Introduction
The incidence of short stature due to growth hormone deficiency (GHD) is estimated to be as 1/4,000–1/10,000 live births and about 3–30% have a genetic background. The genetic forms of GHD may occur as isolated GHD (IGHD) or as a component of multiple pituitary hormone deficiency (MPHD). IGHD appears in sporadic or familial forms, occurring with various inheritances; autosomal recessive (IGHD IA and IB), autosomal dominant (IGHD II), and X-linked (IGHD III) [].
The GH1 gene, encoding growth hormone (GH) is localized in the chromosome 17q23.3 region and consists of five exons []. Mutated GH1 causes IGHD; however, some types (especially IGHD II) may present with MPHD []. Biallelic pathogenic variants in the GH1 gene cause IGHD IA and present with severe short stature, usually becoming apparent in the first months of life and occurring mainly due to gross deletions of different sizes or, less frequently, by insertions, frameshift, or nonsense variations. In the IGHD IA, endogenous GH secretion is undetectable and anti-GH antibodies may develop during the use of recombinant human GH (rhGH). The IGHD IB results from biallelic nonsense, splice-site, and frameshift alterations in the GH1 gene, leading to low but bio-inactive GH, and presents with mild to severe short stature. The autosomal dominant form (IGHD II) occurs due to splice-site, missense, nonsense, and splice enhancer mutations and may have variable expressivity []. This monoallelic form is associated with MPHD, in which hormone deficiencies may develop over time []. Herein, we present 5 patients from four unrelated families, diagnosed with IGHD or MPHD associated with different types of GH1 gene variants.
Subjects and Methods
Subjects
Patients with the clinical diagnosis of IGHD/MPHD (105 patients from 102 families) who were followed by the departments of Pediatric Endocrinology and Medical Genetics at the Istanbul Faculty of Medicine were included in this project. Clinical and laboratory findings were recorded from the medical files, and treatment outcomes and long-term follow-up results were documented. All auxological parameters were expressed in standard deviation score (SDS) according to national standards [].
Hormonal Workup
GH stimulation tests (clonidine and L-Dopa) were performed after overnight fasting according to standard methods []. Human GH was measured by a solid-phase, two-site chemiluminescent immunometric assay (Immulite 2000 system, Siemens AG, Berlin and Munich, Germany). IGF-I and IGFBP-3 levels were measured by immunoradiometric assay (Immunotech Beckman Coulter Inc., Prague, Czech Republic). Luteinizing hormone (LH), follicle-stimulating hormone, free thyroxine, and thyroid-stimulating hormone (TSH) were studied by electrochemiluminescence immunoassay (Cobas, Roche Diagnostics, Mannheim, Germany). Adrenocorticotropic hormone (ACTH), cortisol, and testosterone were analyzed by immunochemiluminescence assay (ICMA) (Immulite 2000 system, Siemens AG, Berlin and Munich, Germany).
Genetic Analyses
Genomic DNA was extracted from the peripheral blood cells of the patients and parents by using standard techniques. Next-generation sequencing-based in-house designed targeted gene panel for GHD and MPHD included 25 genes (BMP4, FGF8, FGFR1, GH1, GHR, GHRH, GHSR, HESX1, HHIP, IGFALS, IGF1, IGF1R, IGFBP3, IGSF1, LHX3, LHX4, OTX2, POU1F, PROKR2, PROP1, SHH, SHOX, SOX3, STAT5B, WDR11) with 99.2% coverage. Sequencing was performed on the Ion Torrent PGM sequencer (Thermo Fisher Scientific, Waltham, MA, USA). Data analysis and variant calling were carried out via Torrent Suite and Ion Reporter system (Thermo Fisher Scientific, Waltham, MA, USA). All alterations were classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines []. Segregation analysis using Sanger sequencing was performed in the families for clinically relevant variants classified as pathogenic or likely pathogenic according to ACMG criteria. Alterations were further searched via open data sources (dbSNP, ClinVar) and HGMD (HGMD®Professional 2022.4) [].
