occurs due to defect in electron transport in the mitochondrial membrane from acyl CoA to ubiquinone (CoQ10). This requires the activity of electron transport flavoprotein (ETF) present in the mitochondrial matrix and the electron transport flavoprotein: ubiquinone oxidoreductase (ETF- QO) which is present on the inner mitochondrial membrane. ETF accepts hydrogen from FAD linked dehydrogenases. This includes acyl CoA dehydrogenases of beta oxidation as well enzymes of branched chain amino acid metabolism and choline metabolism. The electron from reduced ETF is transferred to ETF-QO and further to coenzyme Q. ETF has alpha and beta subunits encoded by ETFA and ETFB, respectively. ETF-QO is encoded by ETFDH. Most cases are attributed to pathogenic variants in ETFDH.
The clinical spectrum of MAD deficiency can be broadly divided into three types. Type I / II (neonatal onset with and without congenital anomalies, respectively) which present in neonatal period with severe metabolic decompensation (hypoglycemia, metabolic acidosis and hyperammonemia) and have high mortality. Congenital anomalies if present may include facial dysmorphism (high forehead, depressed nasal bridge, low set ears), polycystic kidneys (may present with Potter sequence) and hypospadias/chordee in male individuals. Type III is the late onset form (most common presentation) and can present from infancy to adulthood. Affected individuals present with chronic musculoskeletal symptoms of muscle pain/weakness and exercise intolerance (Grunert et al., 2014). About 20% have episodic metabolic decompensation after stressors.
Investigations
The biochemical features of FAODs are due to deficient energy production (because of reduced production of acetyl CoA and ketone bodies) and changes secondary to disruption of other metabolic pathways due to lack of ATP, such as the gluconeogenesis and urea cycle pathways. There is accumulation of free fatty acids and the respective fatty acyl CoA intermediates upstream to the enzyme block. This results in the formation of dicarboxylic acid and hydroxy dicarboxylic acids from the omega oxidation pathway. There is also conversion of fatty acyl CoA esters to their corresponding acylglycines and acylcarnitines which bind to the carnitine and get excreted in the urine leading to secondary depletion of carnitine. The measurement and analysis of these metabolites in the patients thus helps in the diagnosis of fatty acid disorders.
Routine blood investigations might show hypoglycemia, hyperammonemia, elevated transaminases, elevated creatine phosphokinase with hyperuricemia, and absence of ketones in the urine. Though hypoketotic hypoglycemia is a hallmark, ketone bodies may be synthesized in FAODs, especially in medium chain fatty acid oxidation defects and also when there is presence of residual enzyme activity. But the concentration of ketone bodies is lower than expected for the degree of hypoglycemia. Studies report that up to 29% of MCAD deficiency patients have ketonuria (Ruiz-Sala et al., 2021). The presence of hypoglycemia with the inappropriately low ketone bodies should raise a strong suspicion of FAOD. There may be associated metabolic acidosis. Specific disease-related metabolites should be quantitatively assessed. Plasma carnitine levels are low in primary carnitine transporter defects and in other subtypes, the levels are secondarily reduced due to renal loss of carnitine in the urine. Analysis of acylcarnitine levels should be analyzed through tandem mass spectrometry (TMS) in plasma or whole dried blood spot on filter paper. A clue to the enzyme deficiency can be obtained depending on specific acylcarnitines which are detected to be elevated. Measurement of urinary organic acids and acylglycines through gas chromatography mass spectrometry (GCMS) is also helpful in the diagnosis. Table 2 shows the plasma and urine metabolites that are elevated in the various FAODs.
Confirmation of the diagnosis in the presence of clinical manifestations and specific metabolite abnormalities is by the identification of homozygous or compound heterozygous variants in the specific gene through molecular testing. Next-generation sequencing (NGS)-based methods such as multigene panel tests or whole-exome sequencing (WES) are preferred as these disorders have overlapping phenotypes.
