Introduction
Cadmium (Cd) is a toxic heavy metal that poses a health risk for humans due to its wide spectrum of deleterious effects and a long elimination half-life of 20–30 years., Chronic exposure to Cd, through consumption of contaminated food and water, inhalation of tobacco smoke, and the occupational exposure is related to various adverse effects, including osteoporosis, diabetes, cardiovascular disease, renal dysfunction, or even cancers., After absorption, Cd is bound to various sulfhydryl-containing molecules (e.g. albumin, low-molecule-weight thiols, metallothionein (MT), and transferrin) in the blood and the formed complexes are mainly taken up by the liver., In the liver, Cd stimulates the synthesis of MT,, a protein that acts as the precursor of Cd detoxification and Cd is bound to MT to form the Cd-metallothionein (Cd-MT) complex, which is then released to the circulation system. Due to the small size of MTs, the complex is easily filtered through the glomerulus and reabsorbed by the proximal tubular cells, where the Cd-MT complex is endocytosed and degraded in lysosomes into amino acids and free Cd ions.,– When the intracellular loading with Cd ions exceeds the buffering capacity of cytoplasmic MT, liberated Cd ions can immediately start to affect the cell structure and functions, causing pathological changes in renal tubules.,, Furthermore, Cd is mainly excreted through the urine, and the amount of Cd excreted daily in urine is very low. Thus, with the redistribution of Cd, the kidney is the major target organ for Cd toxicity.
Chelating agents have been explored as a major therapy to reduce Cd-induced toxicity for many years.– Dithiocarbamates (DTC) have been shown to effectively mobilize Cd from animal tissues as indicated by their significantly enhanced biliary excretion of Cd, but not impacting the urinary excretion., An in vivo study has also shown that the combination of 2,3-dimercapto-1-propane sulfonic acid (DMPS) and N-acetyl cysteine (NAC) exhibited a limited benefit in the treatment of Cd intoxication. However, none of these chelators are approved because of their ineffectiveness in Cd removal from the kidneys, or severe side effects, such as flushing, agitation, localized burning at the infusion site. To develop a safe and specific antidote for Cd is critical to meet the need in clinical application and public health interventions.
Sodium (S)-2-(dithiocarboxylato ((2S, 3R, 4R, 5R)-2, 3, 4, 5, 6-pentahydroxyhexyl) amino)-4 (methylthio) butanoate (GMDTC) (Figure 1(a)) is a newly developed DTC derivative designed by our research group. It has strong chelating ability for toxic heavy metals, including Cd2+, mercury (Hg2+), chromium (Cr6+), and lead (Pb2+), without significant effects on the status of the essential metals such as sodium (Na+), and potassium (K+), among others.– Particularly, GMDTC was suggested to chelate with Cd2+ to form GMDTC-Cd complex at a ratio of 2:1 based on results of complexometric titration and high performance liquid chromatography (HPLC) detection. It has been reported in our previous study that GMDTC is characterised by its very low toxicity, the acute lethal dose (LD50) of which is more than 5,000 mg/kg or 10,000 mg/kg body weight, correspondingly, via intraperitoneal injection or oral in mice and rats. In addition, as high as 94% of Cd2+ accumulated in the kidneys of Cd2+-laden rabbits was effectively removed and excreted through urine resulting from safe doses of GMDTC (0.25 mmol/kg and 1 mmol/kg body weight, equal to 108 mg/kg and 433 mg/kg body weight, respectively) treatment for 4 weeks. However, it is not entirely clear by what mechanism(s) GMDTC removes accumulated Cd from the kidney, although glucose reabsorption pathway has been proposed as GMDTC contains an open chain glucose moiety. The renal glucose reabsorption pathway is the physiological process of renal glucose reabsorption from the glomerular filtrate through proximal tubule epithelial cells into the blood mediated by active sodium-coupled glucose cotransporters (SGLTs) and passive glucose transporters (GLUTs). Briefly, glucose filtered at the glomerulus is firstly transported by SGLTs into proximal tubule epithelial cells against a concentration gradient coupled with the inward diffusion of sodium ions which is maintained by an Na+/K+ ATPase pump; once the concentration of glucose in epithelial cells has been accumulated to a level above the interstitial one, intracellular glucose diffuses out to the plasma via the facilitative glucose transporters GLUTs in the basolateral membranes.,
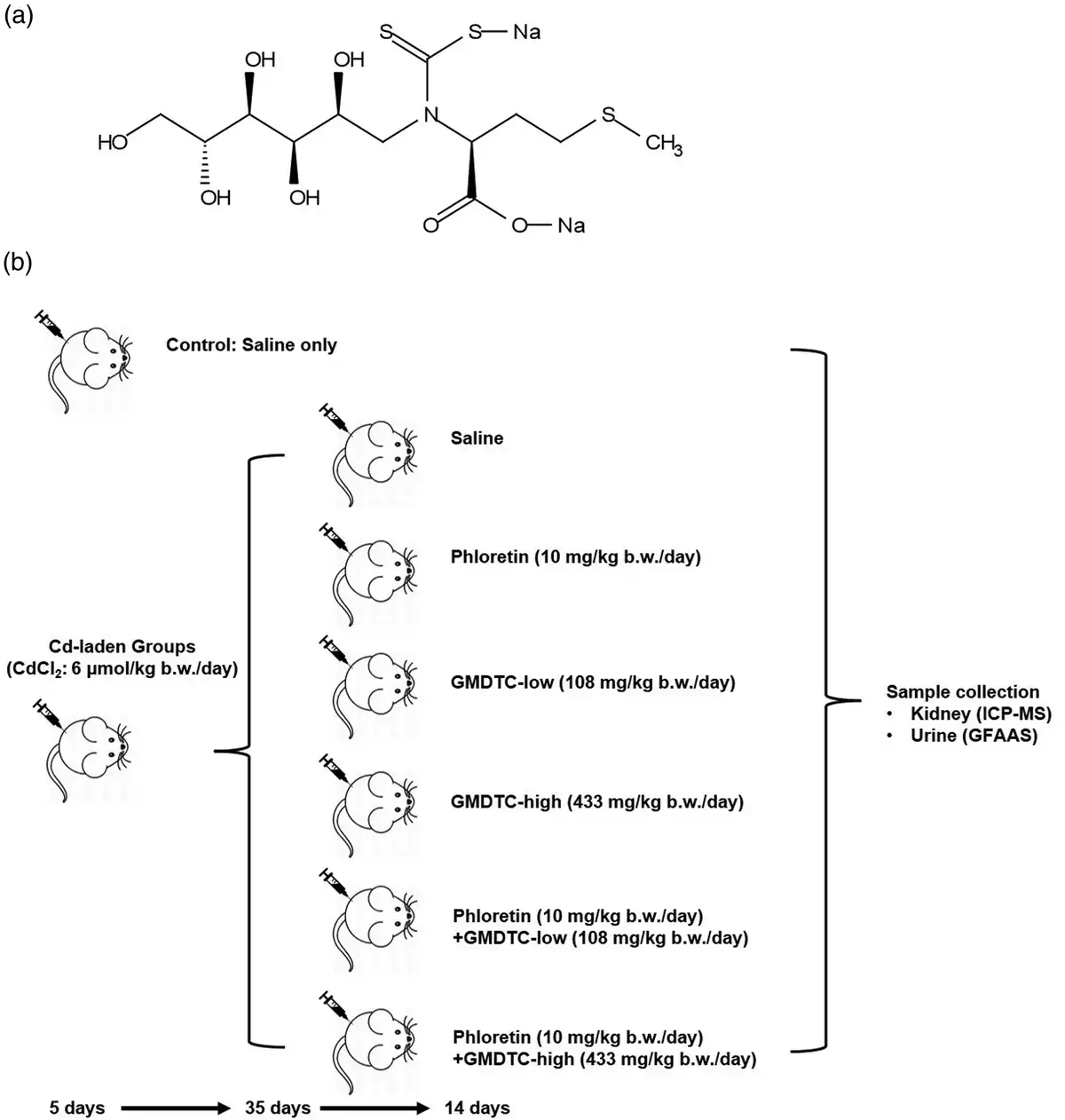
Figure 1
GMDTC Chemical Structure and Schematic Procedure of Mice Treatment. (a) Chemical structure of Sodium (S)-2-(dithiocarboxylato((2S,3 R,4R,5 R)-2,3,4,5,6 pentahydroxyhexyl)amino)-4-(methylthio)butanoate (GMDTC). (b) Grouping and experimental steps of Cd2+-laden mice in the study of the impact of inhibiting renal glucose transporter GLUT2 on GMDTC’s effect in removing Cd2+ from kidney.
