INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a progressive neurological disease characterized by upper and lower motor neuron signs and symptoms, as well as muscle weakness and atrophy. These symptoms reflect motor system degeneration, including within motor neuron groups (, ), axons (), and at the neuromuscular junction (). Extramotor symptoms are also seen in ALS. Most notable among these are cognitive deficits, which are seen in up to 50% of ALS patients with an estimated 15% having frank dementia (, ). Additional extramotor symptoms may include abnormalities in energy metabolism (), autonomic function (), parkinsonism (), and supranuclear gaze palsy (), among others.
In the United States, ALS is more common in men and the white population, with a prevalence of 4.7–5.0 per 100 000 persons and the highest prevalence between the ages of 60 and 69 years (). The onset of symptoms may appear at younger ages particularly in patients with a positive family history, comprising 5%–10% of all ALS patients (). Therapeutic options remain limited and survival remains poor in both familial and sporadic forms with disease durations often between 20 and 48 months (). Poor survival has especially been noted in older patients and with bulbar symptom onset (), as well as in patients with hexanucleotide expansion (G4C2) in chromosome 9 open reading frame 72 (c9orf72) on chromosome 9 (). In the latter group, microsatellite expansion leads to a toxic accumulation of sense and antisense RNA transcripts and cytoplasmic dipeptide aggregates via non-AUG initiated translation (). This group comprises ∼50% and ∼10% of familial ALS (fALS) and sporadic ALS (sALS) cases, respectively ().
The pathologies underlying ALS remain unknown in many cases and multiple mechanisms underlying disease progression have been described. These include the cellular accumulation of ubiquinated proteins (), including proteins involved in RNA metabolism (), mitochondrial dysfunction (), activation of pro-apoptotic signaling (), cytoskeletal instability with abnormal accumulation of neurofilaments and fragmentation of organelles (, ), and dysregulation of glutamate transport with resulting excitotoxicity (, ). The initial pathologic events in genetically driven cases of ALS are better understood. In addition to patients with c9orf72 expansion, ∼20% of patients with fALS have mutations in superoxidase dismutase 1 (SOD1), likely leading to both oxidative stress and binding of misfolded, mutant SOD1 protein to cellular proteins, causing toxicity (). “non-cell autonomous” mechanisms of injury are also recognized as contributors to neuronal dysfunction and death (, ). The transition of neuroprotective microglia (M2 state) to activated, pro-inflammatory microglia (M1 state) that results from a dialogue between injured neurons, the macrophage/microglial system, and regulatory T-cells has been proposed as one of the non-cell autonomous mechanisms of neuron injury in ALS ().
The pathologic accumulation of TAR DNA binding protein 43 kDa (TDP-43) (, ) is now well recognized to contribute to the pathology of ALS. Further, models of spread via prion-like () or other mechanisms involving local and regional diffusion () have been proposed and remain under investigation. TDP-43 accumulation is not specific to ALS and may be seen in ∼50% of cases with frontotemporal lobar degeneration (FTLD) without ALS (), as well as in hippocampal sclerosis of aging, or CARTS (cerebral aging with TDP-43 and sclerosis) (, ), and Alzheimer disease (AD) (). Heterogeneous TDP-43 pathology has also been described in ALS (, ), including more extensive inclusion pathology reported in the extramotor regions of patients with c9orf72-associated ALS (c9ALS) (). The role of TDP-43 pathology in other clinical features of ALS, such as variability in the survival of patients or rate of progression, is not as well understood.
The aim of this study was to determine the clinical significance of whole brain and spinal cord TDP-43 neuropathology in an autopsy cohort of patients with ALS. The study was guided by the hypotheses that TDP-43 pathologic burden would be greatest in patients with a longer duration of clinical disease, in patients with cognitive impairment, and in patients with c9orf72 expansion. In addition, we investigated whether the application of knowledge discovery methods (e.g. cluster analysis) would reveal clinico-pathologic subgroups of ALS patients that might not be readily appreciated by standard univariate and multivariate regression analyses. The study was undertaken in 57 fALS and sALS patients with clinically diagnosed and pathologically confirmed ALS.
MATERIALS AND METHODS
Case Identification and Clinical Assessment
All of the ALS patients whose tissue was examined in this study (n = 57) came to autopsy at Houston Methodist Hospital. This was a convenience sample of patients and representative of the ALS patients seen at our institution with pathologically confirmed ALS. No patients were excluded. The majority of these patients were evaluated in the clinic of 2 of the study authors (SHA, JWA). Pathologic materials and reports were retrieved, and clinical characteristics were reviewed and collated independently (SHA, JWA, EC) from the pathologic review, with the approval of the Institutional Review Board at Houston Methodist Hospital (IRB 2-0114-0013).