Newborn screening
Metabolic decompensations are associated with high mortality, and this can be prevented by early initiation of dietary management and supportive care during times of acute stress. Screening for FAOD can be done through tandem mass spectrometry (TMS) for acylcarnitine profile for early identification of asymptomatic newborns in dried blood spot (DBS) samples. The screening test is not a determinant of disease status and positive results can arise due to other non-specific conditions. The presumptively positive newborn screening should be followed up with confirmatory diagnostic molecular testing. The American College of Medical Genetics and Genomics (ACMG) has recommended screening for CTD, VLCAD, LCHAD, TFP and MCAD deficiency. However, newborn screening has some drawbacks. Patients identified to have SCAD deficiency were found to remain asymptomatic in spite of severe enzyme deficiency. Normal newborns of mothers with CTD deficiency were falsely identified to be affected.
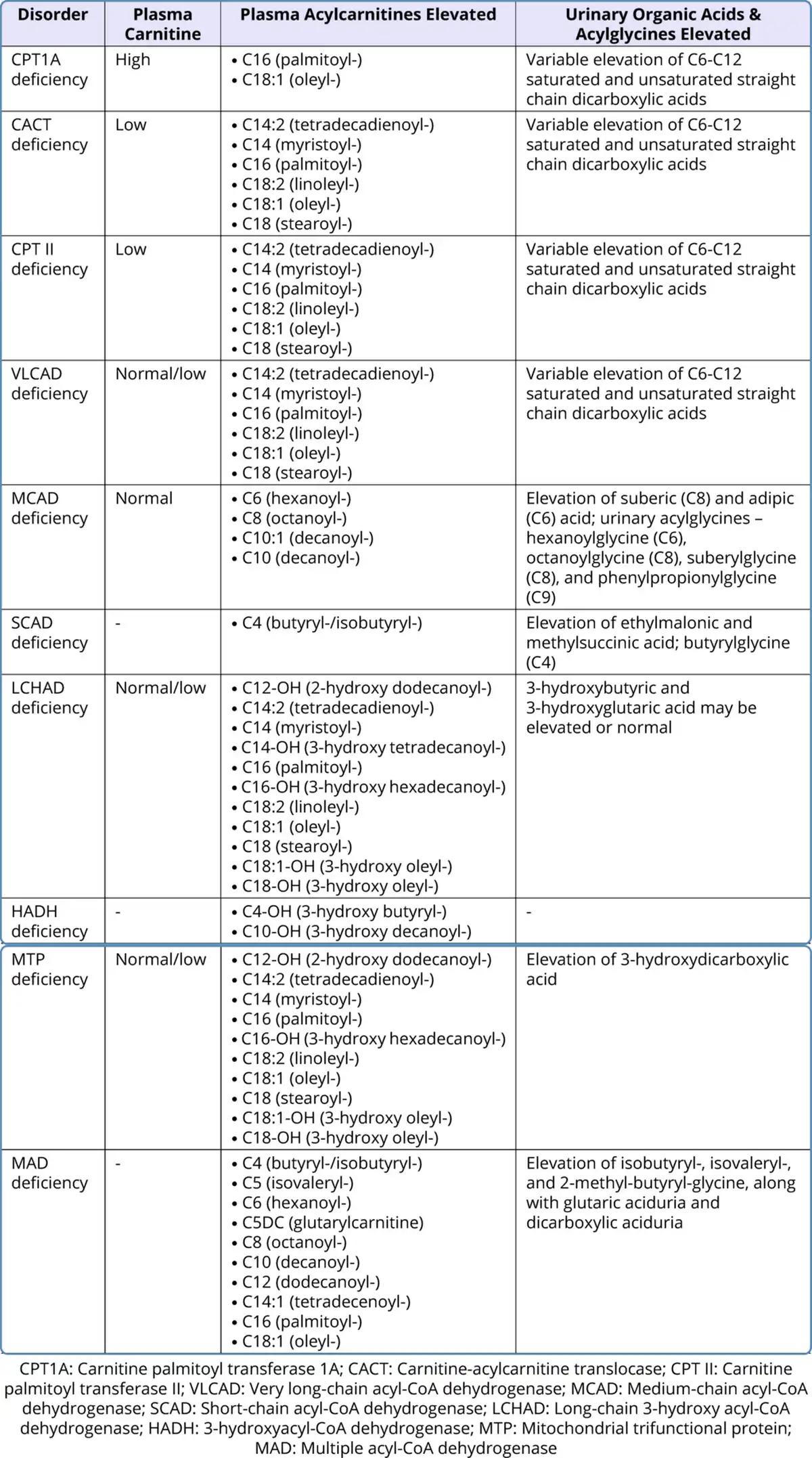
Table 2
Metabolic Assays for Fatty Acid Oxidation Defects
Management
The mainstay of treatment is through dietary management by removing lipids from the diet or through replacement of accumulating lipids with those that can bypass the block.
