INTRODUCTION
Autosomal dominant tubulointerstitial kidney disease (ADTKD) is a group of familial renal diseases characterized by shared features of autosomal dominant inheritance, minimal abnormalities in urinary sediment, and progressive renal failure. A renal histology always displays interstitial fibrosis and tubular atrophy, yet with almost normal glomeruli, which represent a common pathological feature of chronic tubulointerstitial renal diseases, irrespective of etiology. Renal medullary cysts can be found in some patients. Due to the variable clinical and non-specific pathological features mentioned above, medullary cystic kidney disease (MCKD), familial juvenile hyperuricemic nephropathy (FJHN), and hereditary interstitial kidney diseases have been used to describe inherited tubulointerstitial kidney diseases.[] Later genetic linkage studies have identified that type 2 MCKD and FJHN both result from the allelic mutation of the UMOD gene. Moreover, investigators have found that neither medullary cysts nor hyperuricemia denotes the pathognomonic characteristics of the diseases.[] Due to the misleading nature of these names and the discovery of more pathogenic variants, such as mucin-1 (MUC1), renin (REN), hepatocyte nuclear factor 1 beta (HNF1β), and the alpha subunit of the endoplasmic reticular membrane translocon (SEC61A1), Kidney Disease: Improving Global Outcomes (KDIGO) proposed in 2014 to use “Autosomal Dominant Tubulointerstitial Kidney Disease (ADTKD)” to name this group of diseases and suggested subclassing pathogenic genes to increase the detection of pathogenic genes. ADTKD caused by UMOD pathogenic variants is called ADTKD–UMOD.
The prevalence of ADTKD–UMOD is challenging to estimate accurately due to difficulties in identifying this rare kidney disease and the lack of routine genetic testing. Recent studies have indicated that ADTKD–UMOD is the second most common genetic kidney disease, followed by autosomal dominant polycystic kidney disease (ADPKD) and Alport syndrome, among chronic kidney disease (CKD) patient.[] ADPKD is characterized by the progressive enlargement of renal cysts, while Alport syndrome is characterized by persistent glomerular hematuria and renal pathology findings of thickening and thinning, with lamellation in the glomerular basement membrane (GBM). The distinct clinical and pathological features of ADPKD and Alport syndrome have garnered significant attention, leading to rapid advancements in research on their pathogenesis and treatment. In contrast, ADTKD–UMOD lacks specific clinical and pathological manifestations and relies heavily on genetic testing, which is not routinely conducted, resulting in an underestimation of its prevalence. The unclear understanding of its pathogenesis makes it challenging to establish a causal link between newly identified gene mutations and non-specific clinical and pathological phenotypes, further complicating the definitive diagnosis of ADTKD–UMOD.
CLINICAL CHARACTERISTICS AND RENAL BIOPSY
Although the clinical laboratory and histological manifestations of ADTKD are largely nonspecific, there are still some clinical features [Table 1], suggesting a suspicious diagnosis for ADTKD–UMOD.
Table 1
Common clinical features in patients with ADTKD–UMOD
Clinical features |
|---|
Autosomal dominant inheritance |
Chronic kidney disease |
Bland urinary sediment with no or minimal proteinuria |
Early onset of hyperuricemia/gout |
Decreased uric acid excretion fraction |
Normal or small-sized kidneys |
First, most patients have a positive family history of either CKD and/or hyperuricemia compatible with the autosomal dominant inheritance mode. A negative family history can also be observed in about 10% of patients, which may indicate the presence of de novo mutations[] or a missed diagnosis of CKD in other family members. The rate of decline in the renal function is highly variable among and within ADTKD–UMOD families.[] The age at onset of end stage renal disease (ESRD) ranges from 25 to more than 70 years, with a median age of 50 years old.[] Interfamilial differences in the ESRD age were associated with the location of the pathogenic variants.[] As for intrafamilial variability, modifying factors, such as environmental or epigenetic changes and modifier genes interacting with UMOD, may provide a partial explanation.[]
Consistent with the pathogenesis of tubulointerstitial nephropathy, urinalyses in patients with ADTKD reveal bland sediment and absent-to-minimal proteinuria.[]
The early onset of hyperuricemia/gout is a distinctive feature of tubulointerstitial nephropathy with UMOD pathogenic variants. The prevalence of hyperuricemia/gout among patients with ADTKD-UMOD is up to 60% or higher in different cohorts.[] The median age is before age 40 and usually before age 30[] at the diagnosis of hyperuricemia/gout, which is earlier than the onset of CKD. Laboratory results typically report a decreased uric acid excretion fraction (UAEF), and UAEFs lower than the 25th percentile values corresponding to Glomerular filtration rate (GFR) were observed in about 70% of patients (below 0.05 in most patients).[]
Medullary cysts were previously considered a distinct feature of Type 2 MCKD, which is caused by UMOD mutations. However, various cohort studies have repeatedly suggested that only a minority of patients exhibit cysts on an ultrasound and that the localization of the cysts is not just observed in the medulla.[] The kidney’s size is initially normal under an ultrasound and declines as CKD progresses.