Clinical variables examined included () disease duration, defined as the period between the date or month of first symptoms and the date of autopsy, () age at death, () site of disease onset (lower extremity, upper extremity, bulbar, or other), () cognitive status (cognitively intact or cognitively impaired groups, with the majority of the latter having mild frontal–subcortical dysfunction), () positive or negative family history, () Appel ALS (AALS) score as a measure of clinical severity (), and () the monthly rate of change between the first and last AALS scores. The designation of cognitively intact or cognitively impaired was based on formal neuropsychological testing in 36 patients, Montreal Cognitive Assessment (MoCA) score in 3 patients (with 1 patient having clinical cognitive impairment and a MoCA of 22/30, the other 2 being cognitively intact), an interview with a caregiver in one patient, and clinical designation by author SHA in 1 patient. For 16 patients there was insufficient evaluation or documentation of clinical status to make a determination.
Histological and Immunohistochemical Procedures
At the time of autopsy the brain was removed, tissue samples were submitted per research protocol, and the remaining hemispheres and tissue slices were fixed in 20% formalin for at least 1 week prior to further sampling. Formalin-fixed tissue samples were obtained from multiple areas, including frontal lobe and frontal hemispheric white matter, typically including agranular frontal cortex, parietal and temporal association isocortices, striate and parastriate cortex of the occipital lobe, anterior cingulate cortex and corpus callosum, amygdala and entorhinal cortex and adjacent white matter, hippocampal formation, subiculum, presubiculum and parahippocampal cortex, basal forebrain and hypothalamus, thalamus, caudate, putamen and globus pallidus, claustrum and insula, midbrain, pons, medulla, cerebellum, and cervical, thoracic, lumbar, and sacral spinal cord. Sections placed in cassettes were further fixed in 10% formalin until processing and embedding.
Protocol for tissue examination at autopsy included staining of sections with hematoxylin and eosin (H&E) and TDP-43 immunostain (Proteintech, 10782-2-AP, rabbit polyclonal antibody, 1:200; Rosemont, IL). Immunostaining was performed on brain tissue that was fixed in 20% formalin following brain removal, typically within 12–24 hours of the patient’s death due to our research protocol. Formalin-fixed paraffin embedded tissue was sectioned at 4–5 µm, mounted on charged slides and dried at 60 °C. Immunostaining was performed on the BenchMark ULTRA© platform (Ventana Medical Systems, Inc., Tucson, AZ) with appropriate positive and negative controls. The majority of slide materials used in this study were created during the course of the various autopsies and archived. Additional tissue blocks added during the course of the study, and older archival materials, were also stained using this immunohistochemistry protocol to ensure comparability across all cases. A limited number of cases (n = 27) had sections stained with anti-phospho-(409/410)-TDP43 antibody (rabbit, 1:1000; Proteintech, 22309-1-AP) for spinal cord and medulla regions-of-interest (ROIs). While phospho-TDP-43 was not used for analysis in this study, as it was not available for the large majority of cases, the correlation of inclusion density between the 2 Proteintech antibodies in this subset of patients was strong (p = 0.005).
Study-specific immunofluorescence studies were also performed. Samples were incubated with primary antibody at 4 °C following deparaffinization and rehydration steps, antigen retrieval procedures using 10 mM citrate buffer, and a blocking step using 2.5% horse serum. After several washes with phosphate buffered saline, secondary antibodies were applied for 1 hour at room temperature, including Alexa Fluor 568 Anti-Rabbit IgG (1:200; Life Technologies, Thermo Fisher Scientific, A11036; Waltham, MA) and Alexa Fluor 488 anti-Mouse IgG (1:200; Life Technologies, Thermo Fisher Scientific, A11001; Waltham, MA). To identify patients with c9ALS, all cases had sections of cerebellum screened for TDP-43-negative, p62-positive inclusions by immunofluorescence (49 cases, using purified mouse anti-p62 Ick ligand diluted to 1:50; BD Biosciences; San Jose, CA) or immunohistochemistry (8 cases, using purified mouse anti-p62 Ick ligand, BD Biosciences, run on a Leica Bond platform, Buffalo Grove, IL). In cases with characteristic p62-immunoreactive lesions, antibodies for poly-GA (C9ORF72/C9RANT, MABN889, EMD Millipore, 1:200, mouse monoclonal; Billerica, MA) and poly-GP (C9ORF72/C9RANT, ABN455, EMD Millipore, 1:1000, rabbit polyclonal; Billerica, MA) were used. As negative controls, 2 p62-immunonegative cases were run concurrently and were negative for poly-GA and poly-GP. In all 15 cases with p62-immunoreactive lesions, the poly-GA and poly-GP antibodies confirmed the presence of dipeptide repeats, thus confirming c9orf72 expansion in those patients.