To assist the clinical development of GMDTC as a detoxifying agent for Cd-induced toxicity, here we utilized the CRISPR/Cas9 genome editing technology and constructed glucose transporter 2 (GLUT2−/−) and sodium-dependent glucose transporter 2 (SGLT2−/−) knockout (KO) human kidney proximal tubule (HK-2) cell lines, two key glucose transporters. We compared the protective effects of GMDTC between these KO cell lines and parental cells upon Cd exposure and examined the involvement of glucose reabsorption pathway in GMDTC-mediated removal of Cd from cells. We further evaluated this in vitro effect in an animal model. Our results supported the involvement of glucose reabsorption pathway in Cd excretion from the kidney by GMDTC, mechanistically supporting the therapeutic potential of GMDTC used for kidney damage and other health effects caused by Cd exposure.
Materials and methods
Chemicals and reagents
GMDTC (purity 95%–97%, molecular weight 433 g/mol) was synthesized by WuXi AppTec (Tianjin, China). Representative HPLC chromatogram of GMDTC is shown in Supplementary Figure 1. Cadmium chloride (CdCl2, purity ≥99.99%, molecular weight 183.32 g/mol) was purchased from Alfa Aesar (Tewksbury, MA, USA), and phloretin (purity ≥99%) was from Sigma-Aldrich (St. Louis, MO). The chemicals were freshly prepared and dissolved in 0.9% saline and passed through 0.22 μm filter prior to treating cells or animals. The alamarBlue™ Cell Viability Reagent was obtained from ThermoFisher Scientific (Waltham, MA, USA). The human kidney proximal tubule HK-2 (CRL-2190) cell line was obtained from American Type Culture Collection (Manassas, VA, USA).
Cell culture
HK-2 cells were cultured in DMEM/F12 medium containing a 1:1 mixture of Dulbecco’s Modified Eagle Medium (DMEM) and Ham’s F12. Cell culture media were supplemented with 10% fetal bovine serum (FBS), and 1% penicillin-streptomycin (100 U/mL penicillin and 100 μg/mL streptomycin). Cells were incubated at 37°C in an atmosphere of 5% CO2 and 95% humidity.
Targeting strategy
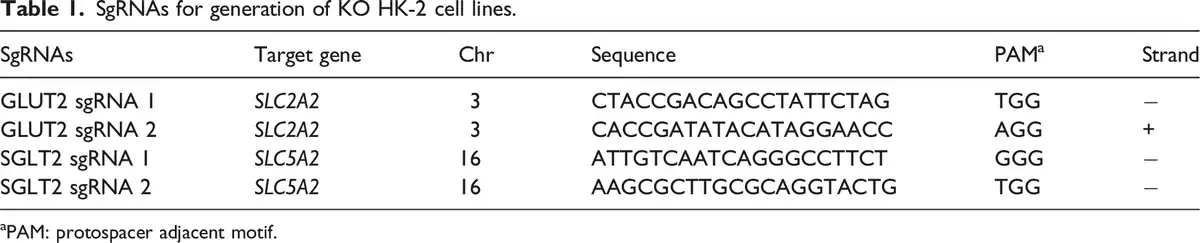
The target genes are solute carrier family 2 member 2 (SLC2A2) and solute carrier family 5 member 2 (SLC5A2), which encode GLUT2 and SGLT2 proteins, respectively. We used CRISPR/Cas 9 gene editing system to generate GLUT2−/− or SGLT2−/− KO HK-2 cell lines. A total of 4 single-guide RNAs (sgRNAs) (Table 1), two for each target gene, were designed and purchased from Thermo Fisher Scientific (Waltham, MA, USA) with high on-target scores.
Analysis of off-target effects
The Cas-OFFinder algorithm (http://www.rgenome.net/cas-offinder/) was used to identify potential off-target sites (NGG PAMs with up to 3 mismatches) in Homo sapiens (GRCh38/hg38). Cas-OFFinder outcomes were further filtered to identify the most problematic off-targets with the fewest mismatches. It is reported that any sgRNAs with more than 3 mismatches have a low risk of causing off-target effects, and 96% of 2 bp mismatches do not function. Seed region (the 10–12 bp closest to the PAM site) is not the most important for binding and any mismatches in this region, and further reduces the possibility of off-target effects. Therefore, problematic off-target sites were defined as off-targets with up to 1 mismatch in the seed region and up to 2 mismatches in the non-seed region with an NGG PAM. Approximately 400 base pairs of sequence flanking each off-target site was amplified by PCR for further cleavage efficiency analysis to determine off-target effect. The potential off-target sites are shown in (Supplemental Table 1).
Cell transfection
Briefly, HK-2 cells were seeded in 24-well plates at a density of 1.0 × 104 cells/well for further transfection using Lipofectamine™ CRISPRMAX™ Transfection Reagent (Invitrogen, Carlsbad, CA, USA). SgRNAs (240 ng of each) were mixed with TrueCut™ Cas9 Protein v2 (Invitrogen, Carlsbad, CA, USA) to form the ribonucleoprotein (RNP) complex, and the transfection procedure was performed according to the manufacturer’s instructions. After 2 days incubation at 37°C, cells were rinsed with PBS and genomic DNA was extracted for cleavage efficiency analysis with T7 endonuclease I assay.
