Tirzepatide (TZP) is a once-weekly, glucose-dependent insulinotropic polypeptide/glucagon-like peptide-1 (GIP/GLP-1) receptor agonist (RA) that was recently approved in the United States for the treatment of people with type 2 diabetes and is in development for chronic weight management and nonalcoholic steatohepatitis indications (). In multiple phase 3 clinical trials, TZP produced substantial reductions in glycated hemoglobin A1c (HbA1c) and body weight, enabling many people with type 2 diabetes to achieve normalization of glucose control ().
TZP is a single linear peptide that is engineered from the GIP sequence and includes a C20 fatty diacid moiety (). Immune responses to the therapeutic protein can elicit antidrug antibodies (ADA) and potentially affect pharmacokinetics, pharmacodynamics, safety, and efficacy (). Similar to other peptide/protein drugs, including GLP-1 RA therapeutics, TZP may generate ADA, resulting in hypersensitivity reactions or neutralization of drug activity (). ADA may neutralize TZP activity and patients may develop cross-reactive neutralizing antibodies (NAb) to native GIP (nGIP) or nGLP-1.
These analyses evaluated treatment-emergent (TE) ADA in TZP-treated participants across 7 phase 3 trials and their potential effect on pharmacokinetics, efficacy, and safety outcomes.
Materials and Methods
SURPASS Clinical Trial Program
The SURPASS registration clinical trials assessed the efficacy and safety of TZP across the diabetes treatment continuum, from monotherapy, to add-on to 1 to 3 oral glucose-lowering agents, to add-on to basal insulin (). Study details, including comparator treatments and concomitant medications, for the 7 SURPASS phase 3 clinical trials can be found in Table 1 and in “References” (). All except SURPASS Japan-Combo were multicenter, randomized, parallel-group, phase 3 trials that compared the efficacy and safety of once-weekly TZP 5, 10, and 15 mg vs placebo or active comparator in adults with type 2 diabetes. SURPASS Japan-Combo was a multicenter, randomized phase 3 trial examining TZP as an add-on treatment to other type 2 diabetes medications. Primary study end points occurred at week 40 (SURPASS-1, -2, and -5) or week 52 (SURPASS-3, -4, Japan-Mono, and Japan-Combo). Baseline characteristics for the TZP-treated population can be found in Supplementary Table S1 (). Trials were conducted in accordance with the Declaration of Helsinki and the Council for International Organizations of Medical Sciences International Ethical Guidelines, the International Conference on Harmonisation Good Clinical Practices Guideline, and other applicable laws and regulations. All protocols were approved by the appropriate local or central institutional review board. Participants provided written consent before undergoing any procedure.
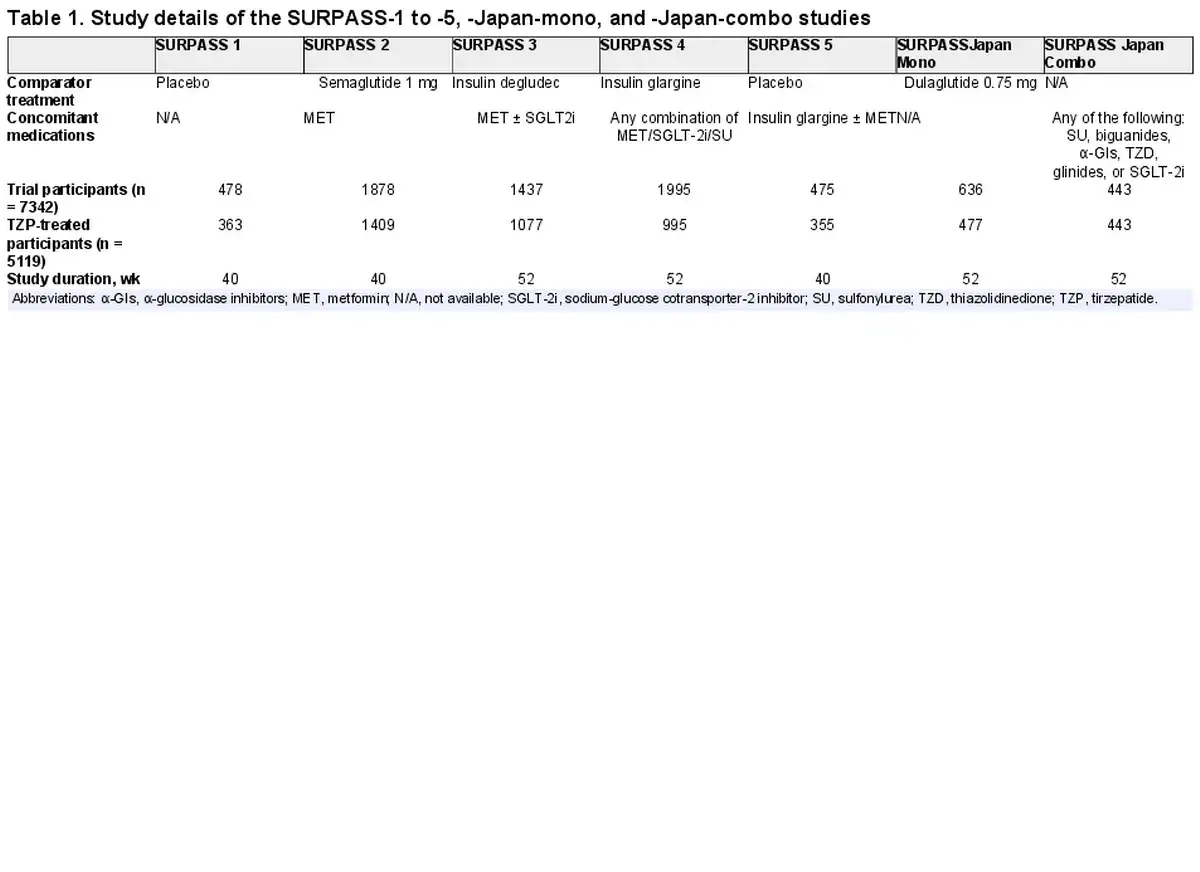
Multitiered Immunogenicity Testing Strategy
In TZP-treated participants, ADA were assessed at baseline and throughout the course of the study up to end point. Participant samples were analyzed and validated using a 4-tiered approach (Fig. 1). In tier 1, ADA were screened using a ligand-binding assay. Using a similar ligand-binding method, tier 2a confirmed the presence of ADA, tier 2b identified cross-reactive ADA binding to nGIP, tier 2c identified cross-reactive ADA binding to nGLP-1, and tier 3 measured ADA titer. Cell-based assays were used to identify NAb against TZP activity on GIPR (tier 4a) and GLP-1R (tier 4b). Novel in silico classifications were used to detect cross-reactive NAb against nGIP and nGLP-1, as detailed elsewhere.