Qualitative and Quantitative Neuropathological Assessments
Several neuropathologists at our institution (MDC, SZP, ALR, and HT) performed these autopsies and applied a common protocol for submission of research and diagnostic sections. Autopsy reports describing brain and spinal cord findings had been previously issued for each of the patients studied here. Given that the descriptions of TDP-43 pathology in those reports were presented in a narrative, descriptive microscopic format, pathologic inclusions were quantified for this study, blinded in as much as possible to those original reports. TDP-43 pathology was quantified for each patient in 73 ROIs, including within spinal cord levels, medulla, pons, midbrain, cerebellum, amygdala, entorhinal and parahippocampal cortices, hippocampus, basal forebrain, hypothalamus, cingulate cortex, corpus callosum, thalamus, basal ganglia, insula and claustrum, and all cerebral lobes. Only minor variance in sections taken at the time of autopsy and available for examination was present. Nonetheless, for some cases a relatively small ROI was not identified (e.g. hypothalamus). To address this situation, archival wet tissue was retrieved for 38 cases and additional sections were submitted and stained for TDP-43.
The entire ROI available for examination was screened and the focus of greatest TDP-43 pathology, if pathology was present, was identified. If no TDP-43 pathology was identified, this ROI was recorded as negative. Recorded variables per patient included () extent of TDP-43 pathology, recorded as the percentage of all ROIs positive for pathology, regardless of inclusion density; () density of TDP-43 pathology per ROI, recorded as the number of pathologic inclusions in 3 high-power microscopic fields (HPFs) at 400× magnification (0.19625 mm2 per HPF) in the area of greatest pathology; () severity of white matter TDP-43 pathology (glial inclusions, neurites), recorded as the maximum density of TDP-43 inclusions across components of the corticospinal tract, temporal lobe white matter, corpus callosum, or cerebellar white matter, and () limbic TDP-43 pathology, recorded as the average value of TDP-43 inclusion densities across components of basal forebrain, dentate gyrus, hippocampal sectors CA1–CA4, entorhinal and parahippocampal cortices, amygdala, and cingulate cortex. One author (MDC) performed analysis on all cases and salient pathologic features and representative examples of quantitation were reviewed and agreed upon by 2 authors (MDC, SZP). As described elsewhere (), H&E sections and non-TDP-43 pathologies (tau, beta-amyloid, and alpha-synuclein) were also available for review to examine other degenerative disease processes.
Statistical Methods
Prior to any statistical analyses, the ROIs were condensed into 34 final study ROIs. For instance, 4 different ROIs in basal forebrain were condensed into a single, final ROI for analysis per patient (“basal forebrain”), reflecting the maximum value of TDP-43 pathology in the entire region. The resulting final ROIs are shown in Table 1.

Quantile regression (Stata, Version 14, College Station, TX) was employed with and without adjustment of c9orf72 status to determine extent of TDP-43 neuropathology and density of TDP-43 pathology (see above) on independent predictor variables, including duration of disease, c9orf72 status, rate of progression, cognitive status, and site of onset. Model-building strategies were assessed to identify whether multiple variable models could be developed; the results indicated that either univariate or bivariate models adjusted for c9orf72 status were the only reliable models.
Hierarchical cluster analysis was also performed on patients and region-specific variables reflecting TDP-43 expression. Canberra distance function was used for geometric distance, whereas the unweighted pair-group method using arithmetic averages (UPGMA) was used for distance during agglomeration. After cluster analysis, tests for equality of means across cluster groups for continuous variables were determined using one-way ANOVA, and Bartlett’s equality of variance tests were also performed. ANOVA tests exhibiting heteroscedasticity were performed using the W-test. Tests of association between categorical variables and the cluster categories were performed using chi-squared (χ2) tests.
RESULTS
Demographic and Clinical Characteristics
Demographic and clinical characteristics have been summarized in Table 2. Briefly, the 57 patients in this study (hereafter referred to as ALS01 to ALS57) comprised 35 men and 22 women with a mean age of 63.3 years at death (SD = 9.23 years; ranging from 39 to 86 years). The median duration of disease was 1118 days or 3.1 years (ranging from 315 to 4886 days, interquartile range, 1113.3 days).