T7 endonuclease 1 (T7E1) assay
The genomic sequences containing on and off target regions mediated by each sgRNA were polymerase chain reaction (PCR) amplified using the primers listed in Table 2. PCR products were purified using E.Z.N.A® Cycle Pure Kit (Omega Bio-tek, Norcross, GA, USA). The purified PCR products were then mixed with 1 μL T7E1 buffer and were denatured and annealed for heteroduplex formation. Reaction amplicons were treated with 0.5 μL T7 endonuclease 1 (New England Biolabs, Ipswich, MA, USA) at 37°C for 15 min, following the addition of 1.7 μL of 0.22 M ethylene diamine tetraacetic acid (EDTA) to stop the reactions. Finally, the digested products were analyzed with 1.5% agarose gel electrophoresis and images were attained by Bio-Rad Gel Doc imaging system. Band intensities were calculated using ImageJ. Cleavage efficiency was calculated according to the equation: Fraction Cleaved = sum of cleaved band intensities/(sum of the cleaved and parental band intensities); Cleavage Efficiency = 1– [(1–Fraction Cleaved) ½].

Clone isolation and tracking of indels by decomposition analysis
The transfected cells were subject to single-cell sorting into 96-well plates to obtain single clones using FACSAriaTMIII Cell Sorter (BD bioscience, USA). When the clonal cells in each well reached confluency, genomic DNAs encompassing the CRISPR/Cas9 target sites were extracted and PCR amplified. The purified PCR products were then submitted for Sanger sequencing. Sequence traces of target-specific PCR products attained from Sanger sequencing were analyzed with the TIDE webtool (https://tide.nki.nl/) to determine the spectrum and frequency of targeted mutations generated.
Western blotting
To analyze the protein expression of the gene knockout cell lines, the cells were lysed using the M-PER™ Mammalian Protein Extraction Reagent (Thermo Fisher Scientific, Waltham, MA, USA). The protein concentration was determined using Pierce® BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). Total protein amount of 20 μg for each sample was separated using 10% SDS-PAGE gel electrophoresis and transferred to a PVDF membrane. After blocking, the following primary antibodies were used: Anti-SGLT2 antibody (1:1,500, Abcam, ab137207); Anti-Glucose Transporter GLUT2 antibody [5D1] (1:1,500, Abcam, ab85715); Anti-β-Actin antibody (1:500,000, Sigma-Aldrich, A1978). As secondary antibodies, Goat Anti-Rabbit IgG H&L (HRP) (1:10,000, Abcam, ab205718) and Goat Anti-Mouse IgG H&L (HRP) (1:10,000, Abcam, ab205719) were utilized. The signals were detected with the ClarityTM Western ECL Substrate (Bio-Rad, Hercules, CA, USA).
Cell viability assays
HK-2 cells with or without CRISPR/Cas9 targeted editing were seeded in 96-well plates at a density of 1.0 × 104 cells/well in 100 μL of medium (composition described in the section of ‘Cell Culture’), and then incubated 24 h before treatments. In the GMDTC and Cd toxicity experiments, the cells were treated with various concentrations of GMDTC or CdCl2 for 24 h. In the GMDTC rescue experiment, the cells were pre-treated with 250 μM CdCl2 for 2 h to allow for accumulation of Cd in cells, washed twice with phosphate buffered saline (PBS), replaced with fresh culture medium to eliminate extracellular Cd, and then treated with various concentrations of GMDTC for 24 and 48 h, respectively. Cell viability was measured using the alamarBlue assay. After treatments, 1 mM alamarBlue reagent was added into each well and incubated for 4 h. Fluorescence was recorded on a BioTek microplate reader using a fluorescence excitation wavelength of 560 nm (excitation range is 540–570 nm) and an emission of 590 nm (emission range is 580–610 nm). Cell viability was calculated relative to control.
Animal and treatment
Forty-two pathogen-free male NIH Swiss mice (17–20 g) were purchased from Guangdong Medical Laboratory Animal Center, China (GDMLAC, National certification No. 2006A015). All animals were provided a standard diet and housed in an approved facility with climate control and a 12 h light/12 h dark cycle. The detailed animal grouping and treatment are shown in the Figure 1(b). Six mice were randomly selected and grouped as Control group, which were administered only saline via intraperitoneal (i.p.) injection with the same administration pattern as the chemical treated groups. The other 36 mice were selected as Cd2+-laden groups and were administered CdCl2via i.p. injection for five consecutive days with a dose of 6 μmol/kg body weight. Thirty-five days post treatment, the Cd2+-laden mice were randomly divided into six subgroups and received saline or related chemicals for 14 days. The six subgroups are described as following: Group 1: saline; Group 2: GLUT2 inhibitor phloretin (10 mg/kg body weight per day); Group 3: GMDTC-low (108 mg/kg body weight per day); Group 4: GMDTC-high (433 mg/kg body weight per day); Group 5: phloretin (10 mg/kg body weight per day) + GMDTC-low (108 mg/kg body weight per day); Group 6: phloretin (10 mg/kg body weight per day) + GMDTC-high (433 mg/kg body weight per day). GMDTC and phloretin were administered via i.p. injection and oral gavage, respectively. In the combined treatment groups, phloretin was administered 30 min prior to GMDTC. Typical serum biochemical parameters (alanine aminotransferase, aspartate aminotransferase, creatinine, blood urea nitrogen, total bilirubin, and uric acid), indicating hepatic and renal functions, were analyzed to evaluate impact of high dose (433 mg/kg body weight) GMDTC on mice. As compared to control group, no significant changes were observed in high dose GMDTC group (Supplementary Table 2). During the last 24 h of treatment, urine samples were collected to determine the Cd excretion level. At the end of treatment, mice were euthanized individually in a CO2 chamber and kidney tissues were collected for Cd measurements. A counterpart control group was administrated 0.9% saline instead of CdCl2, GMDTC or phloretin. The doses and duration of chemicals used for animal treatment were previously reported in our study or elsewhere.,, Animal experiments complied with the Guide for the Care and Use of Laboratory Animals, and the Animal Care and Use Committee of GDMLAC in China approved animal test protocols (#00062324).