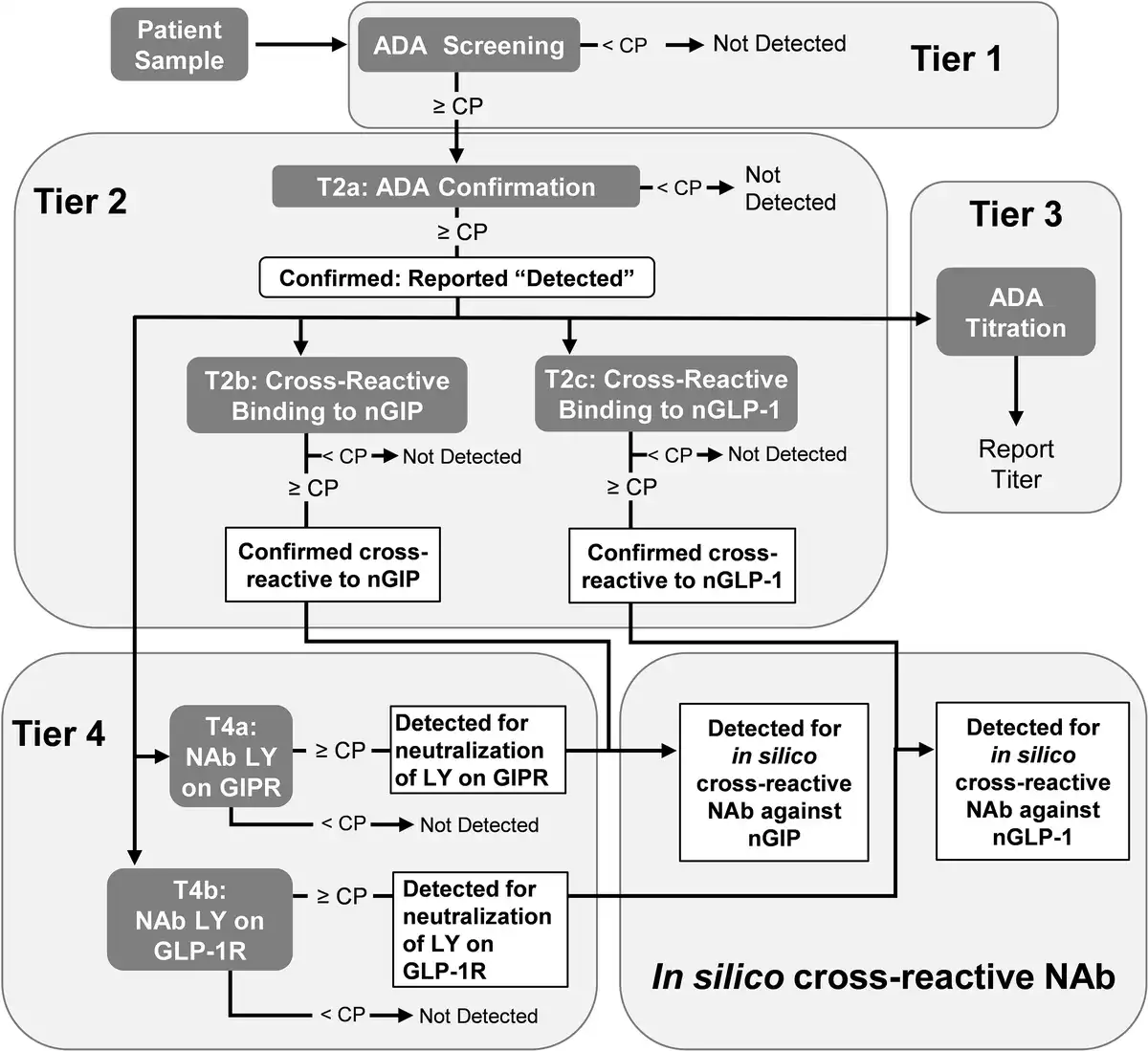
Figure 1
Flowchart of immunogenicity testing strategy. Tier 1 (screening), tier 2 (confirmatory and cross-reactive), tier 3 (titer), tier 4 (neutralizing antibodies), and in silico cross-reactive neutralizing antibodies are displayed in each bubble. ADA, antidrug antibodies; CP, cutpoint; GIP, glucose-dependent insulinotropic polypeptide; GLP, glucagon-like peptide; NAb, neutralizing antibodies; nGIP, native GIP; nGLP, native GLP; R, receptor.
Statistical Analysis
Analysis was carried out on the TZP-treated population of all 7 phase 3 trials, defined as all randomly assigned patients who took at least 1 dose of the study drug. TE ADA-evaluable patients from the TZP-treated population had a baseline ADA assessment and at least one postbaseline ADA assessment. A TE ADA+ patient was identified if an evaluable patient had either of the following:
Baseline status of ADA not present and at least 1 postbaseline status of ADA present with titer 2 or more times the minimum required dilution of the ADA assay, or
Baseline and postbaseline status of ADA present, with the postbaseline titer being greater than or equal to 4-fold than the baseline titer.
A TE ADA-evaluable patient that did not meet the TE ADA+ criteria was classified as a TE ADA– patient.
Titer was assessed when ADA assay result was detected. For occasional detected samples for which the titer assay could not be performed, a titer value was imputed for the purpose of TE ADA determination—1:10 for baseline samples with detected ADA and 1:20 for postbaseline samples with detected ADA.
Descriptive analyses were performed on TE ADA+ and TE ADA– patients, both overall and by study (efficacy results), titer (pharmacokinetics and efficacy results), or dose group (safety results). Comparison between TE ADA+ and TE ADA– is not a randomized comparison and is subject to possible selection biases.
Results
Characterization of Antidrug Antibodies (ADA) and Treatment-emergent ADA in Tirzepatide-treated Patients From all 7 Studies
Across the 7 SURPASS studies, a wide range of patients with different demographics and clinical characteristics were included (Supplementary Table S1) (). As shown in the flow diagram (Fig. 2), there were 5025 (98.2%) TE ADA–evaluable patients from the patients that were randomly assigned to receive TZP (N = 5119). At baseline, 353 (7%) had preexisting ADA (Table 2). The ADA titer at baseline ranged from 1:10 to 1:10 240 (median 1:20). Cross-reactive ADA to nGIP and nGLP-1 were observed at baseline in 97 (1.9%) and 34 (0.7%) patients, respectively. Less than 0.1% of patients had NAb against TZP activity of GIPR or GLP-1R, and no cross-reactive NAb against nGIP or nGLP-1 were detected at baseline.