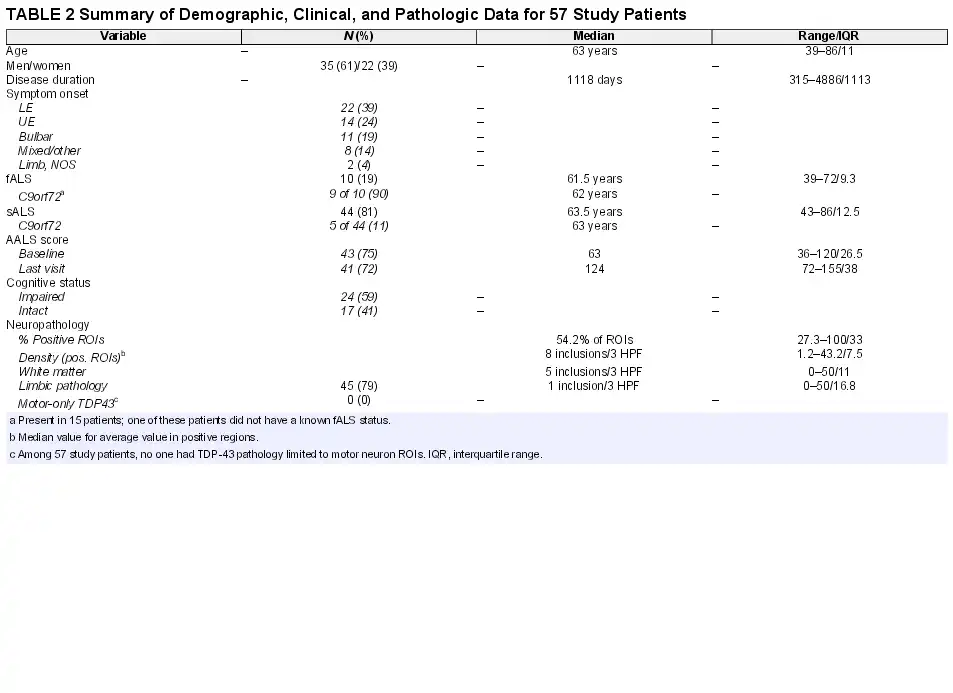
Onset of symptoms was lower extremity in 22 patients (39%), upper extremity in 14 patients (24.5%), bulbar in 11 patients (19%), and in multiple sites in 8 patients (14%). The patients in this last group all had limb symptoms at onset except for one. Two additional patients had a limb onset that was not otherwise specified. Ten patients (17.5%) were designated as fALS, 44 were designated as sALS (77.2%), and 3 patients had no data available regarding family history of ALS. Nine of 10 fALS patients had evidence of c9orf72 expansion by the detection of dipeptide repeats as described above. The genetic basis of the only other fALS patient (ALS45), also having TDP-43 pathology, is not known.
Baseline functional scores at the initial visit were available for 43 patients using the AALS score (), which incorporates scores for 5 domains of bulbar symptoms, respiratory function, muscle strength, lower and upper extremity functions. Similarly, 41 patients had final visit assessments with an AALS score, such that a rate could be calculated as the change in AALS score per month. In the 5 domains of the AALS scale, a score of 30 points represents normal function and a score of 164 points represents maximal dysfunction (). In our autopsy cohort, median AALS baseline (initial visit) score was 63 (ranging from 36 to 120, interquartile range, 26.5) and the median AALS final score was 124 (ranging from 72 to 155, interquartile range, 38). Among patients with documented cognitive status (see above), 24 patients (60%) had mild or greater cognitive impairment and 17 patients were cognitively intact. The large majority of cognitively impaired patients had mild frontal–subcortical dysfunction and not frank dementia/FTD (present in only 3 patients).
Neuropathologic Findings: Topography and Heterogeneity
Several salient points regarding TDP-43 immunoreactive lesions are summarized at the bottom of Table 2. The median value of TDP-43-positive study ROIs was 54.2% (range: 27.3%–100%, interquartile range: 33%) across all patients. The median value of inclusion density (an average of inclusions per 3 HPFs in positive ROIs) was 8 inclusions/3 HPF across all patients (range: 1.2–43.2, interquartile range 7.5). Within white matter ROIs this value was 5 inclusions/3 HPF (range: 0–50, interquartile range: 11) across all patients and within limbic ROIs this value was 1 (range: 0–50, interquartile range 16.8). Every patient studied had glial cytoplasmic inclusions (100%) in at least 1 study ROI (gray or white matter) and 88% of patients (n = 50) had white matter ROI TDP-43 pathology. A strong positive correlation (r = 0.8) was found between inclusion density in ROIs and extent of pathology (% of positive ROIs). The topography of TDP-43 pathology in all patients (n = 57) is depicted as an anatomically based heat map for the 34 study ROIs in Figure 1 (caudal regions are at left, progressing to the right through subcortical structures, mesial temporal lobe, and cortex). Table 3 provides a list of ROIs that were very frequently (76%–100% of patients), commonly (50%–75%), not commonly (26%–49%), and very rarely (<25%) involved by TDP-43 pathology.