General measures: Common elements of the nutritional management of all fatty acid oxidation defects are listed as follows (Rohr, 2015):
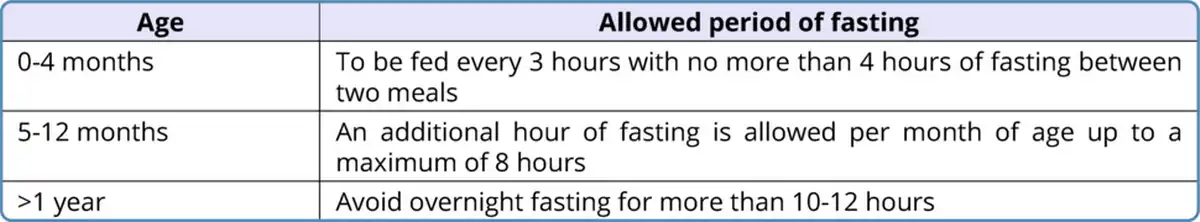
Table 3
Avoidance of fasting in fatty acid oxidation defects
Aggressive treatment during increased metabolic needs: During intercurrent illness when appetite is reduced, carbohydrate rich fluids should be provided via oral or enteral route every 3-4 hours. If the patient is unable to take orally, or when fasting for surgery, intravenous fluids with 10% dextrose should be given at 10-12 mg/kg/min to maintain plasma glucose > 100 mg/dl. Insulin may be given if hyperglycemia develops. Appropriate electrolyte correction needs to be done. If there is acute hyperammonemia, it is treated with sodium benzoate or sodium phenylacetate. Hemodialysis may be done based on the requirement. The goals are to prevent hypoglycemia, suppress lipolysis, and suppress fatty acid oxidation.
Avoidance of fasting: The guidelines given by the Genetic Metabolic Dietitians International are given in Table 3 (Shirley, 2020). Frequent feeds have to be given to prevent fasting. In older children, snacks should be given in the intervals between regular meals. Cornstarch supplementation 1.5-2 g/kg should be given at bedtime.
Carnitine supplementation: Levocarnitine supplementation is important especially in carnitine transporter defects (CTD). In these disorders, levocarnitine has been shown to improve the cardiac and skeletal muscle function to almost near normal within a few months of initiating treatment. The oral levocarnitine dose is 100 mg/kg per day. The role of carnitine supplementation in disorders with secondary carnitine deficiency is controversial as the blocks in the enzyme steps do not directly involve carnitine. The proposed hypothesis for supplementing carnitine is that it might help remove the accumulated toxic metabolites, i.e., acyl CoA intermediates by combining with them to form acylcarnitine which are then excreted in the urine.
Specific management:
Medium chain acyl CoA dehydrogenase (MCAD) deficiency: Breastmilk or standard infant formula is appropriate to meet the nutritional needs of the infant. Medium chain triglycerides (MCT) should be avoided. Excess consumption of coconut oil and infant formulas containing high MCT (such as premature infant formulas) should be avoided.