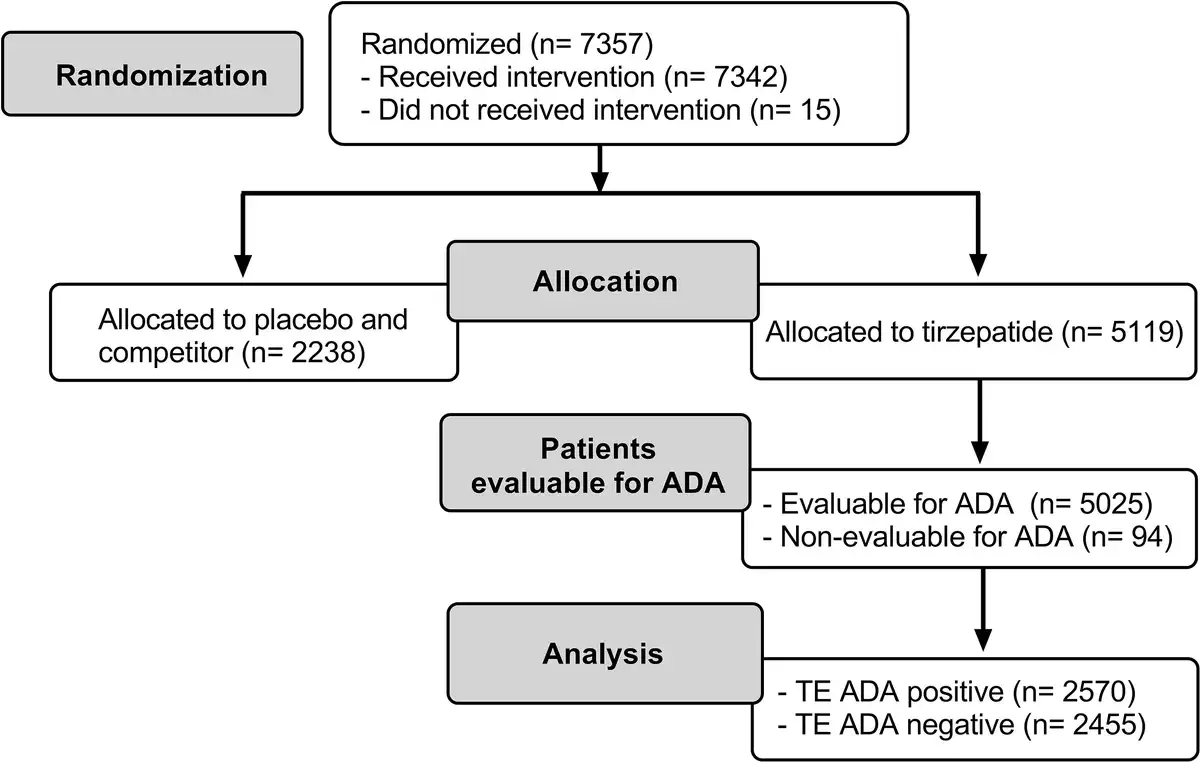
Figure 2
Flow diagram for immunogenicity analysis of randomly assigned participants from SURPASS-1 to -5, -Japan-mono, and -Japan-combo studies. ADA, antidrug antibodies; TE, treatment-emergent.
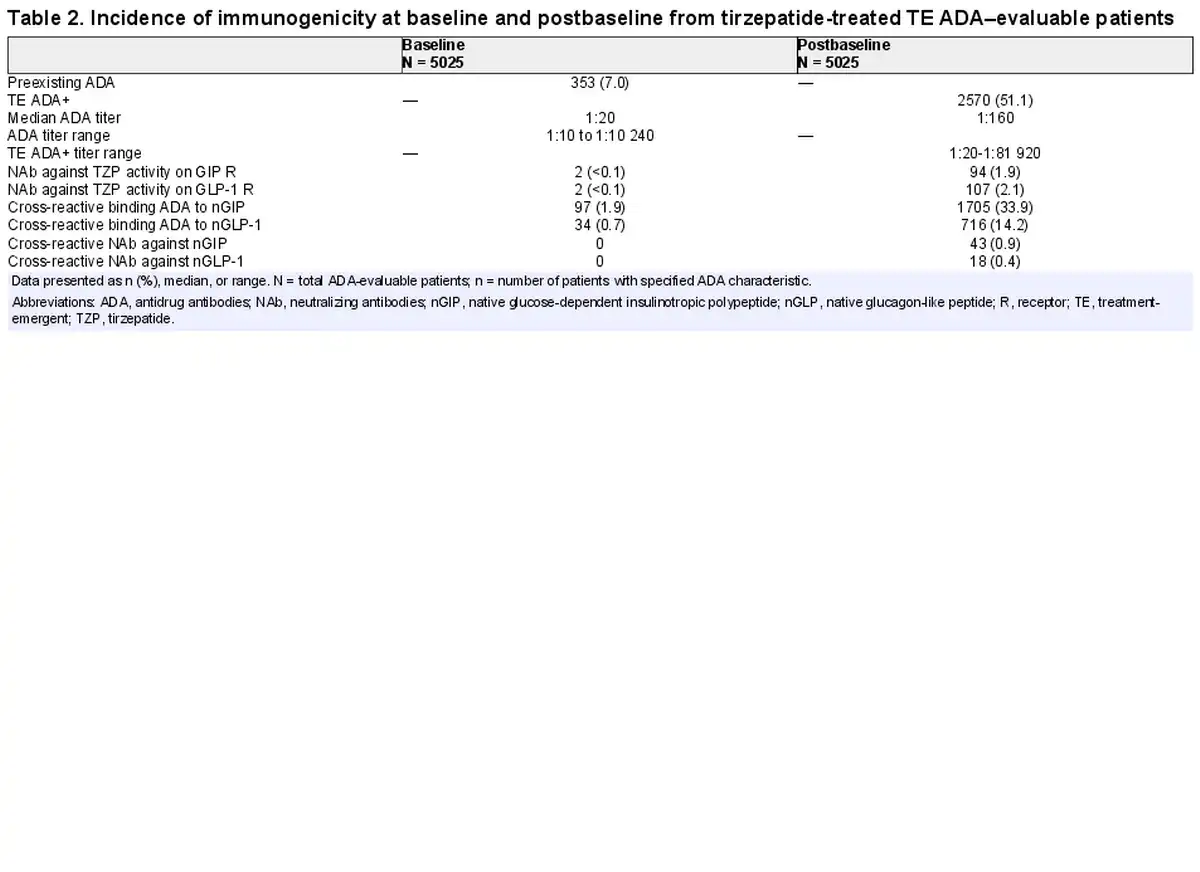
Postbaseline, 2570 (51.1%) TZP-treated TE ADA–evaluable patients were TE ADA+ (see Table 2). There were similar incidences of TE ADA across dose groups (Supplementary Table S2) (). A total of 2403 (47.8%) TE ADA were classified as treatment induced and 167 (3.3%) were classified as treatment boosted. NAb against TZP activity on the GIPR and GLP-1R were observed in 94 (1.9%) and 107 (2.1%) patients, respectively. A total of 1705 (33.9%) and 716 (14.2%) TE ADA+ patients had cross-reactive ADA to nGIP and nGLP-1, respectively. Additionally, cross-reactive NAb against nGIP and nGLP-1 were detected in 43 (0.9%) and 18 (0.4%) patients, respectively.
Time to first TE ADA+ titer followed similar profiles across all phase 3 studies (Table 3). Of the TE ADA–evaluable patients, 4.6% to 10.7% (median 7.2%) developed TE ADA by 12 weeks, 19.9% to 38.2% (median 25.6%) by 24 weeks, and 38.4% to 65.5% (median 50.5%) by 52 weeks.