Measurement of urinary Cd2+ by graphite furnace atomic absorption spectroscopy
Concentrations of Cd2+ in urine were measured by a GFAAS (PerkinElmer Pinnacle 900T, Waltham, MA, USA) as previously described. Briefly, a hollow cathode lamp operated at the current of 6.0 mA and a wavelength of 228.8 nm with a spectral bandpass of 0.8 nm was used. After digested in 0.5 mL 70% nitric acid at 70°C, the urine samples and standard samples were diluted five times with a solution containing 0.5% nitric acid and 0.1% TrionX-100. 10 microliter of the solution was injected into the graphite furnace for analysis. Urinary Cd2+ concentration was calculated using the following equation:
Measurement of Cd2+ in cells and kidneys by inductively coupled plasma mass spectrometry
HK-2 cells with or without CRISPR/Cas9 targeted editing were treated in a similar manner as GMDTC rescue cell viability experiments. The amount of 2 × 106 cells were seeded in a 100-mm plate with 10 mL complete medium for overnight culturing before treatments. On the following day, cells of about 70% confluence were first pretreated with 250 μM CdCl2 for 2 h, washed twice with PBS, and then replaced with fresh culture medium including 0 or 1500 μM GMDTC for another 24 h culturing. After incubation, cells were washed twice with PBS and collected by trypsin treatment. Total cell numbers were checked by TC20 Automated Cell Counter (Bio-rad, Hercules, CA, USA). Cell pellets were collected by centrifuge and digested in 100 μL concentrated 70% nitric acid (HNO3) for 2 h at 100°C. After digestion, mixtures were diluted in 5 mL 2% HNO3, and filtered before analysis of Cd using ICP-MS. In a similar fashion, Cd in the kidney was prepared and analyzed by ICP-MS as described previously. Briefly, the whole right kidney of each animal was collected, weighted (about 200–300 mg) and dried at 80°C for 1.5 h. The dried samples were then digested in 0.5 mL of concentrated 70% HNO3 at 70°C for overnight, followed by dilution in 5 mL 2% HNO3. Cd112 was quantified using authentic metal standards (AACD1, Inorganic Ventures, Lakewood, NJ, USA) and Indium115 (AAIN1, Inorganic Ventures, Lakewood, NJ, USA) was added as internal standard. ICP-MS was performed at the Chemistry Instrumentation Center at the University at Buffalo. Cd2+ content in cells and kidney samples were calculated using the following equation:
Statistical analysis
All data are presented as mean ± SD. One-way ANOVA and post-hoc Tukey Tests were conducted using the GraphPad Prism software (GraphPad Software, San Diego, CA) to analyze the differences between the groups. Mann-Kendall test was used to assess the significance of trend in rescuing effects mediated by GMDTC along with increasing concentrations in Cd-treated cell lines. It was considered statistically significant when the p-value was less than 0.05.
Results
Generation of GLUT2−/− or SGLT2−/− KO HK-2 cell lines with CRISPR/Cas9 gene editing system
We utilized the CRISPR/Cas9 gene editing tool to construct GLUT2−/− and SGLT2−/− KO cell lines. Two sgRNAs were designed for each gene. The gene editing activity of each sgRNA was evaluated using the T7E1 assay. In addition to the multiple bands produced by sgRNA 1 of SGLT2, the cleaved bands by the T7E1 enzyme showed that insertion or deletion mutations (indel) were introduced into the genomes (Supplementary Figure 2(a)). The gene modification efficiencies were 62.90% and 36.43% for GLUT2 sgRNA 1 and 2, respectively, and 27.02% for SGLT2 sgRNA2. It is proposed that pool cells with high cleavage efficiency are suitable for further analysis. Thus, transfected cells with GLUT2 sgRNA 1 and SGLT2 sgRNA 2 were selected to isolate single clones for further validation and proliferation. Then, we analyzed the nucleotide sequences of the PCR products of targeted regions in single clones, and further confirmed the successful indel mutation of the intended GLUT2 and SGLT2 gene locus by both sgRNAs used (Supplementary Figure 2(b) & (c), Supplementary Figure 3(a) & (b)). Next, to investigate the type of mutations in more detail, Sanger sequencing files of PCR products of single clones were analyzed by the TIDE assay. The TIDE analysis results showed that GLUT2−/− single clone 1 (GLUT2−/− S1) exhibited 23 base deletions, and the percentage of sequences with this deletion was 98.4%, while 96.3% of sequences in SGLT2−/− single clone 14 (SGLT2−/− S14) showed eight base deletions (Supplementary Figure 2(d)). Off-target effect is a general concern for the use of CRISPR/Cas9 technology. PCR products of potential off-target sites were subjected to T7E1 enzyme detection (Supplementary Figure 4) and Sanger sequencing (Supplementary Figure 3(c) & D1-3). The results showed that CRISPR/Cas9 does not cause non-specific mutagenesis at high-risk regions as suggested by Cas-OFFinder tool, and each sgRNA is highly specific for intended target site. Furthermore, western blotting results showed that the CRISPR/Cas9 editing had resulted in significant loss of GLUT2 and SGLT2 in GLUT2−/− and SGLT2−/− KO cell lines, respectively Supplementary Figure 2(e)). Though residual protein expression was observed probably due to translation reinitiation leading to N-terminally truncated target proteins or skipping of the edited exon leading to protein isoforms with internal sequence deletions, frameshift mutations could still lead to full disruption of the protein despite no or weak nonsense-mediated decay of target mRNAs. Collectively, these results indicate that GLUT2−/− and SGLT2−/− KO HK-2 cell lines were successfully generated by the CRISPR/Cas9 system.
Compromised protective effects of GMDTC in Cd-treated GLUT2−/− and SGLT2−/− KO HK-2 cell lines
As shown in Supplementary Figure 5(a), at concentrations up to 5,000 μM, GMDTC did not cause significant cytotoxicity on HK-2 cells post 24 h treatment. We choose 1,500 μM GMDTC as the highest dose of GMDTC used in our experiment. At this dose, there was no significant impact on cell viability in all three cell lines, HK-2, GLUT2−/− KO and SGLT2−/− KO cells (Figure 2(a)). We examined the rescue ability of GMDTC in HK-2 cells pretreated with CdCl2 because Cd-induced renal toxicity is largely due to the accumulation of Cd in proximal tubule cells in the kidney. Therefore, as an effective chelator to prevent Cd-induced kidney cell cytotoxicity, the chelator must be able to remove the accumulated Cd2+ from the cell interior. All three cell lines were pretreated with 250 μM CdCl2 (IC50 of 2-h acute CdCl2 treatment, Supplementary Figure 5(b)) for 2 h. After washing the cells and the addition of new medium, various concentrations of GMDTC were administered and cells were further incubated for 24 h. The administration of GMDTC resulted in a significantly dose-dependent improvement of HK-2 cell viability at 24-h post treatment, and HK-2 cell variability with 1,500 μM GMDTC treatment is 1.32-fold higher when compared to HK-2 cells treated with CdCl2 alone (Figure 2(b)). Furthermore, the fold change of HK-2 cell viability, indicating rescue effect of GMDTC treatment, demonstrated a monotonic upward trend along with increasing concentrations of GMDTC (Mann-Kendall trend test p < 0.01). In comparison, the rescuing ability of GMDTC for HK-2 cells from Cd2+-induced cytotoxicity was significantly reduced in GLUT2−/− and SGLT2−/− KO cells at almost all compared doses (Figure 2(b), Supplementary Figure 6). In addition, no dose-dependent effect was observed in both GLUT2−/− and SGLT2−/− KO cells lines (Figure 2(b)). We further measured Cd levels inside of CdCl2-exposed cells with or without GMDTC administration at 1,500 μM by ICP-MS. The amount of Cd in HK-2 cells treated with Cd alone was as high as 327.23 ng/106 cells and was significantly decreased to 234.61 ng/106 cells after 24-h GMDTC rescue treatment (Figure 2(c)). The removal of Cd was 28.3% (Figure 2(d)). Although GMDTC’s effect in removing Cd from the cells was evident in each GLUT2−/− or SGLT2−/− KO cell line (Figure 2(d)), the efficiency was significantly decreased in each KO cell lines, 7.4% in GLUT2−/− KO cells and 14.6% in SGLT2−/− KO cells, respectively, compared to that of HK-2 cells (28.28%).