Long chain fatty acid oxidation disorders: A fat-restricted diet is advised as majority of the fatty acids in the diet are long-chain fatty acids. Breastmilk also has high fat, predominantly containing long-chain fatty acids. The diet should be supplemented with MCT (medium chain triglycerides) which act as substrate for beta oxidation. Breastfeeding may be continued in milder disease or asymptomatic infants, while in severely symptomatic infants, breastfeeding may be discontinued.
For mild to moderate VLCADD, breastfeeding can be continued along with MCT supplemented formula;20% energy needs are met by long-chain fat and 20% from MCT. For severe VLCADD, breastfeeding should be stopped and MCT formula should be started. Diet should be planned such that 10% of the energy needs are obtained from long-chain + 30% from MCT. After infancy, MCT can be supplemented as an oil or powder prescribed by a physician. The MCT may be mixed in non-fat milk and should be drunk at regular intervals or may be mixed with food. MCT supplementation is associated with the reversal of cardiomyopathy in CACT deficiency and VLCAD deficiency.
After the infant formula has been discontinued, the essential fatty acids must be supplemented along with supplementation of fat-soluble vitamins to avoid deficiency. Docosahexaenoic acid (DHA) is supplemented at a dose of 60 mg/day for infants < 20 kg, 100mg/day for children > 20 kg, and 100-200mg/day for adults.
These individuals have a high risk of exercise-induced rhabdomyolysis. The occurrence of rhabdomyolysis and improvement of exercise tolerance can be achieved through additional MCT supplementation 0.15-0.2g/kg mixed with glucose solution prior to exercise and 3:1 carbohydrate: protein snack after the exercise. As the individual is advised for fat restriction, it may be beneficial for them to be taking higher protein intake through lean meat rather than carbohydrates alone as it would help maintain the body composition.
For LCHAD-associated retinopathy, adherence to the strict diet plan along with MCT supplementation decreases the levels of hydroxy acyl carnitines and hydroxy fatty acids. This would help slow down the progression of vision loss. There is no specific treatment to prevent the progression of retinopathy.
c. Multiple acyl CoA dehydrogenase deficiency: High dose riboflavin supplementation at 100-300mg/day should be tried in all patients to assess for riboflavin responsiveness. Supplementation of levocarnitine at 50 - 100mg/kg/day in three divided doses and coenzyme Q10 supplementation of 60- 240mg/day in 2 divided doses are recommended.
Recent advances
Triheptanoin, sold under the brand name 'Dojolvi' was approved by the US Food and Drug Administration (FDA) in 2020 for the treatment of pediatric and adult patients with confirmed disorders of long-chain fatty acid oxidation (Shirley, 2020). It is a triglyceride of three 7-carbon fatty acids. It serves as an alternative fuel source and thus suppresses lipolysis and accumulation of toxic metabolites. Each triheptanoin molecule is hydrolyzed in the small intestine into 3 molecules of heptanoic acids. The heptanoic acid enters the beta oxidation pathway to produce acetyl CoA and propionyl CoA. The propionyl CoA formed enters the citric acid cycle. As per the randomized controlled trial conducted by Gillingham and coworkers (Gillingham et al., 2017), the group receiving triheptanoin was found to have an improvement in the left ventricular ejection fraction (LVEF) by 7.4%, though no difference was found in the occurrence of rhabdomyolysis between the group receiving triheptanoin and those on dietary control with MCT supplementation.
Glycerol phenylbutyrate, sold under the trade name 'Ravicti' is an FDA-approved drug to treat urea cycle disorders. This has been shown to bind as a substrate to the MCAD enzyme and has been hypothesized to have a stabilizing effect on MCAD associated with the p.Lys304Glu (K304E) missense mutation. Phase 2 clinical trial in MCAD patients homozygous for K304E is ongoing (https://go.drug- bank.com/conditions/DBCOND0008334).