Renal biopsies in patients with ADTKD–UMOD typically exhibit interstitial fibrosis, tubular atrophy, thickening, and lamellation of the tubular basement membranes [Table 2]. Microcystic dilatation of the tubules may also be present. Immunofluorescence analyses reveal negative deposition for both complement and immunoglobulin.[] The renal pathology in these patients is considered nonspecific and indistinguishable from chronic tubulointerstitial nephritis due to other etiologies and always serves as evidence to rule out glomerular diseases. However, there have been instances where kidney biopsies from individuals with UMOD variants have displayed glomerular injuries consistent with secondary focal segmental glomerulosclerosis (FSGS),[] which can be misleading. Notably, an abnormal accumulation of uromodulin within the thick ascending limb (TAL) and distal tubules has been reported in the kidneys of ADTKD–UMOD patients through immunofluorescence staining with anti-uromodulin antibodies. These deposits, which are visible to varying degrees in periodic acid-schiff (PAS) staining, often exhibit halos (80.4% of cases). The distinct appearance of these deposits could aid in diagnosing patients with ambiguous UMOD mutations.[] However, further studies with larger sample sizes are warranted to validate these findings.
Table 2
Common renal histological findings in patients with ADTKD–UMO
Histological findings |
|---|
Interstitial fibrosis |
Tubular atrophy |
Thickening and lamellation of tubular basement membranes |
Possibly microcystic dilation of the tubules |
Negative immunofluorescence for both complement and immunoglobulins |
ADTKD, autosomal dominant tubulointerstitial kidney disease. |
THE UMOD GENE AND UROMODULIN
It has been confirmed that ADTKD–UMOD is caused by pathogenic variants in the UMOD gene.[] The UMOD gene is located on chromosome 16p12.3–16p13.11 and consists of 11 exons (the first of which is noncoding) over a genomic region of about 20 kb.[]UMOD expression is typically detected in the kidney.[] It encodes uromodulin (previously known as Tamm–Horsfall glycoprotein), which is the most abundant protein in normal human urine. RNA sequencing and immunofluorescence demonstrate that UMOD exclusively transcripts and produces protein in the TAL and distal convoluted tubule (DCT).[] In TAL cells, uromodulin mainly distributes in the apical membrane, corroborating the fact that uromodulin levels in urine are a thousand times higher than in serum.[] Uromodulin is an 85-kDa glycoprotein with a very high carbohydrate content (approximately 30%) and has a tendency to aggregate in solution.[] The precursor protein comprises 640 amino acids and contains 48 cysteine residues stabilized by 24 disulfide bridges.[] The first 24 N-terminal amino acids constitute a signal peptide that directs its entry into the secretory pathway and is then cleaved. The maturation of the protein mainly comprises the formation of 24 intramolecular disulfide bonds, tertiary folding inside the endoplasmic reticulum (ER), and glycosylation in the Golgi. Uromodulin is a glycosylphosphatidylinositol (GPI) protein.[] with a GPI anchoring site at position 614. The mature protein includes one epidermal growth factor-like (EGF-like) domain, two calcium-binding EGF-like domains, a D8C domain containing eight conserved cysteines, a fourth EGF-like domain, one zona pellucida (ZP)-like domain.[] and a C-terminal hydrophobic stretch that functions as a signal for the attachment of a GPI membrane anchor. There are eight potential Asn-linked glycosylation sites that are consistent with the high carbohydrate content.[]
In vivo and in vitro experiments have proven that uromodulin has various physiological functions. It increases the activity of the Na-K-2Cl cotransporter (NKCC2)[] and the surface expression of the potassium channel (ROMK)[] in TAL cells, thus influencing the urinary concentration capacity and sodium transport of the medulla. Uromodulin also prevents kidney stones by inhibiting the aggregation of calcium crystals in renal tubules[] and reducing calcium excretion.[] Studies in UMOD-knockout mice have demonstrated that uromodulin plays a role in protecting against urinary tract infections by competitively binding to E. coli Type 1 lectin FimH, which impedes epithelial adhesion and virulence.[] Additionally, uromodulin exerts a protective effect during acute kidney injury through binding to lymphokines,[] regulating the number and activity of kidney mononuclear phagocytes,[] and inhibiting the activation of the classical complement pathway by binding to complement 1q and collectin-11.[]
UMOD PATHOGENIC VARIANTS AND PATHOPHYSIOLOGY IN ADTKD–UMOD
Thus far, 135 UMOD pathogenic variants in ADTKD-UMOD patients have been reported, predominantly missense changes and 6 in frame deletion mutations (4%). About 90% of these mutations occur in exons 3 and 4 and are clustered in the N-terminal half of the protein containing the EGF-1, EGF-2, EGF-3, and D8C domains.[] Of all pathogenic variants, about 60% involve cysteine residues, either by the substitution or deletion of a cysteine or the insertion of an extra cysteine.[] As mentioned above, the disulfide bonds between the 48 conserved cysteine residues play an important role in the structure of protein, and these UMOD mutations may lead to protein misfolding.