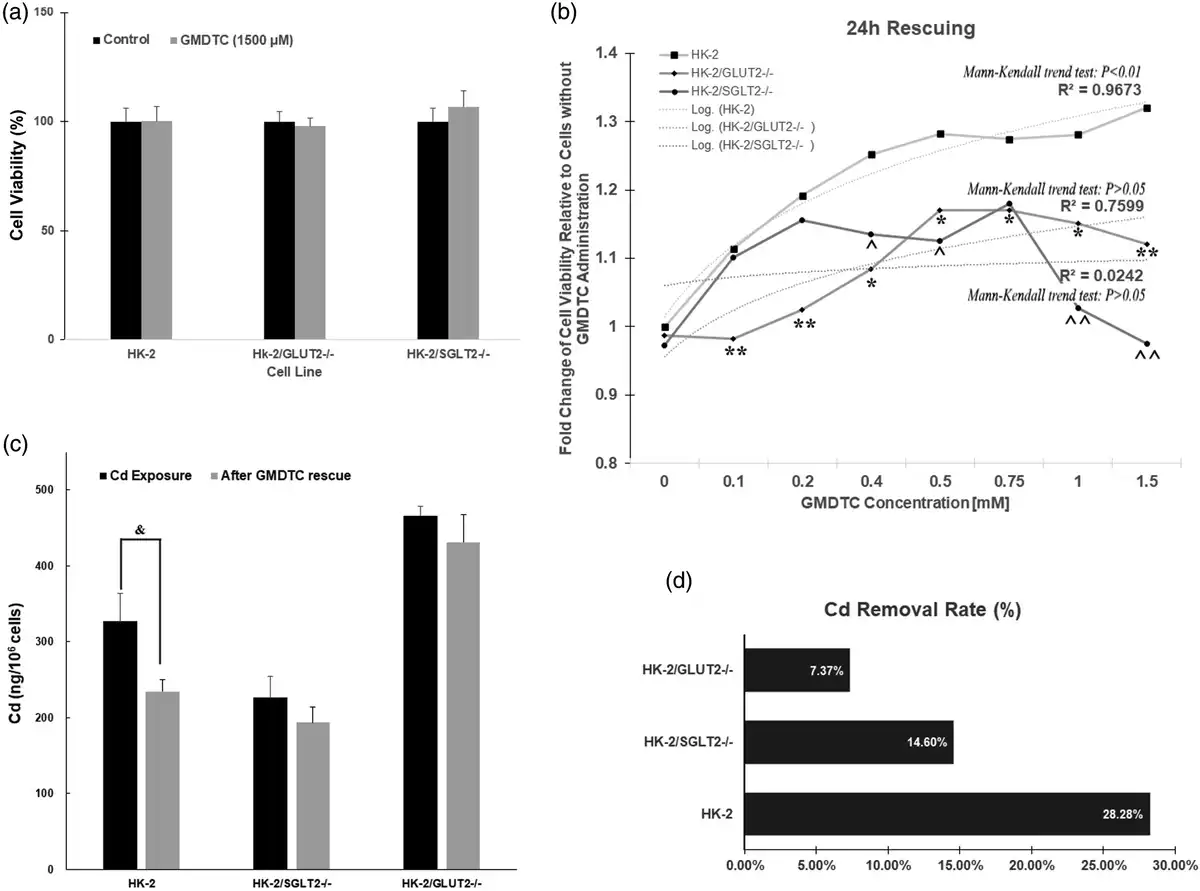
Figure 2
Compromised protection of GMDTC to Cd-induced cytotoxicity cannot protect cells from cadmium-induced cytotoxicity in KO cells. (a) HK-2, HK-2/GLUT2−/− and HK-2/SGLT2−/− cells treated with 1500 μM GMDTC exhibited no significant difference on cell viability compared to control after 24 h. (b) Cell viability analysis of all three cell lines, HK-2, HK-2/GLUT2−/− and HK-2/SGLT2−/− cells post GMDTC treatment. (c) Cd levels among three cell lines with Cd exposure and after GMDTC administration; (d) Cadmium removal ratio after GMDTC rescue. Data are mean ± SD of 3 independent experiments. HK-2 vs. HK-2/GLUT2−/−cells: “*”p < 0.05, “**”p < 0.01; HK-2 vs. HK-2/SGLT2−/−: “”p < 0.05, “”p < 0.01.
The involvement of renal glucose reabsorption pathway in GMDTC-mediated Cd removal in vivo
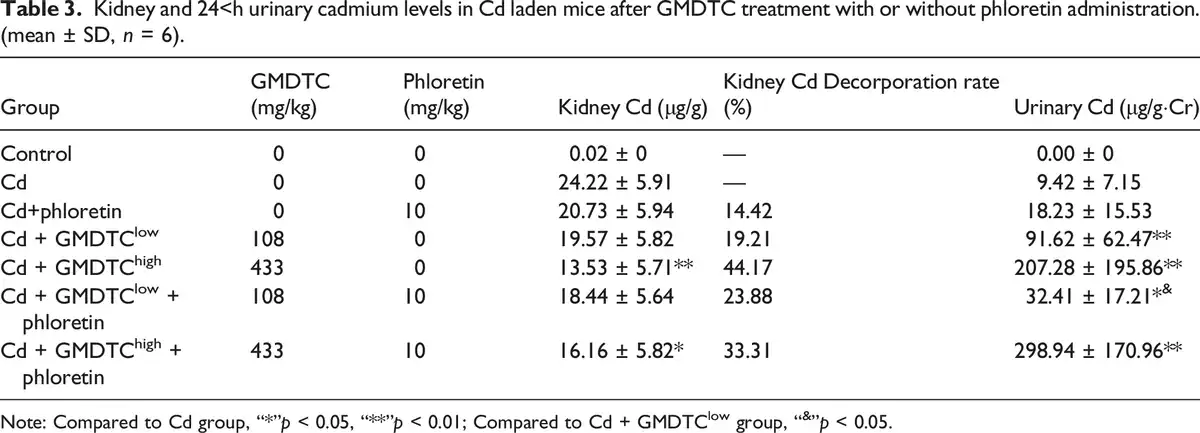
To further examine the role of kidney glucose reabsorption pathway in GMDTC-mediated cleaning Cd in vivo, we treated Cd-laden mice with GLUT2 inhibitor (phloretin) in addition to GMDTC administration. As expected, GMDTC treatment had a significant effect in removing Cd from the kidneys, and significantly increased the excretion of Cd in the urine (Table 3). This effect was observed more significantly in animals given high dose of GMDTC. At low dose of GMDTC treatment group, when the activity of GLUT2 was inhibited by phloretin, the Cd level in kidney was largely comparable between phloretin + GMDTClow + Cd group and the GMDTClow + Cd group. The excretion of Cd from urine was significantly decreased in phloretin + GMDTClow + Cd group (Table 3). The Cd level in the kidneys was increased in phloretin + GMDTChigh + Cd group compared to the GMDTChigh + Cd group while the difference was not significant (p = 0.0543) due to high variance of the data (Table 3). However, urinary Cd level in phloretin + GMDTChigh + Cd group was further increased, which was significantly higher than the urinary level in the GMDTChigh + Cd group (Table 3).