Conclusion
Most of the individuals affected by FAODs, especially the defects of long chain and very long chain fatty acids had significant mortality in the past. With the advent of newborn screening, earlier diagnosis and prompt treatment have improved patient outcomes. It has also identified a large number of patients who might never have developed symptoms. Follow up and monitoring is needed for asymptomatic patients as well along with measures to prevent episodes of metabolic decompensation (especially with MCAD deficiency).
Conflict of Interests: None
References
1.
Bender DA, Mayes PA. Metabolism of Glycogen. In: Rodwell VW, Bender DA, Botham KM, Kennelly PJ, Weil P. eds. Harper's Illustrated Biochemistry, 31e. McGraw-Hill Education;2018.2.
Bennett MJ, Santani AB. Carnitine Palmitoyltransferase 1A Deficiency. 2005 Jul 27 [Updated 2016 Mar 17]. In: Adam MP, Feldman J, Mirzaa GM, et al, editors. GeneReviews(r) [Internet]. Seattle (WA): University of Washington, Seattle;1993–2024. Available from: https://www.ncbi.nlm.nih.gov/sites/books/NBK1527/3.
Boles RG, Buck EA, Blitzer MG, et al. Retrospective biochemical screening of fatty acid oxidation disorders in postmortem livers of 418 cases of sudden death in the first year of life. J Pediatr. 1998;132:924–933.4.
Crefcoeur L, Ferdinandusse S, van der Crabben SN, Dekkers E, Fuchs SA, Huidekoper H, et al. Newborn screening for primary carnitine deficiency: who will benefit? - a retrospective cohort study. J Med Genet. 2023; 60(12):1177–1185.5.
Gillingham MB, Heitner SB, Martin J, Rose S, Goldstein A, El-Gharbawy AH, et al. Triheptanoin versus trioctanoin for long-chain fatty acid oxidation disorders: a double blinded, randomized controlled trial. J Inherit Metab Dis. 2017;40(6):831–843.6.
Grünert SC, Eckenweiler M, Haas D, Lindner M, Tsiakas K, Santer R, et al. The spectrum of peripheral neuropathy in disorders of the mitochondrial trifunctional protein. J Inherit Metab Dis. 2021;44(4):893–902.7.
Guerra IMS, Ferreira HB, Melo T, Rocha H, Moreira S, Diogo L, et al. Mitochondrial Fatty Acid ß-Oxidation Disorders: From Disease to Lipidomic Studies-A Critical Review. Int J Mol Sci. 2022;23(22):13933.8.
Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid ß-oxidation. J Inherit Metab Dis. 2010;33(5):469–477.9.
Innes AM, Seargeant LE, Balachandra K, Roe CR, Wanders RJ, Ruiter JP, et al. Hepatic carnitine palmitoyltransferase I deficiency presenting as maternal illness in pregnancy. Pediatr Res. 2000;47(1):43–4510.
Jones P, Patel K, Rakheja D. Fatty Acid Oxidation Defects: Carnitine palmitoyltransferase 1 deficiency. In: A Quick Guide to Metabolic Disease Testing Interpretation: Testing for Inborn Errors of Metabolism. Second Edition. Elsevier Inc.; 2020. p. 137–140.11.
Karall D, Brunner-Krainz M, Kogelnig K, Konstantopoulou V, Maier EM, Moslinger D. Clinical outcome, biochemical and therapeutic follow-up in 14 Austrian patients with Long-Chain 3-Hydroxy Acyl CoA Dehydrogenase Deficiency (LCHADD). OrphanetJ Rare Dis 2015;10:21.12.
Kerner J, Hoppel C. Fatty acid import into mitochondria. Biochim Biophys Acta. 2000;1486(1):1–17.13.
Lawlor DP, Kalina RE. Pigmentary retinopathy in long chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. Am J Ophthalmol. 1997;123(6):846–848.14.