UMOD mutants rather than a UMOD deficiency detected in mutant UMOD cellular and mouse models have demonstrated that ADTKD–UMOD is caused by a gain of toxic functioning.[] A variety of in vivo and in vitro experiments have established that the pathogenic effect of UMOD pathogenic variants is the ER retention of misfolded uromodulin.[] Mutant UMOD gene-transfected cells revealed a significantly reduced maturation of uromodulin compared with that in the wild type. The uromodulin mutants co-localized with the ER resident protein Calnexin, indicating that uromodulin mutants are retained in the ER.[] Likewise, transgenic mouse models showed impaired mutant uromodulin trafficking to the plasma membrane with ER retention and a decrease in urinary excretion. Moreover, the mouse model presented urinary concentrating defects and progressive tubulointerstitial damage with inflammatory cell infiltration, fibrosis, and tubular atrophy.[] However, the pathogenetic mechanisms downstream of mutant uromodulin ER retention are not fully understood. The TAL cells of transgenic mice depicted mutant uromodulin accumulation in the ER, leading to ER stress and unfolded protein response (UPR) as a stress response to decrease the ER load.[] The early induction of inflammation before any functional or histological kidney damage was detected in a mutant UMOD transfected mouse model, which may contribute to the disease’s onset.[] A study in a mouse model demonstrated that blocking tumor necrosis factor α (TNF-α) can delay the disease’s progression.[] Autophagy deficiency[] and impaired mitochondrial homeostasis may also contribute to the disease’s onset.[] In addition, intracellular uromodulin accumulation may damage TAL cells, leading to the loss of NKCC2 and less urinary concentration and mild volume depletion. Subsequently, the proximal tubular reabsorption of uric acid would increase, and hyperuricemia occurs [Figure 1].[]
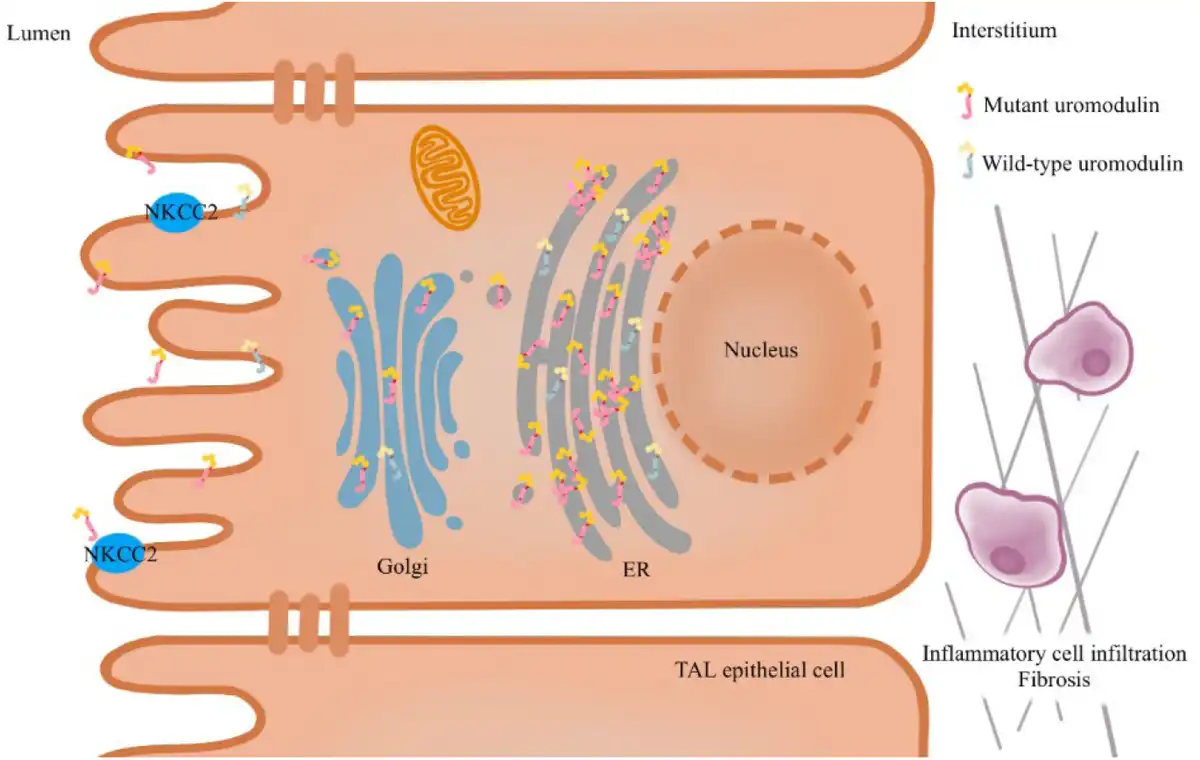
Figure 1.