Multiplex ligation-dependent probe amplification (MLPA) was performed in cases with normal gene panel results. SALSA MLPA Probe mix P216 Growth Hormone Deficiency (MRC Holland, P216 B1 GHD mix) comprised of seven genes including GHRHR (exons 1, 2, 3, 4, 5, 6, 7, 9, 10a, 11, 12, 13); LHX3 (exons 1, 2, 3, 4, 5, 6, 7); LHX4 (exons 1, 2, 3, 4, 5, 6); POU1F1 (exons 1, 2b, 3, 4, 6); PROP1 (exons 1, 2, 3); GH1 (exons 1, 3a, 4, 5, and upstream region); HESX1 (exons 1, 2, 3, 4). Coffalyser (version 140,721.1958; MRC-Holland) was used for the analyses.
Results
Clinical Findings
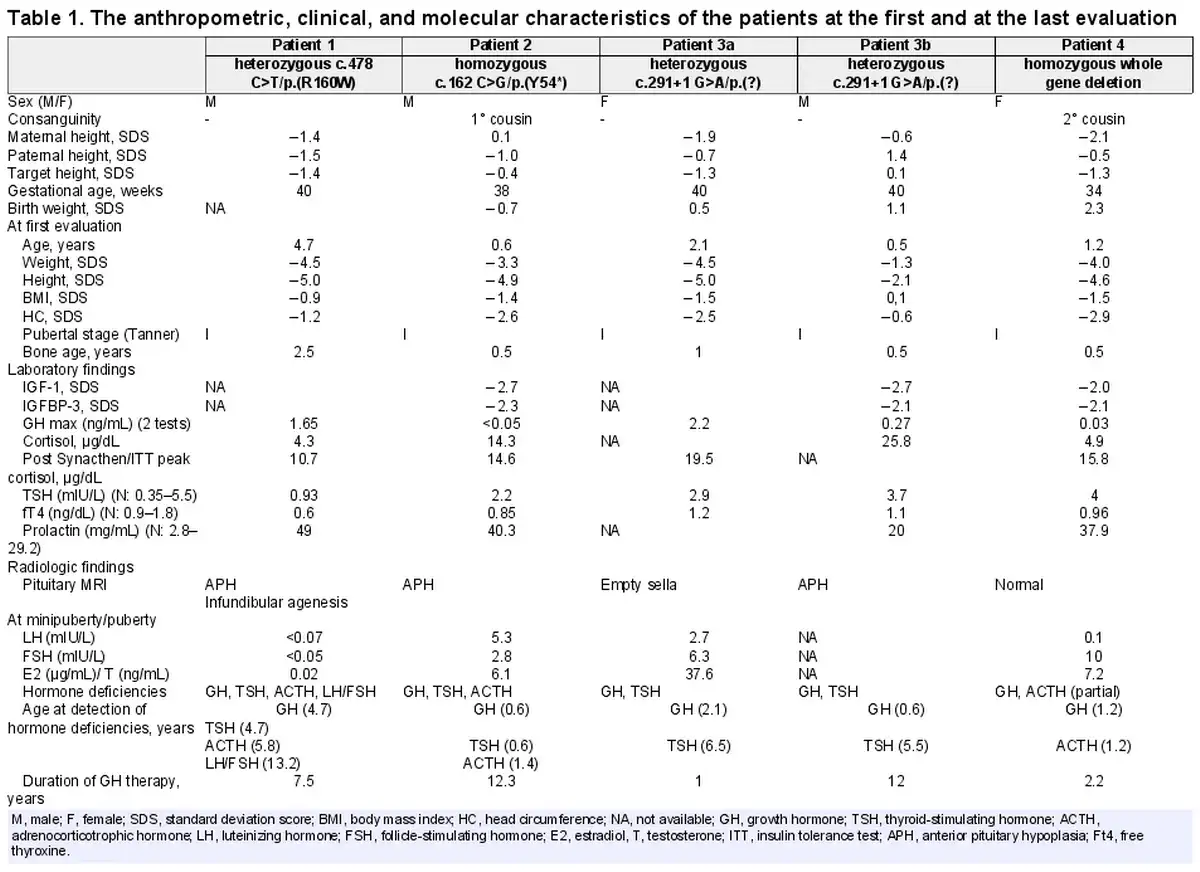
The clinical and biochemical characteristics of 5 patients from four unrelated families were evaluated. The demographic data, and clinical and laboratory findings of patients at the first and last evaluations are illustrated in Table 1.