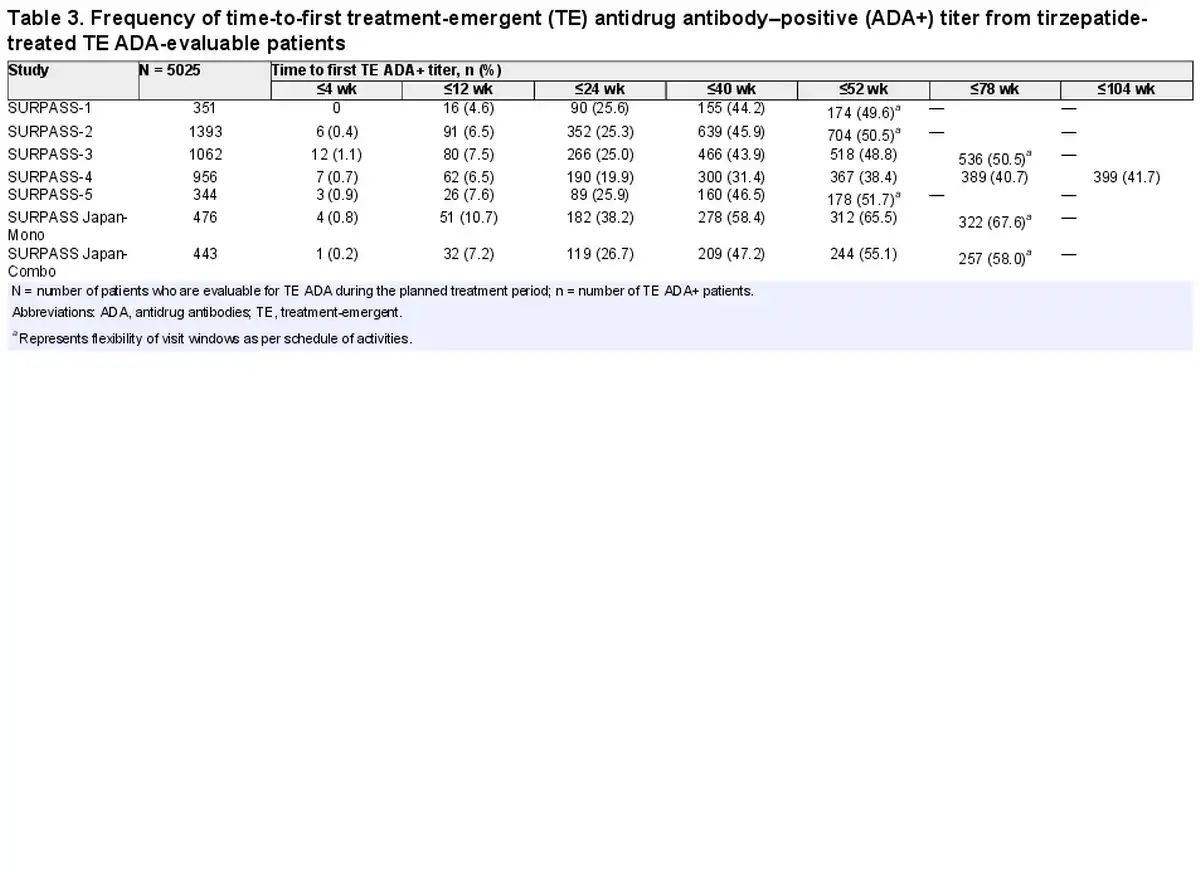
Maximum postbaseline ADA titers in TZP-treated TE ADA+ patients from all 7 phase 3 studies ranged from 1:20 to 1:81 920 (median 1:160; see Table 2) during the planned treatment period (Supplementary Fig. S1A) (). Eighty-six (3.3%) out of 2570 TE ADA+ patients across all phase 3 studies showed maximum ADA titers greater than or equal to 1:5120. Median ADA titers peaked at week 40 (median of 1:160), plateaued up to week 52, and then declined to lower levels (Supplementary Fig. S1B) ().
Comparison of Antidrug Antibodies in Tirzepatide-treated Patients and in Placebo-treated Patients From 2 Studies
There were 923 (96.9%) TE ADA–evaluable patients, including 228 placebo, from 953 TZP-treated population patients. At baseline, 77 (8.3%) patients had preexisting ADA, with similar incidence between TZP and placebo. The preexisting ADA titers ranged from 1:10 to 1:640 (median 1:20). Among them, 19 (2.1%) patients had cross-reactive ADA to nGIP, 8 (0.9%) patients had cross-reactive ADA to nGLP-1, and 1 (0.1%) patient had NAb against TZPactivity on GLP-1R. No NAb against TZP activity on GIPR, or cross-reactive NAb to nGIP or nGLP-1, were detected at baseline.
Postbaseline, 695 TZP-treated patients were evaluable for TE ADA during the planned treatment period. Of these, 352 (50.6%) were TE ADA+. The maximum TE ADA+ titers ranged from 1:20 to 1:40 960 (median 1:160). Among them, 327 (47.1%) were classified as treatment induced, and 25 (3.6%) were classified as treatment boosted. Cross-reactive ADA to nGIP were identified in 226 (32.5%) patients, and 92 (13.2%) patients were positive for cross-reactive ADA to nGLP-1. NAb against TZP activity on GIPR were found in 8 (1.2%) patients, and 7 (1.0%) patients were positive for NAb against TZP activity on GLP-1R. Cross-reactive NAb against nGIP were observed in 5 (0.7%) patients, and 4 (0.6%) patients were positive for cross-reactive NAb against nGLP-1.
Among the 228 TE ADA–evaluable placebo-treated patients, 11 (4.8%) were TE ADA+ during the treatment period. Among them, 8 (3.5%) patients were positive for cross-reactive ADA to nGIP, 1 (0.4%) patient was positive for cross-reactive ADA to nGLP-1, and 1 (0.4%) patient was positive for NAb against TZP activity on GIPR. No patient was positive for NAb against TZP activity on GLP-1R or cross-reactive NAb to nGIP or nGLP-1.
Overall, the percentage of TE ADA+ patients was higher in the TZP groups compared with placebo.
Treatment-emergent Antidrug Antibodies Had no Effect on Tirzepatide Pharmacokinetics
No overt pattern or trend was evident in the graphical comparison of TZP clearance in patients with ADA detected compared to those with ADA not detected (Fig. 3A). The range of TZP clearance values were similar between groups with lowest to highest observed ADA titer (Fig. 3B). No relationship between NAb and TZP concentration was detected (Supplementary Fig. S2) ().
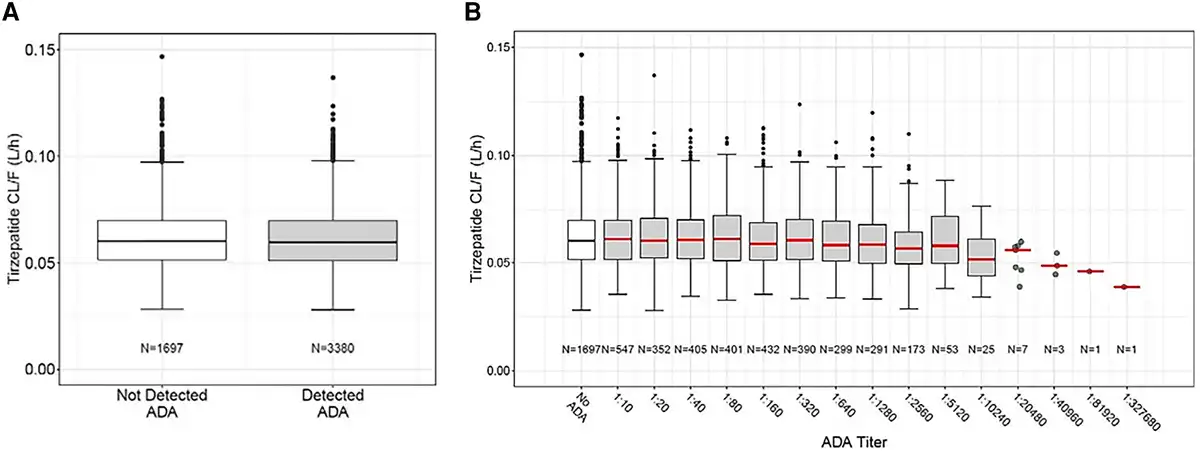
Figure 3
Tirzepatide clearance by ADA status. Tirzepatide apparent clearance (CL/F) was not affected by A, ADA status or B, ADA titer. ADA titer 1:327 680 shown in B was observed at week 78 and was the maximum reported titer in one patient who had been randomly assigned to the tirzepatide 5 mg group. This patient was not TE ADA evaluable as no baseline sample had been collected. The patient's first ADA sample was collected 19 minutes after the first dose of tirzepatide 2.5 mg and was reported as 1:163 840. The second ADA sample for this patient was collected at week 41 during treatment with tirzepatide 5 mg and was reported as 1:40 960. Although all 3 ADA titers collected from this patient during tirzepatide treatment were high, the maximum titer was only 1 dilution higher than the first ADA sample. This patient did not experience any hypersensitivity or injection site reaction and showed glycated hemoglobin A1c-lowering similar to other tirzepatide-treated patients. ADA, antidrug antibodies; CL/F, apparent clearance.