Pathophysiology of ADTKD–UMOD. The pathogenic variants in UMOD result in the misfolding of uromodulin, leading to its retention in the ER and causing ER stress and UPR. Additionally, autophagy defects and impaired mitochondrial homeostasis play a role in the development of the disease. The aggregation of uromodulin in the ER results in reduced urine secretion and impairs the function of TAL cells, ultimately leading to a decrease in NKCC2. Subsequently, interstitial inflammatory infiltration and fibrosis occur. ER, endoplasmic reticulum; UPR, unfolded protein response; NKCC2, Na-K-2Cl cotransporter; TAL, thick ascending limb; ADTKD–UMOD, autosomal dominant tubular kidney disease-UMOD.
DIAGNOSIS
When to suspect ADTKD–UMOD
A diagnosis of ADTKD should be suspected in any patient who has a positive family history of CKD compatible with the autosomal dominant inheritance model (at least two affected family members in two successive generations), bland urinary sediment with absent or minimal proteinuria, and a normal- or small-sized kidney on an ultrasound. The presence of hyperuricemia/gout further suggests that ADTKD is caused by the mutation of UMOD pathogenic variants.
ADTKD can also be suspected in patients who exhibit CKD with bland urinary sediment but do not have a positive family history of CKD. Such cases may be due to de novo variants or a missed diagnosis in other family members. However, ADTKD should be considered in such cases only if the kidney pathology aligns with tubulointerstitial disease and after ruling out potential drug exposure as well as autoimmune or systemic disease.
Genetic testing to confirm the diagnosis
Genetic testing is currently the only way to establish a diagnosis of ADTKD and its respective subtypes. The immunofluorescence staining of kidney biopsies using anti-uromodulin antibodies may identify abnormal uromodulin deposits.[] However, the technology is not easy and does not apply to all patients. Therefore, a renal biopsy may offer limited diagnostic value.
For patients who have a family history of CKD compatible with an autosomal dominant inheritance fulfilling the clinical characteristics, genetic testing is recommended. A genetic diagnosis can help avoid unnecessary invasive examinations and provide valuable prognostic information. Due to the non-specific clinical presentation of ADTKD–UMOD, distinguishing it from other subtypes of ADTKD can be challenging. In addition, many nephrologists may not be well versed in the clinical features of rare kidney diseases. Therefore, utilizing a kidney disease gene panel that includes UMOD, MUC1, REN, HNF1β, and SEC61A1 or opting for whole-exon sequencing, can be a cost-effective strategy to prevent missed diagnoses. Although about 90% of UMOD pathogenic variants occur in exons 3 and 4, it is necessary to sequence all 11 exons of the UMOD gene to capture all possible mutations.
Notably, not all disease-causing mutations and genes have been identified, and MUC1 mutations cannot be detected through standard Sanger sequencing or next-generation sequencing methods. Therefore, a negative genetic test result does not rule out the possibility of ADTKD.
DIFFERENTIAL DIAGNOSIS
In cases where patients have a family history of CKD and bland urinary sediment, it is important to consider other subtypes of ADTKD. ADTKD–REN and ADTKD–MUC1 are common subtypes of ADTKD, aside from ADTKD–UMOD. Patients with pathogenic variants in the REN gene typically exhibit progressive CKD, childhood anemia, mild hyperkalemia, and early-onset hyperuricemia. On the other, patients with pathogenic variants in the MUC1 gene usually present with progressive CKD without other distinguishing features.[] Due to the variability and non-specific nature of the symptoms, distinguishing between these diseases based on clinical presentation alone can be challenging.
In addition, atypical ADPKD caused by a DNAJB11 missense variant can be challenging to distinguish from ADTKD. Individuals carrying this variant may exhibit non-enlarged cystic kidneys and progressive interstitial fibrosis, representing a unique phenotypic blend of ADPKD and ADTKD.[]
Nephronophthisis (NPHP) is an autosomal recessive kidney disease resulting from genetic variants in genes responsible for primary renal cilium and basal body components. Similar to ADTKD–UMOD, patients with NPHP exhibit progressive CKD characterized by a bland urinalysis and normal kidney size. However, NPHP typically manifests at an earlier age, with ESRD commonly occurring in the early teenage years. Furthermore, affected children often display extrarenal symptoms involving the retina, skeleton, brain, and liver.[] Importantly, NPHP is inherited in an autosomal recessive manner, distinguishing it from ADTKD.
MANAGEMENT
Currently, there is no effective treatment for ADTKD–UMOD. The main approach to managing this condition involves supportive care and addressing complications related to CKD, such as anemia, metabolic bone disease, metabolic acidosis, and electrolyte imbalances. Individuals with ADTKD typically have normal blood pressure and minimal protein in their urine, which means that they may not require treatment with angiotensin-converting enzyme (ACE) inhibitors. In cases where hypertension and high uric acid levels are present, losartan may be a preferred medication, as it can help increase the excretion of uric acid.[] There is no conclusive evidence to suggest that ACE inhibitors can slow the progression of CKD in individuals with ADTKD. Importantly, diuretics may worsen hyperuricemia and dehydration in ADTKD patients, so they should be used cautiously. For individuals with ADTKD–UMOD and a diagnosis of gout, there is a heightened risk of future gout attacks and tophi formation. Treatment with allopurinol may help prevent further episodes.[] However, the impact of allopurinol on slowing the progression of renal failure in these patients remains uncertain.