Patient 1
Patient 1 was the first child of non-consanguineous parents, and he was born by spontaneous vaginal delivery (SVD) at 40 gestational weeks (GW). He was referred to pediatric endocrinology for the evaluation of short stature, at 4.7 years of age. At first evaluation, he had a mildly depressed nasal bridge with no other obvious dysmorphic features. Height and body mass index was −5.0 SDS and −0.9 SDS, respectively. His neurodevelopmental milestones were normal for his age. Baseline laboratory investigations including complete blood count, serum biochemistry, and urine analysis were unremarkable. Endocrine workup revealed TSH deficiency, with a low-normal TSH and free thyroxine. After maintaining euthyroidism with L-thyroxine treatment, GH stimulation tests were performed and GHD was detected with a peak serum GH level of 1.65 ng/mL. He was started on rhGH. Other pituitary hormone levels including serum ACTH and cortisol were normal except for high serum prolactin. Magnetic resonance imaging (MRI) of the hypothalamic-pituitary region showed anterior pituitary hypoplasia (APH) and infundibular agenesis. During regular follow-up, at age 5.8 years, ACTH deficiency was detected, and hydrocortisone therapy was initiated. Gonadotropin deficiency was observed at 13.2 years of age with a Tanner stage I puberty, and testosterone replacement was administered.
Patient 2
Patient 2 was referred to the pediatric endocrinology clinic due to growth failure and recurrent hypoglycemia at the age of 0.5 years. He was born at term via SVD and his parents were first-degree cousins. At first evaluation, frontal bossing, depressed nasal bridge, and midfacial hypoplasia were detected and he had proportionate short stature with a height SDS of −4.9. His neuromotor development was normal. Central hypothyroidism was detected with normal ACTH and cortisol levels. He was also started on rhGH due to GHD. A pituitary MRI revealed APH. During regular follow-ups, ACTH deficiency was detected at 1.4 years, and hydrocortisone therapy was commenced. At the last control, his height was −1.4 SDS, attaining a height gain of 3.5 SDS after 12.3 years of rhGH therapy.
Patients 3a and 3b
Patient 3a presented with the complaint of short stature at the age of 2 years. She was born at term via SVD with a birth weight of 3,500 g (0.4 SDS). Her parents were not related. At presentation, her height, weight, and head circumference were 70.0 cm (−5.0 SDS), 6,900 g (−4.5 SDS), and 44.5 cm (−2.5 SDS), respectively. She had proportionate short stature and dysmorphic features, including a prominent forehead, a flat and depressed nasal bridge, and maxillary hypoplasia. Developmental milestones were normal for her age. GHD was detected with a peak serum GH level of 2.2 ng/mL. Empty sella was observed in pituitary MRI. She was put on rhGH treatment. At age 6.5 years, she was diagnosed with central hypothyroidism and L-thyroxine was administered. She had spontaneous menarche at the age of 12.5 years. Her adult final height was −0.6 SDS after receiving rhGH. At a recent evaluation, she had significantly low IGF1 (9.85 ng/mL) and IGFBP3 (0.9 μg/mL) levels and she is on regular follow-up by adult endocrinology due to GHD and central hypothyroidism.
Patient 3b was the first child of patient 3a and was admitted due to growth failure at 6 months of age. His parents were unrelated and he was born via cesarian section at 40th GW. He had jaundice requiring 2 days of phototherapy in the newborn period. At first evaluation, his height and body mass index were −2.1 SDS and 0.1 SDS, respectively. He had midface hypoplasia, large anterior fontanelles, a protruding forehead, depressed nasal bridge. Systematic examination and neurodevelopment were normal. GHD was detected and pituitary MRI revealed APH. At the age of 5.5 years, central hypothyroidism was observed and L-thyroxine treatment was commenced. At the last evaluation at 12.7 years of age, his height was −0.8 SDS.