Treatment-emergent Antidrug Antibodies Had no Effect on the Glycated Hemoglobin A1c Lowering Effect of Tirzepatide
The primary efficacy end point of each phase 3 study was HbA1c change from baseline at week 40 or week 52, depending on the study. Graphical comparison of TZP-induced HbA1c change from baseline showed no apparent pattern relative to either TE ADA status (Fig. 4A) or maximum TE ADA+ titer (≥1:5120 vs <1:5120) (Fig. 4B). Similar results were observed after assessing TE ADA status and ADA titer in each individual study (Supplementary Fig. S3) (). Additionally, comparison of the HbA1c change from baseline showed no apparent effect relative to NAb status against TZP activity on GIPR or GLP-1R (Table 4).
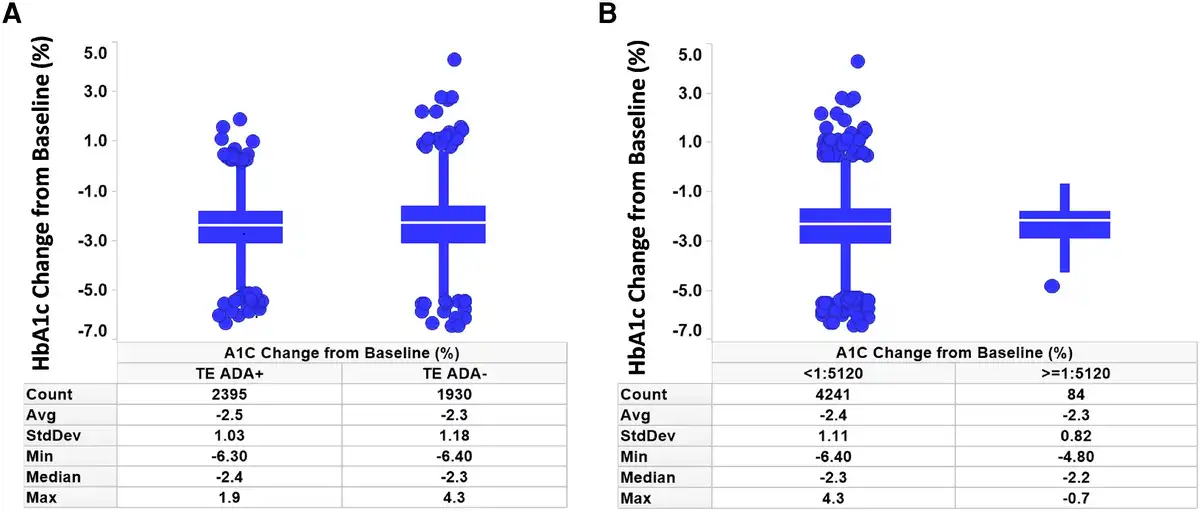
Figure 4
HbA1c assessment by ADA and titer status. Change from baseline in HbA1c was not affected by TE A, ADA status or B, ADA titer less than 1:5120 vs 1:5120 or greater. ADA, antidrug antibodies; HbA1c, glycated hemoglobin A1c; Count, number of patients; TE, treatment-emergent.
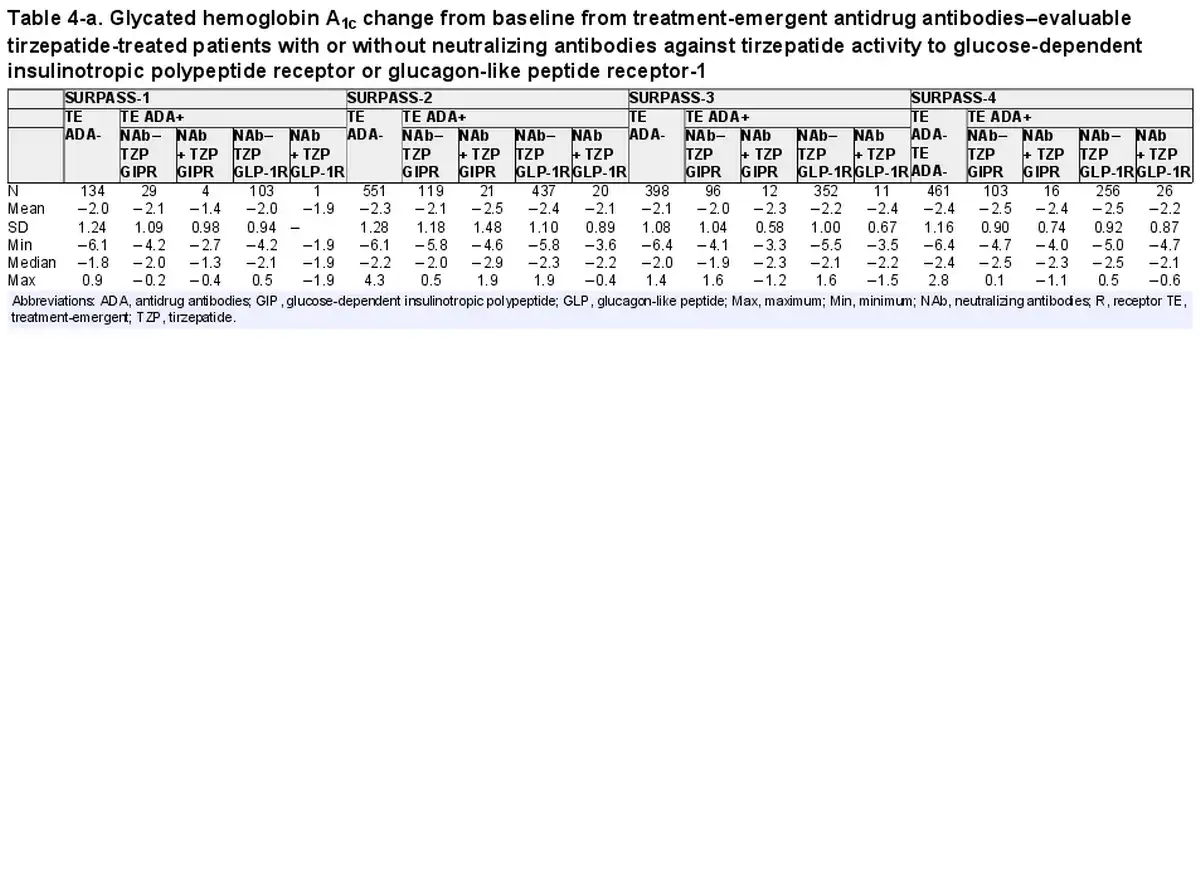
Table 4-a. Glycated hemoglobin A1c change from baseline from treatment-emergent antidrug antibodies–evaluable tirzepatide-treated patients with or without neutralizing antibodies against tirzepatide activity to glucose-dependent insulinotropic polypeptide receptor or glucagon-like peptide receptor-1
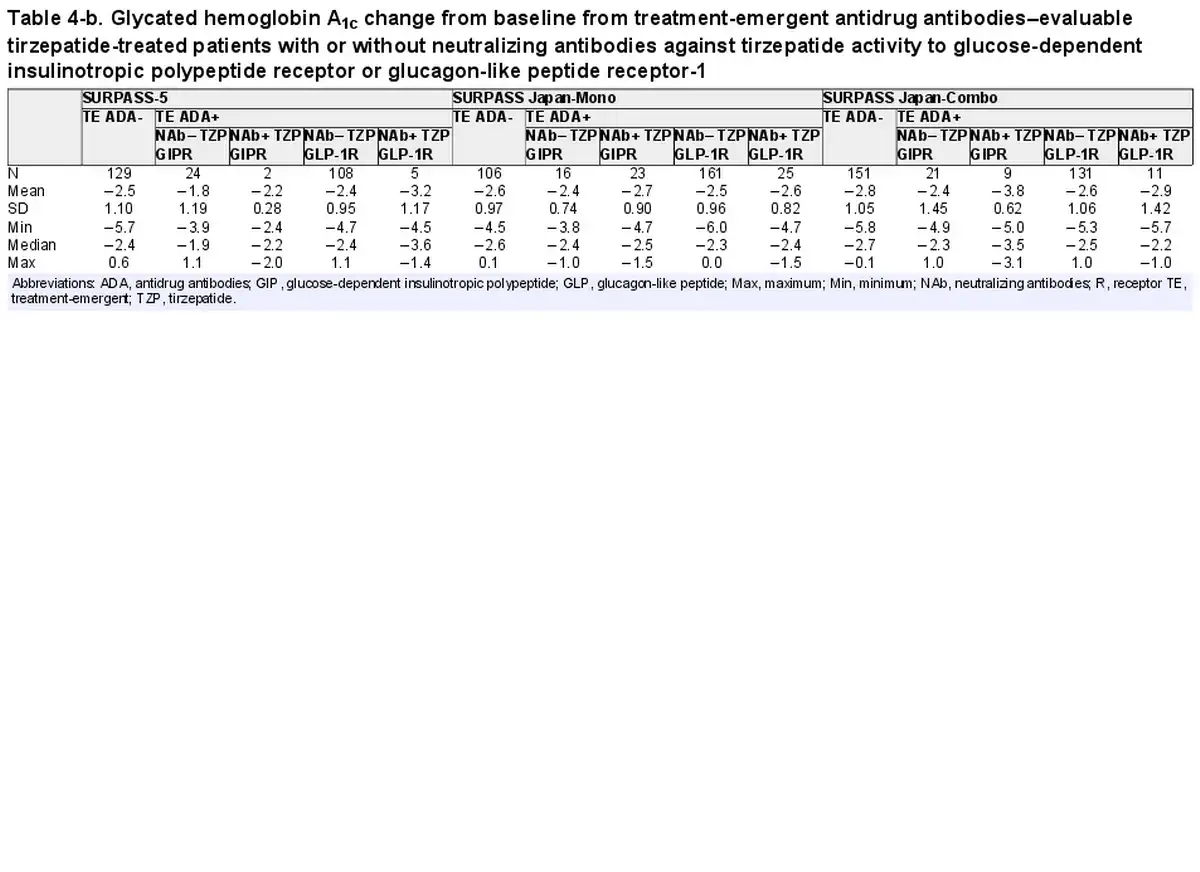
Table 4-b. Glycated hemoglobin A1c change from baseline from treatment-emergent antidrug antibodies–evaluable tirzepatide-treated patients with or without neutralizing antibodies against tirzepatide activity to glucose-dependent insulinotropic polypeptide receptor or glucagon-like peptide receptor-1