Several studies have investigated treatments targeting the pathogenesis of ADTKD–UMOD. In vitro assays have shown that 4-phenylbutyrate treatment can reduce the ER retention of mutants and reverse the apoptosis caused by uromodulin accumulation in cells transfected with mutated UMOD.[] However, in vivo trials have not yielded the same results.[] Another study using a mutant UMOD-transfected mouse model found that blocking TNF-α can slow the disease’s progression,[] which aligns with the early inflammation detected in the kidneys of the mouse model. A recent study reported that a small molecule called BRD4780, which targets TMED9, can help clear misfolded proteins in a cellular model of ADTKD–UMOD.[] Notably, these studies have been limited to animal- or cell-based experiments, highlighting the need for further investigation before clinical application.
Patients with ESRD caused by ADTKD-UMOD are considered suitable candidates for kidney transplantation because the transplanted kidney is not at risk of disease recurrence.[] It is recommended that unaffected family members undergo genetic testing to determine their eligibility to be living kidney donors.
GENETIC COUNSELING
Once a family member is identified with ADTKD–UMOD, genetic counseling should be considered to assist with future family planning. Comprehensive pedigree analysis and family tracing can help identify other clinically affected individuals as well as those who are at risk but not yet affected. It is important for adults in these families to undergo genetic testing and be informed of the risk of inheriting the disease. As there is currently no effective treatment available, genetic testing before the onset of symptoms is not recommended for minors. However, genetic screening should be mandatory for potential kidney donors.
QUALITY OF LIFE (QOL)
A cross-sectional survey concerning QOL in patients with ADTKD–UMOD revealed a high prevalence of depression among affected individuals.[] In the affected family, older members inevitably develop end-stage renal disease, require renal replacement therapy, and face potentially fatal adverse cardiovascular events. Younger family members, including those unaffected by the disease, may be concerned about their prognoses. ADTKD–UMOD primarily affects the kidneys, with few extrarenal manifestations. Thus, CKD is the most significant factor affecting QOL, leading to physical discomfort, unemployment, dependency, and financial burdens. Studies have shown that the QOL in CKD patients before starting renal replacement therapy is higher than in hemodialysis patients but lower than in healthy individuals.[] Genetic counselors and nephrologists play a crucial role in helping patients and their families understand the disease and its inheritance pattern. There is no evidence of ADTKD–UMOD recurring after transplantation, making pre-emptive transplantation before dialysis dependence a potential option. However, further prospective studies are required to assess the prognostic impact of preemptive transplantation.
FUTURE RESEARCH DIRECTIONS
Considerable effort is required to enhance early diagnosis, advance an understanding of the pathophysiology, and identify potential therapeutic targets for ADTKD–UMOD. The nonspecific clinical features of the disease often result in delayed or missed diagnoses, leading to unnecessary and invasive kidney biopsies. Increasing disease awareness among nephrologists and establishing clear diagnostic criteria could broaden the clinical and genetic understanding of the disease, opening new avenues for clinical trials. The development of non-invasive biomarker-based blood or urinary tests would be highly beneficial. Notably, reduced uromodulin excretion in urine has been observed in ADTKD–UMOD patients,[] suggesting a promising new diagnostic approach that requires further validation in larger cohorts. The pathogenic mechanisms following misfolded protein ER retention remain poorly understood, highlighting the need for research to establish effective cell and animal models to investigate profibrotic pathways and identify potential therapeutic targets.
Acknowledgement
All authors significantly contributed to the accomplishment of review. We would like to acknowledge authors of papers included in this review for their efforts for the original studies.
Financial support and sponsorship
This review did not receive any specific grant from funding agencies in the public, commercial or not-for-profit sectors.
Authors contribution
Tian XY is responsible for writing the article. Chen YQ is responsible for selecting the topic, designing the framework, guiding the writing, and revising the article. All authors reviewed and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Conflicts of interest
Not applicable.
Data availability statement
No additional data.
How to cite: Tian XY, Chen YQ. Autosomal Dominant Tubulointerstitial Kidney Disease–UMOD: A Monogenic Renal Disease that Cannot Be Ignored. Integr Med Nephrol Androl. 2024;11:e24-00009. doi: 10.1097/IMNA-D-24-00009
REFERENCES
1.
Thompson GR, Weiss JJ, Goldman RT, Rigg GA. Familial occurrence of hyperuricemia, gout, and medullary cystic disease. Arch Intern Med. 1978;138(11):1614–1617.2.
Stavrou C, Koptides M, Tombazos C, et al. Autosomal-dominant medullary cystic kidney disease type 1: clinical and molecular findings in six large Cypriot families. Kidney Int. 2002;62(4):1385–1394.3.
Massari PU, Hsu CH, Barnes RV, Fox IH, Gikas PW, Weller JM. Familial hyperuricemia and renal disease. Arch Intern Med. 1980;140(5):680–684.4.