Patient 4
Patient 4 was born via cesarian section at 34th GW with a birth length and weight of 0.2 SDS and 2.3 SDS, respectively. Her parents were second-degree cousins. At 1.2 years old, she was admitted to the due to growth failure and hypoglycemia. She had a doll face, midface hypoplasia, large-unclosed anterior fontanelles, raised forehead, and a depressed nasal bridge. Her height was −4.6 SDS and she had a proportionate short stature. Developmental milestones were normal for age. She had GHD with a GH peak of 0.03 ng/mL and partial ACTH deficiency with low peak cortisol levels. She was started on rhGH and hydrocortisone therapies. The pituitary MRI was normal.
Molecular Genetic Findings
Patient 1
A heterozygous missense variant, c.478C>T/[p.(Arg160Trp), (rs377600944)], in exon 5 of the GH1 gene (NM_000515.4) was detected in the panel. This variant was considered to be a variant of uncertain significance (VUS) according to ACMG criteria, due to the fact that it has an extremely low frequency in gnomAD population databases []. There were two records as VUS in ClinVar and one VUS and one likely benign record in LOVD for this variant []. This variant was not present in HGMD but was suggested as “benign” in a previous study in the literature []. Sanger sequencing confirmed this variant in Patient 1 and his mother. MLPA analysis of this patient was normal.
Patient 2
A novel nonsense homozygous variant in exon 2 of the GH1 gene c.162C>G/p.(Tyr54*) was detected in the GHD gene panel. This variant was confirmed by Sanger sequencing and segregation analysis revealed that parents were heterozygous. This variant was not present in HGMD and it was “likely pathogenic” according to ACMG criteria. Due to the “truncating” effect of this nonsense variant, in silico analysis tools were reported as “deleterious” and it was not present in the gnomAD [].
Patients 3a and 3b
The molecular analysis yielded a heterozygous, previously reported splice-donor site alteration; c.291+1G>A/p.(?) variant in exon 5 of the GH1 gene []. The segregation analysis showed that patient 3b who had also MPHD was also heterozygous for this variant.
Patient 4