Discussion
Cd is one of the most toxic heavy metals that human beings can be exposed to at work and in the industrial environment., Once absorbed, Cd mainly circulates to the kidneys and accumulates in the proximal tubular cells throughout life with a half-life of several decades., Once the accumulation of Cd reaches certain levels, it can lead to chronic damage to the kidneys and cause irreversible deleterious consequences.,, However, there is no effective treatment for cadmium induced toxicity, since existing chelating agents have limited safety and deficiency in the clinic.– Therefore, to protect against Cd-induced renal toxicity, treatments aiming at limiting Cd accumulation and enhancing its removal from the kidneys have attracted great interest. Our previous work showed GMDTC was low in toxicity in vivo and exhibited a strong chelating ability with toxic heavy metals, including Cd, and a high efficiency in removing Cd from kidneys. In addition, given the hydrophilic characters of GMDTC and the formed GMDTC-Cd complexes, we have hypothesized that it is likely that they are transported through active transport mechanisms, the renal glucose transportation pathway, instead of a passive transport mechanism. In this study, we constructed cell models by knocking out glucose transporters (GLUT2 and SGLT2) and animal model by inhibiting the GLUT2 to demonstrate the impact of renal glucose transporters in GMDTC-mediated removal of Cd in vitro and in vivo.
Chelators, the main type of metal removal agents, have been studied intensively as potential therapeutic drugs for Cd-induced renal toxicity.– However, there are currently no approved chelation therapies available due to their severe side effects and disappointing ineffectiveness in removing Cd from the kidney., Although Fulgenzi et al. have reported that EDTA chelation therapy significantly increased urinary elimination of Cd based on a study of 379 patients with neurodegenerative diseases, EDTA could cause nephrotoxicity especially with repeated high-dose treatment (above 75 mg/kg) in subjects with a previous history of kidney damage and increase the risk of renal dysfunction. Dimercaprol (British Anti-Lewisite, BAL) and its analogues meso-2, 3-dimercaptosuccinic acid (DMSA) are also used as antidotes for heavy metal poisoning. In contrast, BAL may increase burdens of kidney and liver, decrease the level of patients’ survival and even enhance nephrotoxicity. A randomized trial, in which 396 children were given DMSA at the dose of 1,050 mg/body surface area (m2)/day for 7 days, showed that DMSA did not lower blood Cd in children with background exposure. Combination therapy with chelating agents and other substances has been proposed in the management of heavy metal toxicity., But overall, there is a general dearth of studies to develop chelators that may be employed for the treatment of Cd toxicity.
Our data showed GMDTC administration could significantly reduce intracellular Cd level in HK-2 cells pre-treated with Cd, and the removal of Cd in 24 h was 28.3%. This reduction of Cd level inside of HK-2 cells led to a reduced toxicity and an increase of cell viability in a dose-dependent manner. In GLUT2−/− or SGLT2−/− KO HK-2 cell models, cells were similarly pretreated with Cd before GMDTC administration. However, GMDTC treatment under most concentrations could not improve the cell viability when compared to KO cells treated with Cd alone, as observed in the GMDTC rescuing experiment. The reduced effects of GMDTC in protecting cytotoxicity from Cd exposure was supported by the evidently reduced removal ratio of Cd from cells. The removal ratio was reduced to 7.4% and 14.6% in HK-2 cells with GLUT2 or SGLT2 gene knocked out, respectively. In view of the hydrophilic characters of GMDTC and the formed GMDTC-Cd complexes as well as the open chain glucose moiety harbored by GMDTC, we hypothesize the transportation process of GMDTC and GMDTC-Cd is comparable with glucose. Firstly, GMDTC filtered at the glomerulus of nephron is actively transported into the proximal tubule epithelial cells via SGLT2 against a concentration gradient coupled with the inward diffusion of sodium ions which is maintained by an Na+/K+ ATPase pump; in renal tubule cells, GMDTC binds free Cd ions to form GMDTC-Cd complexes; driven by concentration gradient, intracellular GMDTC-Cd diffuses out to the plasma facilitated by the facilitative glucose transporters GLUT2 in the basolateral membranes; the majority of GMDTC-Cd complexes is consequently excreted from the body through urine following glomerular filtration. Although our research measures Cd not GMDTC and GMDTC-Cd complex directly in the constructed model cells and kidneys of mice, the data does suggest that knocking down of the SGLT2 and GLUT2 affects the Cd cytotoxicity and the deposition of Cd, which is compatible with our hypothesis. However, we also noticed that the GMDTC’s effects in removing Cd from the cells were not completely abolished. One possible explanation is that GMDTC can enter cells by multiple mechanisms, such as other glucose transporters (e.g., SGLT1, sodium-dependent glucose transporter 1). Studies have shown that a compensatory increase in SGLT1-mediated glucose reabsorption occurs when SGLT2 is inhibited in mice., In addition, the chemical structure of GMDTC contains not only a glucose motif, but also a methionine group. GMDTC could utilize amino acid transporter entering cells. Considering that there could be more mechanisms involved in the transportation of GMDTC entering the cells, we conducted an animal study by blocking GLUT2 transporter using phloretin, so blocking the transportation route of GMDTC out of the cells. The in vivo results are not as clear as the in vitro data. GMDTC’s effects in removing Cd from the kidney were clearly demonstrated at both low and high dose. However, blocking of the GLUT2 transporter has less impact on the Cd level in the kidneys. Only the high GMDTC dose group showed a higher Cd in the kidney after phloretin treatment with a borderline significant level (p = 0.054), as compared with the high GMDTC group without phloretin treatment. Although, as projected, the urinary Cd level was significantly lower in the low GMDTC dose group post phloretin treatment, phloretin treatment further increased the urinary Cd level in the high GMDTC dose group. One possible reason is that phloretin could partially, not completely, inhibit the function of GLUT2, which was also reported in other study. In addition, along with degrading metabolism of phloretin, the inhibition effect of GLUT2 was further decreased. Though the low GMDTC group demonstrated inhibition of urinary Cd excretion after phloretin treatment, the high GMDTC dose may potentially compensate for or mask the inhibition effect of GLUT2 by phloretin. Thus, the kidney GLUT2 conditional knockout mice will warrant the further validation of the mode of Cd-GMDTC complex transportation. Altogether, while the results are not all consistent, particularly the in vivo data, we believe that the constructed GLUT2−/− and SGLT2−/− knockout cell models provide convincing evidence supporting the involvement of renal glucose transporter in GMDTC-mediated cleaning of Cd from cells.