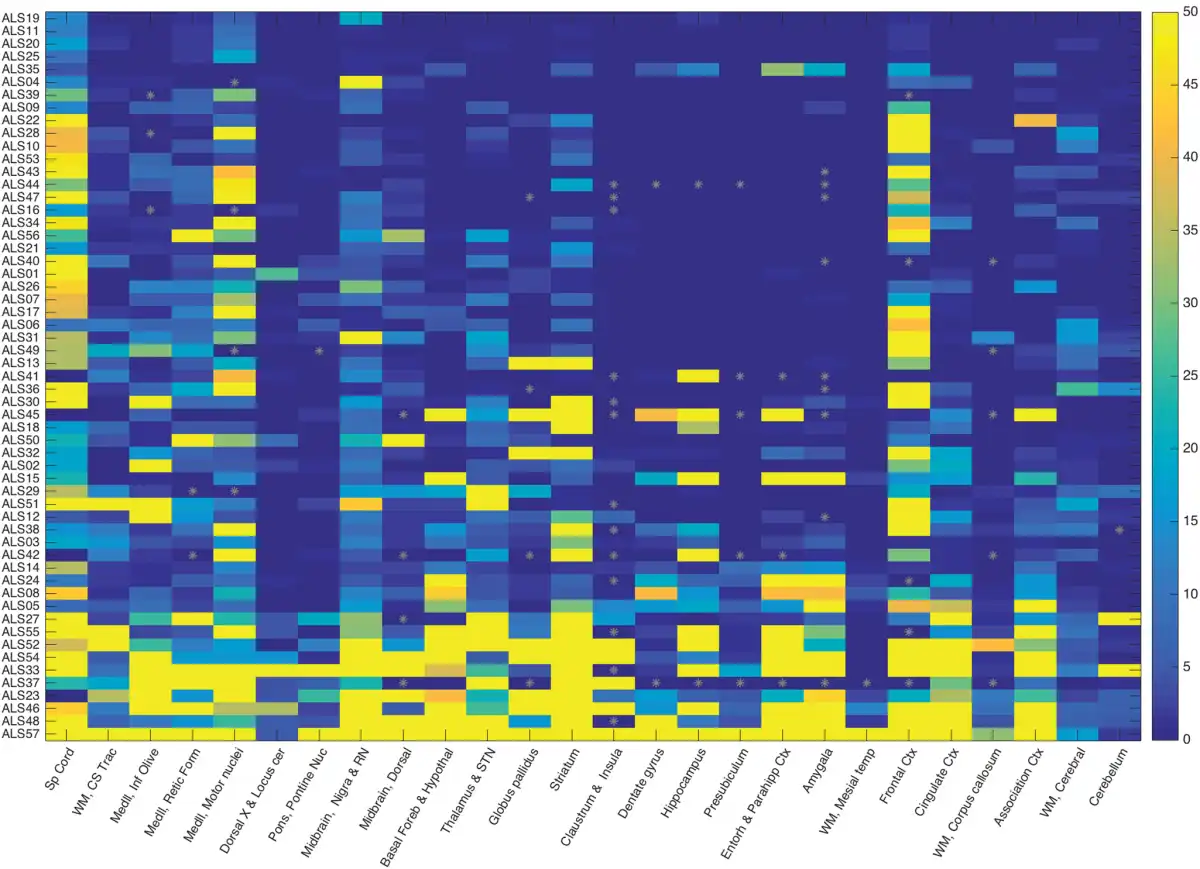
FIGURE 1
Heat map of TDP-43 inclusion density and extent in 57 ALS patients based on caudal (left) to rostral (right) anatomic regions. This heat map shows the extent and density of TDP-43 neuropathology in 57 ALS patients (study ID labels in rows) by ROIs (columns). The ROIs are organized from more caudal regions at left, progressing rightward through brainstem, subcortical, hippocampal/limbic, and cortical regions. The color bar on the right (values 0–50) represents the density of TDP-43 inclusions in 3 microscopic HPFs (see “Materials and Methods”) with the maximum value at 50 (i.e. ≥50 pathologic inclusions). Gray asterisks indicate missing data points. Data points are sorted from patients with the least (top rows) to most extensive (bottom rows) TDP-43 neuropathology across all ROIs. The patients in the top 3 rows (ALS19, ALS11, and ALS20) had durations of 2533, 2922, and 995 days, respectively. The patients in the bottom 3 rows had durations of 2922, 2112, and 791 days, respectively. All 6 were sALS without c9orf72 expansion.