Marsden D, Bedrosian CL, Vockley J. Impact of newborn screening on the reported incidence and clinical outcomes associated with medium- and long-chain fatty acid oxidation disorders. Genet Med. 2021;23(5):816–829.15.
Mathur A, Sims HF, Gopalakrishnan D, Gibson B, Rinaldo P, Vockley J, et al. Molecular heterogeneity in very-long-chain acyl-CoA dehydrogenase deficiency causing pediatric cardiomyopathy and sudden death. Circulation. 1999;99(10):1337–43.16.
McGuinness OP. Overview of Metabolism & the Provision of Metabolic Fuels. In: Kennelly PJ, Botham KM, McGuinness OP, Rodwell VW, Weil P. eds. Harper's Illustrated Biochemistry, 32nd Edition. McGraw Hill Education;2023.17.
Morris AAM, Spiekerkoetter U. Disorders of Mitochondrial Fatty Acid Oxidation & Riboflavin Metabolism. In: Saudubray JM, Baumgartner M, Walter J (eds). Inborn Metabolic Diseases. Springer, Berlin, Heidelberg;2016: 201–213.18.
Nyhan WL, Hoffman GF, Al-Aqeel AI, Barshop BA. Disorders of fatty acid oxidation. In: Nyhan WL, Hoffman GF (eds). Atlas of Inherited Metabolic Diseases, 4th Edition. CRC Press; 2019.19.
Pedersen CB, Kølvraa S, Kølvraa A, Stenbroen V, Kjeldsen M, Ensenauer R, et al. The ACADS gene variation spectrum in 114 patients with short-chain acyl-CoA dehydrogenase (SCAD) deficiency is dominated by missense variations leading to protein misfolding at the cellular level. Hum Genet. 2008;124(1):43–56.20.
Rohr F. Nutrition Management of Fatty Acid Oxidation Disorders. In: Bernstein LE, Rohr F, Helm JR (eds). Nutrition Management of Inherited Metabolic Diseases. Springer International Publishing;2015: pp: 325–335.21.
Ruiz-Sala P, Peña-Quintana L. Biochemical Markers for the Diagnosis of Mitochondrial Fatty Acid Oxidation Diseases. J Clin Med. 2021;10(21): 4855.22.
Shirley M. Triheptanoin: First Approval. Drugs. 2020;80(15):1595–1600.23.
Sim KG, Hammond J, Wilcken B. Strategies for the diagnosis of mitochondrial fatty acid beta-oxidation disorders. Clin Chim Acta. 2002;323(1-2):37–58.24.
Spiekerkoetter U, Sun B, Khuchua Z, Bennett MJ, Strauss AW. Molecular and phenotypic heterogeneity in mitochondrial trifunctional protein deficiency due to beta-subunit mutations. Hum Mutat. 2003;21(6):598–607.25.
Stanley CA, Bennett MJ, Mayatepek E. Disorders of Mitochondrial Fatty Acid Oxidation and Related Metabolic Pathways. In: Fernandes J, Saudubray JM, van den Berghe G, Walter JH (eds). Inborn Metabolic Diseases. Springer, Berlin, Heidelberg. 2006; pp 175–190.26.
Tamai I, Ohashi R, Nezu J, Yabuuchi H, Oku A, Shimane M, et al. Molecular and functional identification of sodium ion-dependent, high affinity human carnitine transporter OCTN2. J Biol Chem. 1998;273(32):20378–20382.27.
Wang Y, Korman SH, Ye J, Gargus JJ, Gutman A, Taroni F, et al. Phenotype and genotype variation in primary carnitine deficiency. Genet Med. 2001;3(6):387–392.28.
Wieser T. Carnitine Palmitoyltransferase II Deficiency. 2004 Aug 27 [Updated 2019 Jan 3]. In: Adam MP, Feldman J, Mirzaa GM, et al, editors. GeneReviews(r) [Internet]. Seattle (WA): University of Washington, Seattle;1993–2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1253/