This study is not without potential issues. Our repeated sequencing data confirms the completely knockout of the SGLT2 and GLUT2 genes in the HK-2 cells. However, western blotting results indicate there are measurable SGLT2 and GLUT2 protein. We could not determine whether these detectable proteins are SGLT2 and GLUT2 protein or not. Though residual protein expression was observed probably due to translation reinitiation leading to N-terminally truncated target proteins or skipping of the edited exon leading to protein isoforms with internal sequence deletions, frameshift mutations could still lead to full disruption of the protein despite no or weak nonsense-mediated decay of target mRNAs. In addition, the knocking down of SGLT2 and GLUT2 in HK-2 cells apparently affect the accumulation of Cd in the cell. The reason is unknown at this time. The animal study did not deliver as clear results as the in vitro experiments, and the specificity of phloretin’s effects in blocking GLUT2 transporter is not satisfied and the blocking effects are not complete. We are in the process of obtaining SGLT2 knockout mice and of generating kidney GLUT2 conditional knockout mice. We expect that these knockout mouse models should provide more defined information regarding the role of glucose transporters in GMDTC-mediated Cd removal.
In conclusion, we show that GMDTC is a relative safe with low toxicity and can remove Cd inside of cells and from kidney, supporting GMDTC as an effective chelator in protecting Cd-caused chronic renal toxicity. The ability of GMDTC to protect cells from toxicity was significantly decreased in GLUT2 and SGLT2 gene KO cells, suggesting that renal glucose transportation pathway is involved in the GMDTC-mediated Cd removal from kidney cells.
Declaration of conflicting interests The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (81872571), Support Scheme of Guangzhou for Leading Talents in Innovation and Entrepreneurship (2019013), Zhuhai Industry-University Research Cooperation Project (ZH22017001210086PWC) and Guangdong Basic and Applied Basic Research Foundation (2021A1515010771).
Supplemental Material Supplemental material for this article is available online.
References
- 1. Genchi G, Sinicropi MS, Lauria G, et al. The effects of cadmium toxicity. Inter J Environ Res Pub Health 2020; 17: 3782.
- 2. Tchounwou PB, Yedjou CG, Patlolla AK, et al. Heavy metal toxicity and the environment. Exper Supplem 2012; 101: 133–164.
- 3. World Health Organization. Preventing disease through healthy environments: exposure to cadmium: a major public health concern. Geneva, Switzerland: World Health Organization, 2019.
- 4. Fu Z, Xi S. The effects of heavy metals on human metabolism. Toxicol Mech Meth 2020; 30: 167–176.
- 5. Zalups RK, Ahmad S. Molecular handling of cadmium in transporting epithelia. Toxicolo Appl Pharmacol 2003; 186: 163–188.
- 6. Hill A, Gailer J. Linking molecular targets of Cd in the bloodstream to organ-based adverse health effects. J Inorg Biochem 2021; 216: 111279.
- 7. Rani A, Kumar A, Lal A, et al. Cellular mechanisms of cadmium-induced toxicity: a review. Inter J Environ Health Res 2014; 24: 378–399.
- 8. Klaassen CD, Liu J, Diwan BA. Metallothionein protection of cadmium toxicity. Toxicol Appl Pharmacol 2009; 238: 215–220.
- 9. Wu X, Cobbina SJ, Mao G, et al. A review of toxicity and mechanisms of individual and mixtures of heavy metals in the environment. Environ Sci Pollut Res 2016; 23: 8244–8259.
- 10. Joseph P. Mechanisms of cadmium carcinogenesis☆. Toxicol Appl Pharmacol 2009; 238: 272–279.
- 11. Sabolić I, Breljak D, Škarica M, et al. Role of metallothionein in cadmium traffic and toxicity in kidneys and other mammalian organs. Biometals 2010; 23: 897–926.
- 12. Klaassen CD, Choudhuri S, McKim JM Jr, et al. In vitro and in vivo studies on the degradation of metallothionein. Environ Health Perspec 1994; 102: 141–146.
- 13. Đukić-Ćosić D, Baralić K, Javorac D, et al. An overview of molecular mechanisms in cadmium toxicity. Curr Opin Toxicol 2020; 19: 56–62.
- 14. Moulis J-M. Cellular mechanisms of cadmium toxicity related to the homeostasis of essential metals. Biometals 2010; 23: 877–896.
- 15. Yang H, Shu Y. Cadmium transporters in the kidney and cadmium-induced nephrotoxicity. Inter J Mole Sci 2015; 16: 1484–1494.
- 16. Gobe G, Crane D. Mitochondria, reactive oxygen species and cadmium toxicity in the kidney. Toxicol Let 2010; 198: 49–55.
- 17. Glicklich D, Shin CT, Frishman WH. heavy metal toxicity in chronic renal failure and cardiovascular disease. Cardiol Rev 2020; 28: 312–318.
- 18. Kim J-J, Kim Y-S, Kumar V. Heavy metal toxicity: an update of chelating therapeutic strategies. J Tra Ele Med Biol 2019; 54: 226–231.
- 19. Rajak C, Singh N, Parashar P. Metal toxicity and natural antidotes: prevention is better than cure. Environ Sci Pollut Res 2020; 27(35): 43582–43598.
- 20. Hogarth G. Metal-dithiocarbamate complexes: chemistry and biological activity. Mini-Rev Med Chem 2012; 12: 1202–1215.
- 21. Tátrai E, Brózik M, Náray M, et al. Combined pulmonary toxicity of cadmium chloride and sodium diethyldithiocarbamate. J Appl Toxicol 2001; 21: 101–105.
- 22. Tandon SK, Prasad S, Singh S. Chelation in metal intoxication: influence of cysteine orN-acetyl cysteine on the efficacy of 2,3-dimercaptopropane-1-sulphonate in the treatment of cadmium toxicity. J Appl Toxicol 2002; 22: 67–71.
- 23. Rahimzadeh MR, Rahimzadeh MR, Kazemi S, et al. Cadmium toxicity and treatment: an update. Caspian J Inter Med 2017; 8: 135–145.
- 24. Tang X, Zhu J, Zhong Z, et al. Mobilization and removing of cadmium from kidney by GMDTC utilizing renal glucose reabsorption pathway. Toxicol Appl Pharmacol 2016; 305: 143–152. DOI: .