All of the patients studied had motor neuron TDP-43 pathology. However, no one among the 57 study patients studied had TDP-43 pathology limited only to motor region ROIs. For those with the lowest global burden of TDP-43 pathology, the greatest burden was frequently but not always within motor regions. In the 8 study patients with a relatively low TDP-43 burden (≤35% of study ROIs involved), additional non-motor ROIs with TDP-43 pathology commonly included red nucleus, substantia nigra, dorsal midbrain, reticular formation of medulla, corticospinal white matter, and striatum (in particular ventral and lateral putamen), and less commonly, inferior olive, pontine reticular formation, pontine nuclei, basal forebrain, cingulate cortex, nonmotor isocortex, amygdala, dentate gyrus and hippocampal formation, entorhinal and parahippocampal cortex, locus ceruleus, thalamus/subthalamic nucleus, and cerebral white matter. Table 4 provides a list of non-motor ROIs with TDP-43 pathology in patients with a low global TDP-43 burden.

In addition there was morphologic heterogeneity of TDP-43 pathology, particularly outside of motor neuron groups. Figure 2 shows representative images of this pathologic heterogeneity. An additional morphologic finding of interest was the presence of “thread-like” TDP-43-immunoreactive inclusions in some patients with c9orf72 expansion in sections through striatum (Fig. 2d). Likewise, several patients had dense oligodendroglial inclusion-rich pathology on p62 (not shown) and TDP-43, reminiscent of glial cytoplasmic inclusions in multiple system atrophy (Fig. 2b, c). As described above, all 57 patients (100%) examined had at least minimal glial inclusion pathology—the examples in Figures 2a–c depict more severe cases of glial cytoplasmic inclusion-rich pathology.
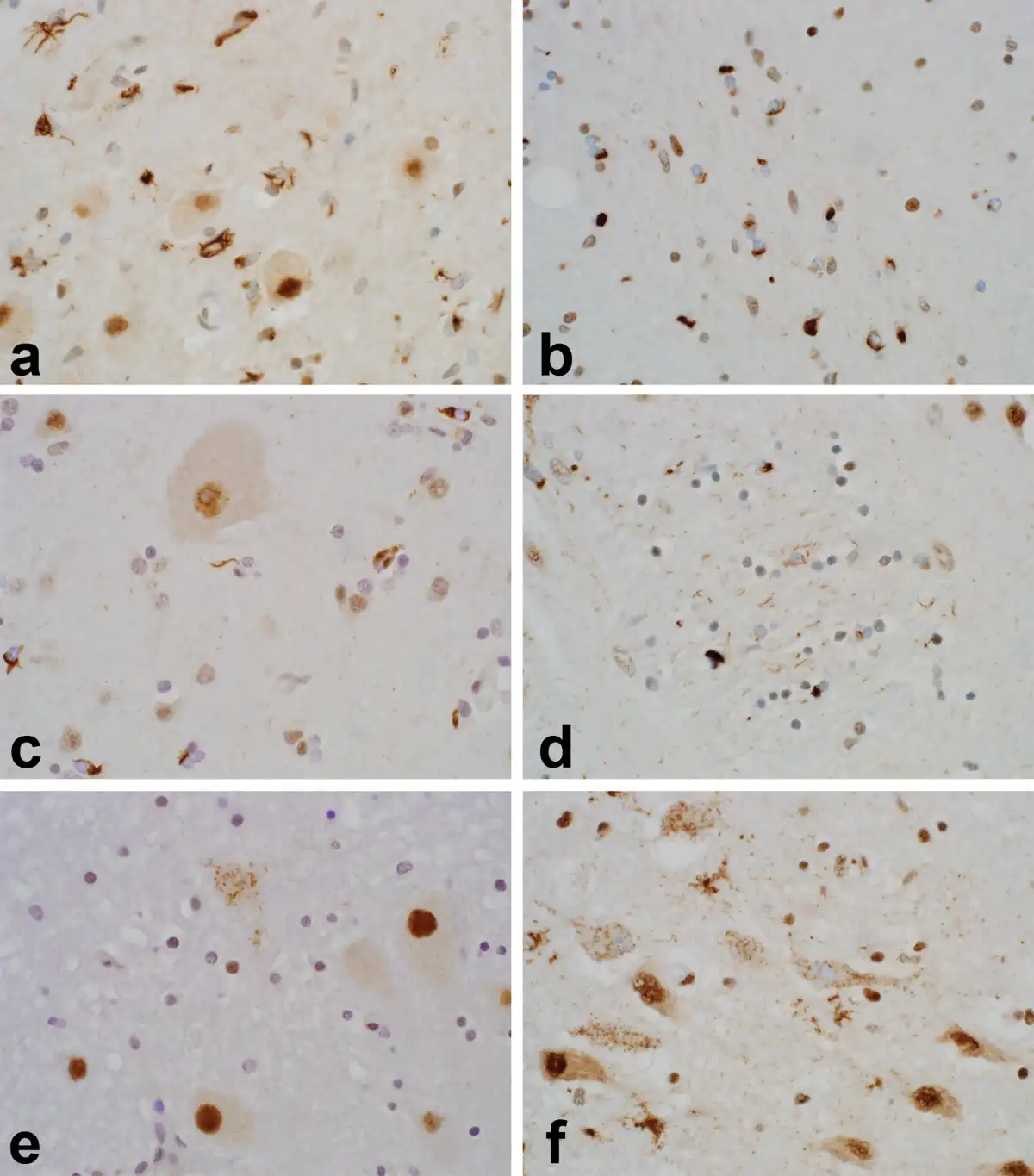
FIGURE 2
Heterogeneity of TDP-43 inclusion pathology in ALS patients. The images emphasize the morphologic and anatomic heterogeneity of TDP-43 pathology in ALS. Several patients had pleomorphic glial inclusions, as shown in inferior olive ((A); note the larger olivary neurons free of inclusion pathology). Other patients had numerous oligodendroglial cytoplasmic inclusions, as shown here in internal capsule (B) and motor cortex ((C); note the larger Betz cell free of inclusion pathology). Patients with c9ALS had more severe striatal TDP-43 pathology and this included several patients with “thread-like” immunoreactive neurites within pencil fibers (D). Pleomorphic neuronal inclusions were also seen in regions not often implicated in ALS. Examples shown here include dense, filamentous inclusions in a neuron of the dentate nucleus (B) and granular inclusions in magnocellular neurons of basal forebrain (F). Five different patients are shown here, including ALS02 (A), ALS57 (B, F), ALS48 (C), ALS52 (D), and ALS36 (E). Images photographed at 600×.