The gene panel was normal for this patient but copy number variation (CNV) analysis revealed a deletion including the GH1 gene region. Hence, a homozygous whole GH1 gene deletion [rsa(GRCh38) 17q23.3(63,917,242–63,919,542) × 0] was further confirmed by MLPA analysis []. The heterozygous deletion was observed in both parents. The pedigrees and electropherograms of the families are demonstrated in Figure 1.
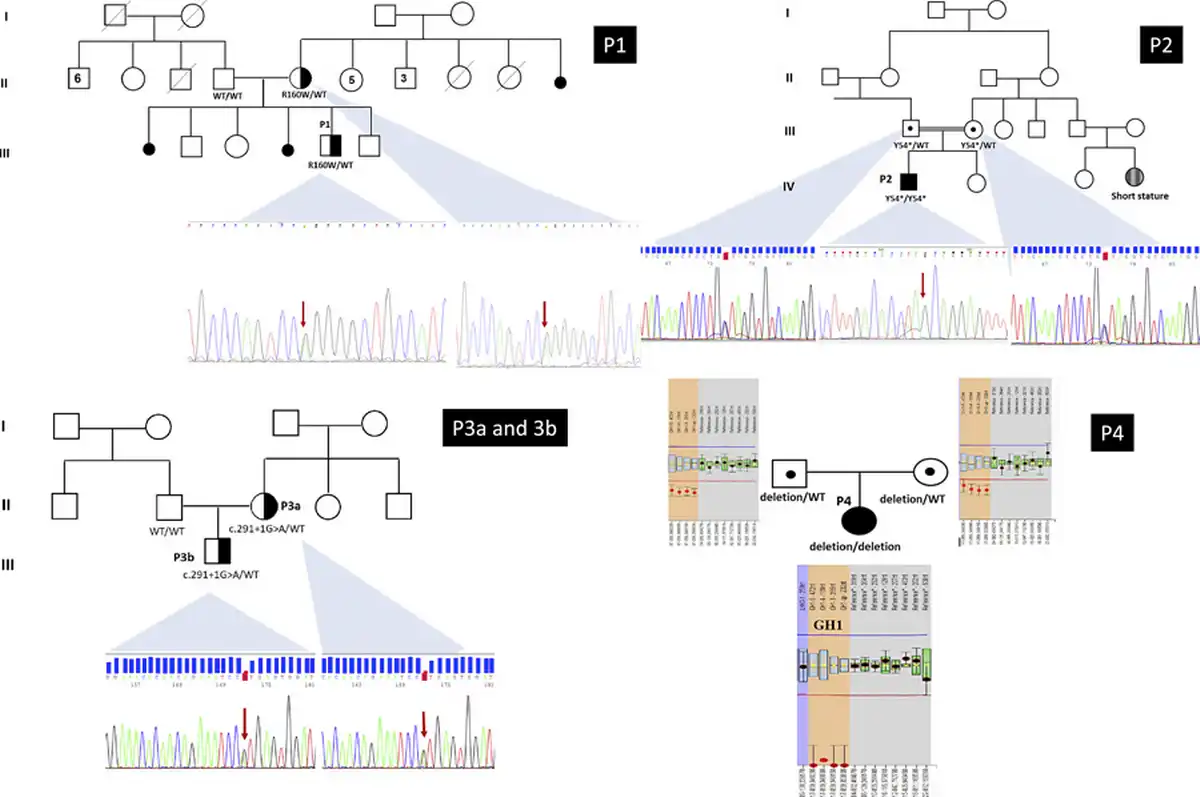
Fig. 1
The pedigrees of the families and electropherograms of the Sanger analysis. P1 GH1 (NM_000515.4 9 c.478C>T/p.(R160W) heterozygous; mother was also heterozygous. The mother has no features of IGHD/MPHD. P2 GH1 (NM_000515.4) exon 2 c.162 C>G/p.(Tyr54*) homozygous; parents heterozygous. P3a and 3b GH1 (NM_000515.4) exon 3 c.291+1 G>A/p.(?) heterozygous. The affected mother also has the same variant. P4 GH1 (NM_000515.4) homozygous whole gene deletion, parents were heterozygous. rsa(GRCh38) 17q23.3(63,917,242–63,919,542) × 0. P patient.
Discussion
GHD most frequently occurs as a sporadic condition, but severe forms may have a genetic basis []. The present study reports the clinical and molecular characteristics of 5 patients with IGHD/MPHD due to the GH1 gene alterations, with one novel nonsense variant. The characteristic phenotype of severe GHD or resistance includes craniofacial disproportion, frontal bossing, truncal obesity, and doll face. All the patients in our cohort had this phenotype in variable severity but this situation did not correlate with the inheritance mode or severity of the variants [].
Depending on the population studied, in patients with severe short stature and IGHD (height less than −4.5 SDS), the prevalence of GH1 or GHRHR gene defects was reported as 20% []. The height SDS of our patients was also less than −4.5 SDS at admission, except for patient 3b who was referred at 0.5 years of age. The reason why he was applied with a relatively better height SDS (−2.1 SDS) may be that he was diagnosed early because of his mother’s diagnosis of MPHD or due to the variable expressivity. The height SDS and endocrinological evaluations of heterozygous parents of patients 2 and 4 were normal (Table 1).