Dahan K, Fuchshuber A, Adamis S, et al. Familial juvenile hyperuricemic nephropathy and autosomal dominant medullary cystic kidney disease type 2: two facets of the same disease? J Am Soc Nephrol. 2001;12(11):2348–2357.5.
Hart TC, Gorry MC, Hart PS, et al. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet. 2002;39(12):882–892.6.
Gast C, Marinaki A, Arenas-Hernandez M, et al. Autosomal dominant tubulointerstitial kidney disease-UMOD is the most frequent non polycystic genetic kidney disease. BMC Nephrol. 2018;19(1):301.7.
Groopman EE, Marasa M, Cameron-Christie S, et al. Diagnostic Utility of Exome Sequencing for Kidney Disease. N Engl J Med. 2019;380(2):142–151.8.
Bollée G, Dahan K, Flamant M, et al. Phenotype and outcome in hereditary tubulointerstitial nephritis secondary to UMOD mutations. Clin J Am Soc Nephrol. 2011;6(10):2429–2438.9.
Bleyer AJ, Kmoch S, Antignac C, et al. Variable clinical presentation of an MUC1 mutation causing medullary cystic kidney disease type 1. Clin J Am Soc Nephrol. 2014;9(3):527–535.10.
Ayasreh N, Bullich G, Miquel R, et al. Autosomal Dominant Tubulointerstitial Kidney Disease: Clinical Presentation of Patients With ADTKD-UMOD and ADTKD-MUC1. Am J Kidney Dis. 2018;72(3):411–418.11.
Devuyst O, Knoers NV, Remuzzi G, Schaefer F. Rare inherited kidney diseases: challenges, opportunities, and perspectives. Lancet. 2014;383(9931):1844–1859.12.
Olinger E, Hofmann P, Kidd K, et al. Clinical and genetic spectra of autosomal dominant tubulointerstitial kidney disease due to mutations in UMOD and MUC1. Kidney Int. 2020;98(3):717–731.13.
Scolari F, Caridi G, Rampoldi L, et al. Uromodulin storage diseases: clinical aspects and mechanisms. Am J Kidney Dis. 2004;44(6):987–999.14.
Liang Y, Chen YQ, Wang SX, Liu Y, Ao J, Zhang H. Clinical and pathological analyses of medullary cystic kidney disease. Chin J Blood Purif. 2011;10(05):270–273.15.
Onoe T, Hara S, Yamada K, et al. Significance of kidney biopsy in autosomal dominant tubulointerstitial kidney disease-UMOD: is kidney biopsy truly nonspecific? BMC Nephrol. 2021;22(1):1.16.
Chun J, Wang M, Wilkins MS, et al. Autosomal Dominant Tubulointerstitial Kidney Disease-Uromodulin Misclassified as Focal Segmental Glomerulosclerosis or Hereditary Glomerular Disease. Kidney Int Rep. 2020;5(4):519–529.17.
Pennica D, Kohr WJ, Kuang WJ, et al. Identification of human uromodulin as the Tamm-Horsfall urinary glycoprotein. Science. 1987;236(4797):83–88.18.
Pook MA, Jeremiah S, Scheinman SJ, Povey S, Thakker RV. Localization of the Tamm-Horsfall glycoprotein (uromodulin) gene to chromosome 16p12.3–16p13.11. Ann Hum Genet. 1993;57(4):285–290.19.
Lee JW, Chou CL, Knepper MA. Deep Sequencing in Microdissected Renal Tubules Identifies Nephron Segment-Specific Transcriptomes. J Am Soc Nephrol. 2015;26(11):2669–2677.20.
Sikri KL, Foster CL, MacHugh N, Marshall RD. Localization of Tamm-Horsfall glycoprotein in the human kidney using immuno-fluorescence and immuno-electron microscopical techniques. J Anat. 1981;132(Pt 4):597–605.21.
Thornley C, Dawnay A, Cattell WR. Human Tamm-Horsfall glycoprotein: urinary and plasma levels in normal subjects and patients with renal disease determined by a fully validated radioimmunoassay. Clin Sci (Lond). 1985;68(5):529–535.22.
Muchmore AV, Decker JM. Uromodulin: a unique 85-kilodalton immunosuppressive glycoprotein isolated from urine of pregnant women. Science. 1985;229(4712):479–481.23.
Rindler MJ, Naik SS, Li N, Hoops TC, Peraldi MN. Uromodulin (Tamm-Horsfall glycoprotein/uromucoid) is a phosphatidylinositol-linked membrane protein. J Biol Chem. 1990;265(34):20784–20789.24.
Serafini-Cessi F, Malagolini N, Hoops TC, Rindler MJ. Biosynthesis and oligosaccharide processing of human Tamm-Horsfall glycoprotein permanently expressed in HeLa cells. Biochem Biophys Res Commun. 1993;194(2):784–790.25.
Bokhove M, Nishimura K, Brunati M, et al. A structured interdomain linker directs self-polymerization of human uromodulin. Proc Natl Acad Sci U S A. 2016;113(6):1552–1557.26.