No Lewy body pathology was identified in any case on H&E-stained slides or by alpha-synuclein immunostains. According to NIH/NIA (2012) criteria (), AD neuropathology was “Low” or “Not” in the vast majority of patients, “Intermediate” in 2 patients, and “High” in only 1 patient (a 72-year-old woman with c9ALS and cognitive impairment). Twelve patients had cerebral amyloid angiopathy (typically focal, mild and in leptomeningeal vessels). No atypical tau immunoreactive lesions of chronic traumatic encephalopathy were reported; however, this was not systematically investigated and further studies of this pathology in our autopsy cohort are underway according to consensus recommendations and methods (). Three patients had tau pathology diagnosed as argyrophilic grain disease. Three patients had incidentally diagnosed pituitary microadenomas.
Clinicopathologic Associations Determined by Regression Analysis
Regression analyses demonstrated that disease duration and rate of progression (% change in AALS score) were not predictors of TDP-43 neuropathology (extent or density), even when adjusting for c9orf72 status. In contrast, a trend between c9ALS and more extensive TDP-43 pathology was seen (Mann–Whitney Z-score = −1.89, p = 0.059). In addition, patients with c9orf72 expansion had significantly decreased survival (t = 3.3, p = 0.0008 on log-transformed duration data) and more severe striatal TDP-43 pathology (p = 0.0029). There was no significant difference in the extent of TDP-43 pathology based on site of symptom onset (lower or upper extremity vs bulbar vs multifocal groups). Although upper extremity and bulbar groups had higher average values of TDP-43 inclusion density compared with lower extremity onset, this was not significant by Kruskal–Wallis testing (p = 0.097).
A nonsignificant trend was identified between TDP-43 pathology and the presence of cognitive impairment (Mann–Whitney test Z-score = −1.8, p = 0.07). There was not a significant difference between cognitively impaired and intact groups identified with respect to the extent of TDP-43 neuropathology when adjusting for c9orf72 status (p = 0.09). The same analyses were repeated and group differences were not significant using the mean density of TDP-43 inclusions in limbic regions (see “Materials and Methods”) (t = 1.4, p = 0.16).
Cluster Analyses and Clinical Associations
Hierarchical cluster analysis revealed a rich cluster structure of 3 patient groups based on the extent and severity of TDP-43 pathology. These 3 groups are shown in Figure 3 and are referred to as “TDP43-limited” (group 1; n = 33 patients), “TDP43-moderate” (group 2; n = 15), and “TDP43-severe” (group 3; n = 9) groups. Not surprisingly, these groups differed significantly with respect to TDP-43 extent and density (p < 0.0005), white matter pathology (p = 0.05), and limbic TDP-43 pathology (p < 0.0005) with all of these measures being highest in subgroup 3 (the TDP43-severe subgroup; please also see Table 5).