IGHD I is characterized by the absence of basal or stimulated GH with normal secretion of other pituitary hormones []. IGHD IA, the most severe form, causes hypoglycemia in infancy and severe growth failure by 6 months of age. Under rhGH therapy, patients develop antibodies to GH and the response to rhGH was blocked. It is generally reported that about 10–15% of subjects with severe IGHD and height less than −4.5 SDS had deletions in the GH1 gene. Frameshift and nonsense GH1 variants have also been detected in patients with IGHD IA []. In our study, patient 4 with homozygous whole GH1 gene deletion and hypoglycemia in infancy was categorized as IGHD IA. Owing to her young age and the recent initiation of rhGH therapy, it is necessary to monitor her for the development of anti-GH antibodies and treatment response to rhGH. The partial ACTH deficiency described in this case was planned to check periodically to see if this was a real pituitary hormone deficiency or a discordant laboratory finding.
Biallelic pathogenic variants in the GH1 and GHRHR genes cause IGHD IB, characterized by low but detectable levels of GH, mild to severe short stature, and a favorable response to rhGH []. In our study, patient 2 was classified as IGHD IB, due to a homozygous nonsense variant and a good height velocity under rhGH. At the last evaluation, his height was −1.4 SDS, and it was suggested that anti-GH antibodies have not appeared. By these findings, TSH and partial ACTH deficiencies in patient 2 were questionable to define as accompanying MPHD. Nevertheless, APH detected on pituitary MRI may still be an explanation for MPHD in this case, especially for TSH deficiency. Despite low peak cortisol after Synacthen test in patient 2, his basal cortisol value was fairly good at first evaluation. As expected in the recessive mode of inheritance in IGHD I, the heterozygous parents of the patients with biallelic GH1 variants (patients 2 and 4) were clinically and biochemically normal except for the unexplained mild growth failure in the mother of patient 4 (height SDS −2.1).
It has been suggested that patients with missense variants may be older and taller at diagnosis than their counterparts with splice-site variants in the GH1 gene [, ]. Patient 1 with a heterozygous missense mutation in our cohort was diagnosed later than other patients, but he had severe growth failure at admission. He had also MPHD including TSH, ACTH, and gonadotropin deficiencies. Although monoallelic variants in the GH1 gene are known to cause IGHD II with or without MPHD, the actual data about the rare missense variant p.(R160W) were still conflicting. In addition, although it was not reported in HGMD, a previous report suggested this variant as “benign.” Hence, this situation weakened the relation of this variant with the IGHD II phenotype. We think that severe short stature and MPHD in patient 1 need to be clarified with further analyses like exome sequencing.
Patients 3a and 3b were heterozygous for the splice site variant and patient 3b had mild growth retardation at admission which is suggested to be due to early diagnosis. These 2 patients were phenotyped as IGHD II. The severity of GHD is highly variable in IGHD. Compatible with the literature, although the peak GH levels were significantly low in our patients with IGHD II the peaks were not as low as compared to IGHD I []. Height SDS responses to rhGH therapy were good in all patients in our study.
The splice sites around exon 3 of the GH1 gene are relatively weak, and splicing enhancers are required to ensure the inclusion of exon 3 in the transcript. One of these exon splice enhancers (ESE1) comprises the first seven bases of exon 3, and augments the use of the upstream 3′ splice site, and suppresses a downstream cryptic splice site. Disruption of ESE1 causes skipping of exon 3 and produces the smaller 17.5-kDa isoform with dominant-negative effects on the production of the 22-kDa isoform of GH. Hamid et al. [] reported guanine to adenine substitution (formerly E3 +1 G/A; defined as c.291+1 G>A in this study), which disrupts ESE1 and causes exon 3 skipping. This situation causes IGHD II [].
It has been suggested that the ratio of mutant (17.5 kDa) and normal GH transcripts (22 kDa) may explain the phenotypic differences within the carriers of the same mutation. The 17.5-kDa human GH variant exhibits a dominant-negative effect for the secretion of the 22-kDa isoform. In mice carrying several copies of a 17.5-kDa isoform, encoding transgene loss of somatotrophs and pituitary hypoplasia is accompanied by the development of MPHD including TSH, prolactin, and gonadotropin (males) deficiencies []. Similarly, patients 3a and 3b in our study had TSH deficiencies in addition to GHD. Patient 3b, who was 12.7 years of age at the last evaluation had normal pubertal progression suggesting normal gonadotroph function, so far.