Effect of Treatment-emergent Antidrug Antibodies on Hypersensitivity and Injection Site Reactions
Of the 5025 TZP-treated TE ADA–evaluable population, 179 (3.6%) experienced hypersensitivity reactions during the planned treatment period; 106 were TE ADA+ and 73 were TE ADA– (Table 5). Of the 106 TE ADA+ patients that experienced 1 or more hypersensitivity reactions, the ADA titer range was 1:10 to 1:10 240 during the treatment period. The events in TE ADA+ patients were mostly mild to moderate in severity. Of the 4 patients who experienced severe or serious hypersensitivity reactions, 3 were TE ADA– and no temporal relationship was identified with ADA titer and onset of urticaria in the TE ADA+ patient. The majority of hypersensitivity reactions first occurred within 16 weeks of receiving TZP and resolved independently of TE ADA status or titer. Six TZP-treated patients discontinued study drug because of hypersensitivity reactions, of which 4 were TE ADA+. The most common hypersensitivity reactions reported were urticaria, eczema, and rash. No anaphylactic reactions were observed in TZP-treated patients.
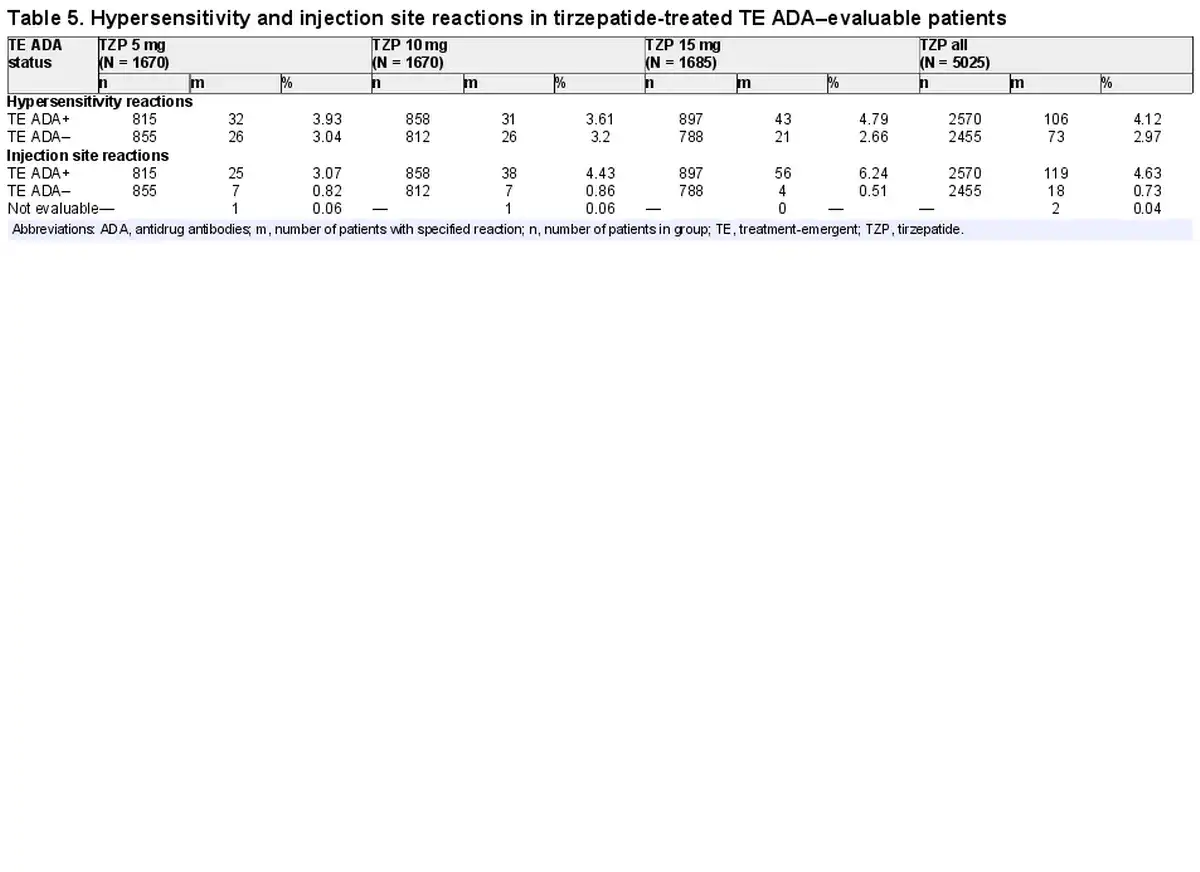
Of the 5025 TZP-treated TE ADA–evaluable population, 137 (2.7%) developed injection site reactions, 119 of which were TE ADA+ and 18 were TE ADA– (Table 5). Of the 119 TE ADA+ patients that experienced 1 or more injection site reactions, the ADA titer range was 1:10 to 1:81 920 during the treatment period. All events were nonserious and nonsevere. Four TZP-treated patients discontinued study drug because of injection site reactions; all were TE ADA+. The majority of reactions first occurred within 16 weeks of receiving TZP and resolved independently of TE ADA status or titer level.