Rampoldi L, Scolari F, Amoroso A, Ghiggeri G, Devuyst O. The rediscovery of uromodulin (Tamm-Horsfall protein): from tubulointerstitial nephropathy to chronic kidney disease. Kidney Int. 2011;80(4):338–347.27.
Serafini-Cessi F, Malagolini N, Cavallone D. Tamm-Horsfall glycoprotein: biology and clinical relevance. Am J Kidney Dis. 2003;42(4):658–676.28.
Serafini-Cessi F, Dall’Olio F, Malagolini N. High-mannose oligosaccharides from human Tamm-Horsfall glycoprotein. Biosci Rep. 1984;4(3):269–274.29.
Hård K, Van Zadelhoff G, Moonen P, Kamerling JP, Vliegenthart FG. The Asn-linked carbohydrate chains of human Tamm-Horsfall glycoprotein of one male. European Journal of Biochemistry. 1992;209(3):895–915.30.
van Rooijen JJ, Voskamp AF, Kamerling JP, Vliegenthart JF. Glycosylation sites and site-specific glycosylation in human Tamm-Horsfall glycoprotein. Glycobiology. 1999;9(1):21–30.31.
Mutig K, Kahl T, Saritas T, et al. Activation of the bumetanide-sensitive Na+, K+, 2Cl- cotransporter (NKCC2) is facilitated by Tamm-Horsfall protein in a chloride-sensitive manner. J Biol Chem. 2011;286(34):30200–30210.32.
Renigunta A, Renigunta V, Saritas T, Decher N, Mutig K, Waldegger S. Tamm-Horsfall glycoprotein interacts with renal outer medullary potassium channel ROMK2 and regulates its function. J Biol Chem. 2011;286(3):2224–2235.33.
Mo L, Huang HY, Zhu XH, Shapiro E, Hasty DL, Wu XR. Tamm-Horsfall protein is a critical renal defense factor protecting against calcium oxalate crystal formation. Kidney Int. 2004;66(3):1159–1166.34.
Serafini-Cessi F, Monti A, Cavallone D. N-Glycans carried by Tamm-Horsfall glycoprotein have a crucial role in the defense against urinary tract diseases. Glycoconj J. 2005;22(7–9):383–394.35.
Wolf MT, Wu XR, Huang CL. Uromodulin upregulates TRPV5 by impairing caveolin-mediated endocytosis. Kidney Int. 2013;84(1):130–137.36.
Orskov I, Ferencz A, Orskov F. Tamm-Horsfall protein or uromucoid is the normal urinary slime that traps type 1 fimbriated Escherichia coli. Lancet. 1980;1(8173):887.37.
Hession C, Decker JM, Sherblom AP, et al. Uromodulin (Tamm-Horsfall glycoprotein): a renal ligand for lymphokines. Science. 1987;237(4821):1479–1484.38.
Liu Y, El-Achkar TM, Wu XR. Tamm-Horsfall protein regulates circulating and renal cytokines by affecting glomerular filtration rate and acting as a urinary cytokine trap. J Biol Chem. 2012;287(20):16365–16378.39.
Tarek M El-Achkar, Ruth McCracken, Yan Liu, et al. Tamm-Horsfall protein translocates to the basolateral domain of thick ascending limbs, interstitium, and circulation during recovery from acute kidney injury. Am J Physiol Renal Physiol. 2013;304(8):F1066–F1075.40.
Micanovic R, Khan S, Janosevic D, et al. Tamm-Horsfall Protein Regulates Mononuclear Phagocytes in the Kidney. J Am Soc Nephrol. 2018;29(3):841–856.41.
Gong K, Xia M, Wang Y, et al. Importance of glycosylation in the interaction of Tamm-Horsfall protein with collectin-11 and acute kidney injury. J Cell Mol Med. 2020;24(6):3572–3581.42.
Rhodes DC. Importance of carbohydrate in the interaction of Tamm-Horsfall protein with complement 1q and inhibition of classical complement activation. Immunol Cell Biol. 2006;84(4):357–365.43.
Wolf MT, Mucha BE, Attanasio M, et al. Mutations of the Uromodulin gene in MCKD type 2 patients cluster in exon 4, which encodes three EGF-like domains. Kidney Int. 2003;64(5):1580–1587.44.
Williams SE, Reed AA, Galvanovskis J, et al. Uromodulin mutations causing familial juvenile hyperuricaemic nephropathy lead to protein maturation defects and retention in the endoplasmic reticulum. Hum Mol Genet. 2009;18(16):2963–2974.45.
Bernascone I, Janas S, Ikehata M, et al. A transgenic mouse model for uromodulin-associated kidney diseases shows specific tubulo-interstitial damage, urinary concentrating defect and renal failure. Hum Mol Genet. 2010;19(15):2998–3010.46.
Kemter E, Prueckl P, Sklenak S, et al. Type of uromodulin mutation and allelic status influence onset and severity of uromodulin-associated kidney disease in mice. Hum Mol Genet. 2013;22(20):4148–4163.47.
Elisabeth Kemter, Birgit Rathkolb, Jan Rozman, et al. Novel missense mutation of uromodulin in mice causes renal dysfunction with alterationsin urea handling, energy, and bone metabolism. Am J Physiol Renal Physiol. 2009;297(5):F1391–F1398.48.