Patients with mutations that sufficiently increase 17.5-kDa levels may cause IGHD II. Characteristics of IGHD II include GHD, and possible development of MPHD and APH with variability in onset, severity, and progression, even among family members who share the same variant. Mullis et al. [] concluded that the pituitary status of IGHD II should be monitored over time because other hormonal deficiencies may evolve []. In our study, all the patients had biochemically defined MPHD with variable hormone deficiencies, however, this was not compatible with the definition of IGHD 1A and IB in relation to genetic results in 2 patients (patients 2 and 4). Three patients had APH (Patients 1, 2, and 3b) and patient 4 with IGHD 1A had a normal pituitary MRI. It has been hypothesized that the variable expression frequently observed in IGHD II is secondary to as yet unidentified “modifying” genes. However, our knowledge about these modifying genes is still lacking []. In conclusion, in this study, we report 2 patients with IGHD I, one with a novel nonsense variant and 3 patients with IGHD II. Expanding our knowledge about the genotype-phenotype correlation of GH1 gene variants by apprehending clinical and molecular data from more cases helps to define a follow-up strategy for the later occurrence of additional pituitary hormone deficiencies and response to rhGH therapy.
Acknowledgments
We thank all the patients and their family members for their participation in the study.
Statement of Ethics
Ethical approval was obtained for the study from The Ethics Committee of Istanbul University, Istanbul Faculty of Medicine (2021/2094). The patients/their parents gave general consent approving anonymous data use for academic purposes and written informed consent was obtained for participation in this study. Written informed consent was obtained from the parents for publication of this study and any accompanying images.
Conflict of Interest Statement
The authors have no conflicts of interest.
Funding Sources
This study was funded by the Scientific Research Projects Coordination Unit of Istanbul University (Project number: TDK-2022-38742).
Author Contributions
All authors contributed to the study’s conception and design. A.P.O., Z.Y.A., S.P., F.B., A.D.A., and F.D. clinically characterized the patient. Z.Y.A., G.T., V.K., G.B., and Z.O.U. performed and analyzed the sequencing data and evaluated the results. A.P.O., Z.Y.A., F.D., and Z.O.U. prepared the draft manuscript. All authors contributed to the discussion of results and edited and approved the final manuscript.
References
- 1. Birla S, Khadgawat R, Jyotsna VP, Jain V, Garg MK, Bhalla AS, et al. Identification of novel GHRHR and GH1 mutations in patients with isolated growth hormone deficiency. Growth Horm IGF Res. 2016;29:50–6.
- 2. Mullis PE. Genetics of isolated growth hormone deficiency. J Clin Res Pediatr Endocrinol. 2010;2(2):52–62.
- 3. Nassar LR, Barber GP, Benet-Pagès A, Casper J, Clawson H, Diekhans M, et al. The UCSC Genome Browser database: 2023 update. Nucleic Acids Res. 202351D1D1188–95.
- 4. Phillips JA 3rd, Hjelle BL, Seeburg PH, Zachmann M. Molecular basis for familial isolated growth hormone deficiency. Proc Natl Acad Sci U S A. 1981;78(10):6372–5.
- 5. Alatzoglou KS, Dattani MT. Genetic causes and treatment of isolated growth hormone deficiency-an update. Nat Rev Endocrinol. 2010;6(10):562–76.
- 6. Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 201543Database issueD789–98.
- 7. Wit JM, Losekoot M, Baumann G. Growth hormone-releasing hormone receptor and growth hormone gene abnormalities. In: Cohen LE, editor. Growth hormone deficiency: physiology and clinical managementChamSpringer International Publishing. 2016. p. 149–75.
- 8. Cullingford DJ, Siafarikas A, Choong CS. Genetic etiology of congenital hypopituitarism. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, et al, editors. EndotextSouth Dartmouth (MA)2000.
- 9. Neyzi O, Bundak R, Gokcay G, Gunoz H, Furman A, Darendeliler F, et al. Reference values for weight, height, head circumference, and body mass index in Turkish children. J Clin Res Pediatr Endocrinol. 2015;7(4):280–93.