Preexisting Antidrug Antibodies (ADA) had no Effect on Treatment-emergent ADA
The TE ADA+ incidence during the planned treatment period was similar between patients with and without preexisting ADA (47.3% and 51.4%, respectively; Supplementary Table S3) ().
In addition, the vast majority of TE ADA+ were classified as treatment induced rather than treatment boosted (47.8% and 3.3%, respectively). As such, preexisting ADA did not predispose patients to develop TE ADA.
Treatment-emergent Antidrug Antibody Incidence by Race, Region, and Ethnicity
Consistent with the TE ADA incidence in the 2 Japanese trials as shown in Table 3 (SURPASS Japan-Mono and SURPASS Japan-Combo with 67.6% and 58.0% TE ADA incidence, respectively), Asian participants by race and Asia by region had the highest TE ADA incidence (Supplementary Table S4) (). However, HbA1c change from baseline in Japanese trials showed no apparent pattern relative to either TE ADA status (Supplementary Fig. S3A) (), maximum TE ADA+ titer (≥1:5120 vs <1:5120) (Supplementary Fig. S3B) (), or NAb status against TZP activity on GIPR or GLP-1R (see Table 4).
Prior Exposure to a Glucagon-like Peptide-1 Receptor Agonist Did not Alter Treatment-emergent Antidrug Antibody Incidence
There were 351 TE ADA–evaluable TZP-treated patients with known prior exposure to a GLP-1 RA. At baseline, 29 (8.3%) patients had preexisting ADA, with similar incidence between dose groups (Supplementary Table S5) (). Post baseline, 187 (53.3%) were TE ADA+ during the planned treatment period. The incidence of preexisting ADA, TE ADA+, cross-reactive antibodies, and NAb seen in patients with prior exposure to GLP-1 RA are comparable to the overall TZP-treated population during the planned treatment period. The maximum ADA titer distribution for TZP-treated TE ADA+ patients who had prior GLP-1 RA ranged from 1:20 to 1:20 480 (median 1:160). Of the 187 TE ADA+ patients with known exposure to GLP-1 RA, 9 patients (4.8%) experienced injection site reactions and 4 patients (2.1%) experienced hypersensitivity reactions. These were comparable to the overall TZP-treated TE ADA+ population.
Discussion
TZP is the first Food and Drug Administration–approved GIP and GLP-1 RA for the treatment of people with type 2 diabetes. Approximately half of TZP-treated patients developed TE ADA during the planned treatment period with low incidence of NAb against TZP activity on GIPR and GLP-1R and cross-reactive NAb against nGIP and nGLP-1. This translated to no discernible effects of TE ADA against TZP on pharmacokinetics or improvement of glycemic control. While hypersensitivity and injection site reactions were more frequently reported in TE ADA+ than in TE ADA– patients, no pattern was detected between the time of the event reporting and ADA status, and most events resolved irrespective of ADA status.
The propensity of a therapeutic protein product to generate an immune response to itself and to related endogenous counterparts or to induce immunologically related adverse events is a regulatory area of interest (). Immunogenicity to therapeutic proteins has the potential to affect the drug's pharmacokinetics, efficacy, and/or safety (). The data show that the development of anti-TZP antibodies had no effect on the drug's pharmacokinetics or efficacy. Although there was a numerical imbalance seen in hypersensitivity and injection site reactions, these events resolved irrespective of TE ADA status and titer, and were mostly mild in severity and most patients continued on treatment. Patient-specific factors such as age, genetics, concomitant medications, and preexisting ADA are known to influence immunogenicity (). At baseline, 7% of the overall population and 8% of patients with prior GLP-1 RA exposure had preexisting ADA, which had no effect on the development of TE ADA. Additionally, NAb have the potential to interfere with the actions of endogenous GIP and GLP-1 (). The present results indicate that the incidence of NAb was low and did not affect the pharmacokinetics or HbA1c-lowering effects of TZP.
ADA development is a substantial concern for patients receiving peptide or protein-based therapies. It is important to note that ADA comparisons between different GLP-1 RA therapies are not possible due to differences in administration, immunogenicity assay sensitivities and specificities, sample handling, timing of sample collection, concomitant medications, and underlying disease, among other factors. Head-to-head trials with GLP-1 RAs using common ADA screening methods would be necessary to directly compare TZP against other available GLP-1 RAs. Approximately 50% of TZP-treated patients developed TE ADA. This is within the range of published studies for other incretin therapies, including dulaglutide (up to 4.1%) (, ), semaglutide (up to 4.3%) (), lixisenatide (up to 71.2%) (), liraglutide (up to 8.7%) (), albiglutide (2.5%) (), exenatide twice-daily (up to 36.7%), and exenatide once-weekly (56.8%) (). The incidence of TE ADA seen in TZP-treated patients may be because TZP is engineered from nGIP (,27). The TZP ADA assay was likely more sensitive than the assays used in prior GLP-1 RA studies to comply with newer Food and Drug Administration guidelines. Therefore, without head-to-head clinical trial data, robust comparisons between incretin therapies cannot be made. Although the incidences of TE ADA were similar between GLP-1–naive and prior GLP-1–treated patients, additional data are needed to explore the effect of switching patients from GLP-1 RAs to TZP.
These analyses have several strengths, namely the study population size, study length, and range of included patient characteristics. These analyses are limited by the different study durations of the 7 SURPASS studies, different inclusion and exclusion criteria, and different concomitant medications.