Ma L, Liu Y, Landry NK, El-Achkar TM, Lieske JC, Wu XR. Point mutation in D8C domain of Tamm-Horsfall protein/uromodulin in transgenic mice causes progressive renal damage and hyperuricemia. PLoS One. 2017;12(11):e0186769.49.
Piret SE, Olinger E, Reed AAC, et al. A mouse model for inherited renal fibrosis associated with endoplasmic reticulum stress. Dis Model Mech. 2017;10(6):773–786.50.
Johnson BG, Dang LT, Marsh G, et al. Uromodulin p.Cys147Trp mutation drives kidney disease by activating ER stress and apoptosis. J Clin Invest. 2017;127(11):3954–3969.51.
Liu M, Chen Y, Liang Y, et al. Novel UMOD mutations in familial juvenile hyperuricemic nephropathy lead to abnormal uromodulin intracellular trafficking. Gene. 2013;531(2):363–369.52.
Rampoldi L, Caridi G, Santon D, et al. Allelism of MCKD, FJHN and GCKD caused by impairment of uromodulin export dynamics. Hum Mol Genet. 2003;12(24):3369–3384.53.
Schaeffer C, Merella S, Pasqualetto E, Lazarevic D, Rampoldi L. Mutant uromodulin expression leads to altered homeostasis of the endoplasmic reticulum and activates the unfolded protein response. PLoS One. 2017;12(4):e0175970.54.
Trudu M, Schaeffer C, Riba M, et al. Early involvement of cellular stress and inflammatory signals in the pathogenesis of tubulointerstitial kidney disease due to UMOD mutations. Sci Rep. 2017;7(1):7383.55.
Kim Y, Li C, Gu C, et al. MANF stimulates autophagy and restores mitochondrial homeostasis to treat autosomal dominant tubulointerstitial kidney disease in mice. Nat Commun. 2023;14(1):6493.56.
Kemter E, Fröhlich T, Arnold GJ, Wolf E, Wanke R. Mitochondrial Dysregulation Secondary to Endoplasmic Reticulum Stress in Autosomal Dominant Tubulointerstitial Kidney Disease - UMOD (ADTKD-UMOD). Sci Rep. 2017;7:42970.57.
Mabillard H, Sayer JA, Olinger E. Clinical and genetic spectra of autosomal dominant tubulointerstitial kidney disease. Nephrol Dial Transplant. 2023;38(2):271–282.58.
Cornec-Le Gall E, Olson RJ, Besse W, et al. Monoallelic Mutations to DNAJB11 Cause Atypical Autosomal-Dominant Polycystic Kidney Disease. Am J Hum Genet. 2018;102(5):832–844.59.
Simms RJ, Eley L, Sayer JA. Nephronophthisis. European journal of human genetics. 2009;17(4):406–16.60.
Hamada T, Ichida K, Hosoyamada M, et al. Uricosuric action of losartan via the inhibition of urate transporter 1 (URAT 1) in hypertensive patients. Am J Hypertens. 2008;21(10):1157–1162.61.
Choi SW, Ryu OH, Choi SJ, Song IS, Bleyer AJ, Hart TC. Mutant tamm-horsfall glycoprotein accumulation in endoplasmic reticulum induces apoptosis reversed by colchicine and sodium 4-phenylbutyrate. J Am Soc Nephrol. 2005;16(10):3006–3014.62.
Ma L, Liu Y, El-Achkar TM, Wu XR. Molecular and cellular effects of Tamm-Horsfall protein mutations and their rescue by chemical chaperones. J Biol Chem. 2012;287(2):1290–1305.63.
Kemter E, Sklenak S, Rathkolb B, et al. No amelioration of uromodulin maturation and trafficking defect by sodium 4-phenylbutyrate in vivo: studies in mouse models of uromodulin-associated kidney disease. J Biol Chem. 2014;289(15):10715–10726.64.
Dvela-Levitt M, Kost-Alimova M, Emani M, et al. Small Molecule Targets TMED9 and Promotes Lysosomal Degradation to Reverse Proteinopathy. Cell. 2019;178(3):521–535.65.
Cormican S, Kennedy C, Connaughton DM, et al. Renal transplant outcomes in patients with autosomal dominant tubulointerstitial kidney disease. Clin Transplant. 2020;34(2):e13783.66.
Bleyer AJ, Kidd K, Johnson E, et al. Quality of life in patients with autosomal dominant tubulointerstitial kidney disease. Clin Nephrol. 2019;92(6):302–311.67.
Perlman RL, Finkelstein FO, Liu L, et al. Quality of life in chronic kidney disease (CKD): a cross-sectional analysis in the Renal Research Institute-CKD study. Am J Kidney Dis. 2005;45(4):658–666.68.
Bleyer AJ, Hart TC, Shihabi Z, Robins V, Hoyer JR. Mutations in the uromodulin gene decrease urinary excretion of Tamm-Horsfall protein. Kidney Int. 2004;66(3):974–977.