- 25. Ge Y, Zheng N, Chen X, et al. GMDTC chelating agent attenuates cisplatin-induced systemic toxicity without affecting antitum'or efficacy. Chem Res Toxicol 2019; 32: 1572–1582.
- 26. Zhong Z, Tang W, Li G, et al. Study on the Specific Complexation of GMDTC and Metal Ion. Chinese J Indus Hyg Occupat Dis 2018; 36: 408–412.
- 27. Bakris GL, Fonseca VA, Sharma K, et al. Renal sodium-glucose transport: role in diabetes mellitus and potential clinical implications. Kidney International 2009; 75: 1272–1277.
- 28. Bailey CJ. Renal glucose reabsorption inhibitors to treat diabetes. Trends Pharmacol Sci 2011; 32: 63–71.
- 29. Mather A, Pollock C. Glucose handling by the kidney. Kidney International 2011; 79: S1–S6.
- 30. Anderson EM, Haupt A, Schiel JA, et al. Systematic analysis of CRISPR-Cas9 mismatch tolerance reveals low levels of off-target activity. J Biotechnol 2015; 211: 56–65.
- 31. Labuhn M, Adams FF, Ng M, et al. Refined sgRNA efficacy prediction improves large- and small-scale CRISPR-Cas9 applications. Nucl Acids Res 2018; 46: 1375–1385.
- 32. Lee EJ, Kim JL, Kim YH, et al. Phloretin promotes osteoclast apoptosis in murine macrophages and inhibits estrogen deficiency-induced osteoporosis in mice. Phytomedicine 2014; 21: 1208–1215. DOI:
- 33. Ying Y, Jin J, Ye L, et al. Phloretin prevents diabetic cardiomyopathy by dissociating Keap1/Nrf2 complex and inhibiting oxidative stress. Front Endocrinol 2018; 9: 774. DOI:
- 34. Lackner DH, Carré A, Guzzardo PM, et al. A generic strategy for CRISPR-Cas9-mediated gene tagging. Nature Commun 2015; 6: 10237.
- 35. Smits AH, Ziebell F, Joberty G, et al. Biological plasticity rescues target activity in CRISPR knock outs. Nature Meth 2019; 16: 1087–1093.
- 36. Lindeboom RG, Supek F, Lehner B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nature Genet 2016; 48: 1112–1118.
- 37. Sinicropi MS, Amantea D, Caruso A, et al. Chemical and biological properties of toxic metals and use of chelating agents for the pharmacological treatment of metal poisoning. Arch Toxicol 2010; 84: 501–520.
- 38. Godt J, Scheidig F, Grosse-Siestrup C, et al. The toxicity of cadmium and resulting hazards for human health. J Occupat Med Toxicol (London, England) 2006; 1: 22–26.
- 39. Satarug S. Dietary cadmium intake and its effects on kidneys. Toxics 2018; 6: 15.
- 40. Zhang H, Reynolds M. Cadmium exposure in living organisms: a short review. Sci Tot Environ 2019; 678: 761–767.
- 41. Kurowska E, Bal W. Recent advances in molecular toxicology of cadmium and nickel. Adv Mole Toxicol 2010; 4: 85–126.
- 42. Badisa VL, Latinwo LM, Odewumi CO, et al. Mechanism of DNA damage by cadmium and interplay of antioxidant enzymes and agents. Environ Toxicol 2007; 22: 144–151.
- 43. Bjørklund G, Crisponi G, Nurchi VM, et al. A review on coordination properties of thiol-containing chelating agents towards mercury, cadmium, and lead. Molecules 2019; 24: 3247.
- 44. Andersen O. Chemical and biological considerations in the treatment of metal intoxications by chelating agents. Mini Rev Med Chem 2004; 4: 11–21.
- 45. Fulgenzi A, Vietti D, Ferrero ME. EDTA chelation therapy in the treatment of neurodegenerative diseases: an update. Biomedicines 2020; 8: 269.
- 46. Zhai Q, Narbad A, Chen W. Dietary strategies for the treatment of cadmium and lead toxicity. Nutrients 2015; 7: 552–571.
- 47. Wu X, Su S, Zhai R, et al. Lack of reversal effect of EDTA treatment on cadmium induced renal dysfunction: a fourteen-year follow-up. Biometals 2004; 17: 435–441.
- 48. Kamenova K, Gluhcheva Y, Dorkov P, et al. Comparative assessment of the effects of meso-2,3-dimercaptosuccinic acid and salinomycin on spleen function of cadmium-exposed mice. Environ Sci Pollut Res 2019; 26: 33304–33310.
- 49. Mehta A, Pant SC, Flora SJ. Monoisoamyl dimercaptosuccinic acid induced changes in pregnant female rats during late gestation and lactation. Repro Toxicol 2006; 21: 94–103.
- 50. Cao Y, Chen A, Bottai M, et al. The impact of succimer chelation on blood cadmium in children with background exposures: a randomized trial. J Pediat 2013; 163: 598–600.
- 51. Flora SJ, Pachauri V. Chelation in metal intoxication. Inter J Environ Res Pub Health 2010; 7: 2745–2788.
- 52. Rieg T, Masuda T, Gerasimova M, et al. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am J Physiol-Ren Physiol 2014; 306(2): F188–F193.
- 53. Powell DR, DaCosta CM, Gay J, et al. Improved glycemic control in mice lacking Sglt1 and Sglt2. Am J Physiol-Endocrinol and Metabol 2013; 304(2): E117–E130.
- 54. Scow JS, Iqbal CW, Jones TW 3rd, et al. Absence of evidence of translocation of GLUT2 to the apical membrane of enterocytes in everted intestinal sleeves. J Surg Res 2011; 167: 56–61. DOI:
Abbreviations
Cd: Cadmium
Cd-MT: Cd-metallothionein
DMEM: Dulbecco’s Modified Eagle Medium
DMPS: 2,3-dimercapto-1-propane sulfonic acid;
DTC: Dithiocarbamates
EDTA: ethylene diamine tetra acetic acid
FBS: fetal bovine serum
GFAAS: graphite furnace atomic absorption spectroscopy
GLUT2: glucose transporter 2;
GMDTC: sodium (S)-2-(dithiocarboxylato((2S,3 R,4R,5 R)-2,3,4,5,6-pentahydroxyhexyl) amino)-4(methylthio)butanoate
ICP-MS: inductively coupled plasma mass spectrometry
KO: knockout
MT: metallothionein
NAC: N-acetyl cysteine
PAM: protospacer adjacent motif
PBS: phosphate buffered saline
RNP: ribonucleoprotein
SGLT2: sodium-dependent glucose transporter 2
sgRNA: single-guide RNA
SLC2A2: solute carrier family 2 member 2
SLC5A2: solute carrier family 5 member 2
T7E1: T7 endonuclease 1
TIDE: tracking of indels by decomposition