In conclusion, approximately half of TZP-treated patients developed TE ADA. Immunogenicity was not associated with any effect on TZP pharmacokinetics or efficacy, and the majority of hypersensitivity or injection site reactions (ADA+) were mild to moderate in severity.
Acknowledgments
The authors would like to thank Ana Hickey, PhD, Eli Lilly and Company, for medical writing and editorial services.
Abbreviations
ADA: antidrug antibodies
GIP: glucose-dependent insulinotropic polypeptide
GLP: glucagon-like peptide
HbA1c: glycated hemoglobin A1c
NAb: neutralizing antibodies
nGIP: native glucose-dependent insulinotropic polypeptide
nGLP: native glucagon-like peptide
RA: receptor agonist
TE: treatment-emergent
TZP: tirzepatide
References
- 1. Min T, Bain SC. The role of tirzepatide, dual GIP and GLP-1 receptor agonist, in the management of type 2 diabetes: the SURPASS clinical trials. Diabetes Ther. 2021; 12(1):143–157.
- 2. Rosenstock J, Wysham C, Frías JP, et al Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): a double-blind, randomised, phase 3 trial. Lancet. 2021; 398(10295):143–155.
- 3. Frías JP, Davies MJ, Rosenstock J, et al Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N Engl J Med. 2021; 385(6):503–515.
- 4. Ludvik B, Giorgino F, Jódar E, et al Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): a randomised, open-label, parallel-group, phase 3 trial. Lancet. 2021; 398(10300):583–598.
- 5. Del Prato S, Kahn SE, Pavo I, et al Tirzepatide versus insulin glargine in type 2 diabetes and increased cardiovascular risk (SURPASS-4): a randomised, open-label, parallel-group, multicentre, phase 3 trial. Lancet. 2021; 398(10313):1811–1824.
- 6. Dahl D, Onishi Y, Norwood P, et al Effect of subcutaneous tirzepatide vs placebo added to titrated insulin glargine on glycemic control in patients with type 2 diabetes: the SURPASS-5 randomized clinical trial. JAMA. 2022; 327(6):534–545.
- 7. Inagaki N, Takeuchi M, Oura T, Imaoka T, Seino Y. Efficacy and safety of tirzepatide monotherapy compared with dulaglutide in Japanese patients with type 2 diabetes (SURPASS J-mono): a double-blind, multicentre, randomised, phase 3 trial. Lancet Diabetes Endocrinol. 2022; 10(9):623–633.
- 8. Kadowaki T, Chin R, Ozeki A, Imaoka T, Ogawa Y. Safety and efficacy of tirzepatide as an add-on to single oral antihyperglycaemic medication in patients with type 2 diabetes in Japan (SURPASS J-combo): a multicentre, randomised, open-label, parallel-group, phase 3 trial. Lancet Diabetes Endocrinol. 2022; 10(9):634–644.
- 9. Sun B, Willard FS, Feng D, et al Structural determinants of dual incretin receptor agonism by tirzepatide. Proc Natl Acad Sci. 2022; 119(13):e2116506119.
- 10. FDA. Guidance for Industry: Immunogenicity Assessment for Therapeutic Protein Products. https://www.fda.gov/media/85017/download.
- 11. Gorovits B, Peng K, Kromminga A. Current considerations on characterization of immune response to multi-domain biotherapeutics. BioDrugs. 2020; 34(1):39–54.
- 12. De Block C, Bailey C, Wysham C, Hemmingway A, Allen SE, Peleshok J. Tirzepatide for the treatment of adults with type 2 diabetes: an endocrine perspective. Diabetes Obes Metab. 2023; 25(1):3–17.
- 13. Mullins G, Hodsdon ME, Li YG, et al“Supplemental tables and figures of ‘Tirzepatide immunogenicity on pharmacokinetics, efficacy, and safety: analysis of data from Phase 3 studies’”, Mendeley Data, V1 (2023),
- 14. FDA. Immunogenicity Testing of Therapeutic Protein Products—Developing and Validating Assays for Anti-Drug Antibody Detection. https://www.fda.gov/media/119788/download.
- 15. EMA. Guideline on Immunogenicity assessment of therapeutic proteins. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-immunogenicity-assessment-therapeutic-proteins-revision-1_en.pdf.
- 16. Milicevic Z, Anglin G, Harper K, et al Low incidence of anti-drug antibodies in patients with type 2 diabetes treated with once-weekly glucagon-like peptide-1 receptor agonist dulaglutide. Diabetes Obes Metab. 2016; 18(5):533–536.
- 17. Wang W, Nevárez L, Filippova E, et al Efficacy and safety of once-weekly dulaglutide versus insulin glargine in mainly Asian patients with type 2 diabetes mellitus on metformin and/or a sulphonylurea: A 52-week open-label, randomized phase III trial. Diabetes Obes Metab. 2019; 21(2):234–243.
- 18. Marso SP, Bain SC, Consoli A, et al Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016; 375(19):1834–1844.
- 19. Sorli C, Harashima SI, Tsoukas GM, et al Efficacy and safety of once-weekly semaglutide monotherapy versus placebo in patients with type 2 diabetes (SUSTAIN 1): a double-blind, randomised, placebo-controlled, parallel-group, multinational, multicentre phase 3a trial. Lancet Diabetes Endocrinol. 2017; 5(4):251–260.
- 20. Ahrén B, Masmiquel L, Kumar H, et al Efficacy and safety of once-weekly semaglutide versus once-daily sitagliptin as an add-on to metformin, thiazolidinediones, or both, in patients with type 2 diabetes (SUSTAIN 2): a 56-week, double-blind, phase 3a, randomised trial. Lancet Diabetes Endocrinol. 2017; 5(5):341–354.
- 21. Ahmann AJ, Capehorn M, Charpentier G, et al Efficacy and safety of once-weekly semaglutide versus exenatide ER in subjects with type 2 diabetes (SUSTAIN 3): a 56-week, open-label, randomized clinical trial. Diabetes Care. 2018; 41(2):258–266.
- 22. Ratner RE, Rosenstock J, Boka G. Dose-dependent effects of the once-daily GLP-1 receptor agonist lixisenatide in patients with type 2 diabetes inadequately controlled with metformin: a randomized, double-blind, placebo-controlled trial. Diabet Med. 2010; 27(9):1024–1032.
- 23. Buse JB, Garber A, Rosenstock J, et al Liraglutide treatment is associated with a low frequency and magnitude of antibody formation with No apparent impact on glycemic response or increased frequency of adverse events: results from the liraglutide effect and action in diabetes (LEAD) trials. J Clin Endocrinol Metab. 2011;96(6):1695–1702.
- 24. Rosenstock J, Reusch J, Bush M, Yang F, Stewart MGroup for the AS. Potential of albiglutide, a long-acting GLP-1 receptor agonist, in type 2 diabetes: A randomized controlled trial exploring weekly, biweekly, and monthly dosing. Diabetes Care. 2009; 32(10):1880–1886.
- 25. Prasad-Reddy L, Isaacs D. A clinical review of GLP-1 receptor agonists: efficacy and safety in diabetes and beyond. Drugs Context. 2015; 4:212283.
- 26. Yu M, Benjamin MM, Srinivasan S, et al Battle of GLP-1 delivery technologies. Adv Drug Deliv Rev. 2018; 130:113–130.
- 27. Coskun T, Sloop KW, Loghin C, et al LY3298176, A novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: from discovery to clinical proof of concept. Mol Metab. 2018; 18:3–14.