- 10. Demir K, Konakçı E, Ozkaya G, Kasap Demir B, Ozen S, Aydın M, et al. New features for child metrics: further growth references and blood pressure calculations. J Clin Res Pediatr Endocrinol. 2020;12(2):125–9.
- 11. Ranke MBH. Growth hormone stimulation tests in Diagnostics of endocrine function in children and adolescent. In: MB R, editor. Heildeberg-Leipzig. Johann Ambrosius Barth J & J. 1996. p. 134–48.
- 12. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and Genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–24.
- 13. Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NST, et al. Human gene mutation database (HGMD®): 2003 update. Hum Mutat. 2003;21(6):577–81.
- 14. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43.
- 15. Landrum MJ, Chitipiralla S, Brown GR, Chen C, Gu B, Hart J, et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 202048D1D835–44.
- 16. Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32(5):557–63.
- 17. Plachy L, Strakova V, Elblova L, Obermannova B, Kolouskova S, Snajderova M, et al. High prevalence of growth plate gene variants in children with familial short stature treated with GH. J Clin Endocrinol Metab. 2019;104(10):4273–81.
- 18. Hamid R, Phillips JA 3rd, Holladay C, Cogan JD, Austin ED, Backeljauw PF, et al. A molecular basis for variation in clinical severity of isolated growth hormone deficiency type II. J Clin Endocrinol Metab. 2009;94(12):4728–34.
- 19. Kale S, Gada JV, Jadhav S, Lila AR, Sarathi V, Budyal S, et al. Genetic spectrum and predictors of mutations in four known genes in Asian Indian patients with growth hormone deficiency and orthotopic posterior pituitary: an emphasis on regional genetic diversity. Pituitary. 2020;23(6):701–15.
- 20. Keselman A, Scaglia PA, Rodríguez Prieto MS, Ballerini MG, Rodríguez ME, Ropelato MG, et al. Type IA isolated growth hormone deficiency (IGHD) consistent with compound heterozygous deletions of 6.7 and 7.6 Kb at the GH1 gene locus. Arq Bras Endocrinol Metabol. 2012;56(8):558–63.
- 21. Mullis PE, Akinci A, Kanaka C, Eblé A, Brook CG. Prevalence of human growth hormone-1 gene deletions among patients with isolated growth hormone deficiency from different populations. Pediatr Res. 1992;31(5):532–4.
- 22. Giordano M. Genetic causes of isolated and combined pituitary hormone deficiency. Best Pract Res Clin Endocrinol Metab. 2016;30(6):679–91.
- 23. Gucev Z, Tasic V, Saranac L, Stobbe H, Kratzsch J, Klammt J, et al. A novel GH1 mutation in a family with isolated growth hormone deficiency type II. Horm Res Paediatr. 2012;77(3):200–4.
- 24. Binder G, Keller E, Mix M, Massa GG, Stokvis-Brantsma WH, Wit JM, et al. Isolated GH deficiency with dominant inheritance: new mutations, new insights. J Clin Endocrinol Metab. 2001;86(8):3877–81.
- 25. Salemi S, Yousefi S, Baltensperger K, Robinson IC, Eblé A, Simon D, et al. Variability of isolated autosomal dominant GH deficiency (IGHD II): impact of the P89L GH mutation on clinical follow-up and GH secretion. Eur J Endocrinol. 2005;153(6):791–802.
- 26. Ryther RC, McGuinness LM, Phillips JA 3rd, Moseley CT, Magoulas CB, Robinson IC, et al. Disruption of exon definition produces a dominant-negative growth hormone isoform that causes somatotroph death and IGHD II. Hum Genet. 2003;113(2):140–8.
- 27. Mullis PE, Robinson IC, Salemi S, Eblé A, Besson A, Vuissoz JM, et al. Isolated autosomal dominant growth hormone deficiency: an evolving pituitary deficit? A multicenter follow-up study. J Clin Endocrinol Metab. 2005;90(4):2089–96.