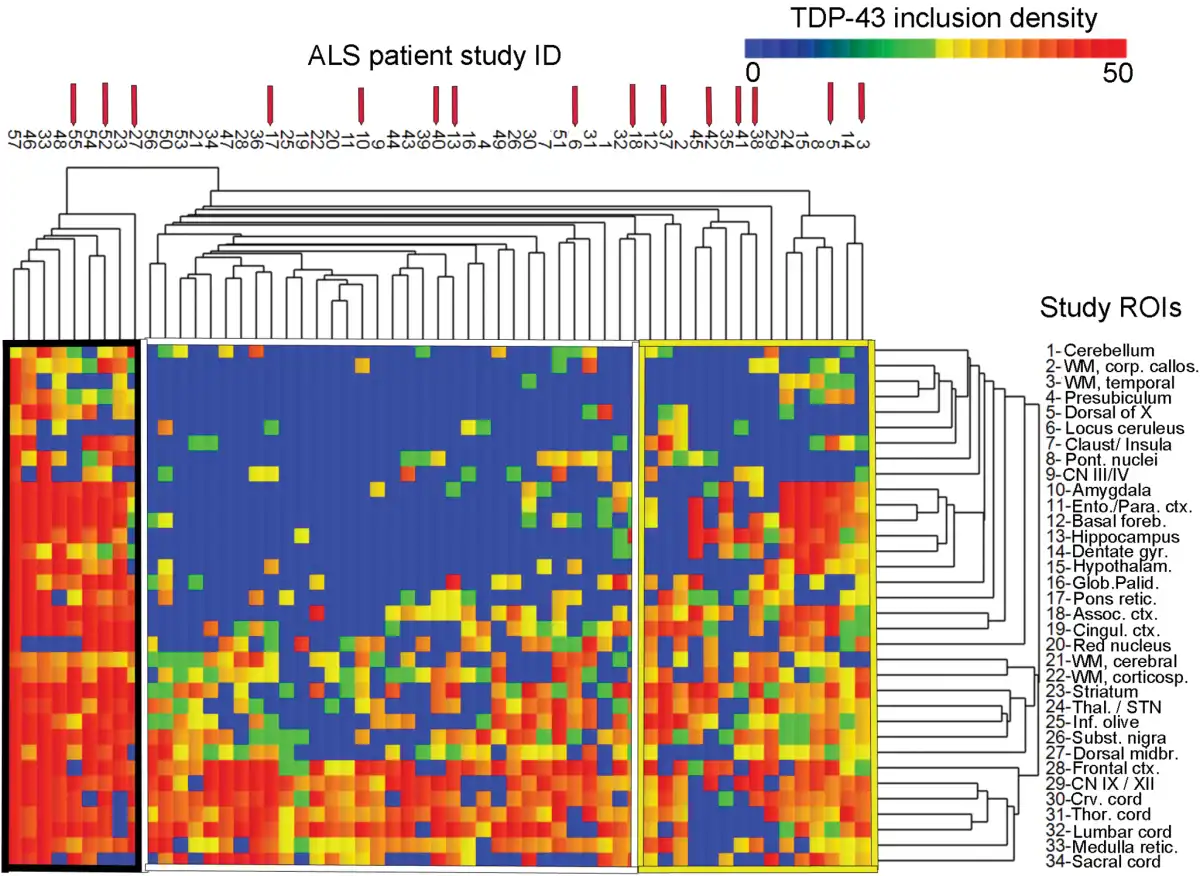
FIGURE 3
Canberra distance-based hierarchical clustering based on TDP-43 inclusion data. Graphical demonstration of “TDP43-limited” (Group 1; middle, white border), “TDP43-moderate” (Group 2; right, yellow border), and “TDP43-severe” (Group 3; left, black border) pathologic subgroups as identified by hierarchical clustering using Canberra distance. The heat map reflects the density of TDP-43 inclusion pathology and a color bar is provided at the top right. Patients are represented with columns, and the study ID of each ALS patient is provided (for instance, the right-most listing to the far right is for patient ALS03). Red arrows above the study ID indicate c9ALS patients (n = 15). Rows indicate each of the 34 study ROIs, which are labeled (Table 1). Table 5 shows the analysis of pathologic and clinical variables using the patients identified in these 3 Groups. A Supplementary Data Table S1 is also available that shows an analysis using only the patients in the 6 right-most columns in Group 2 (TDP43-moderate).
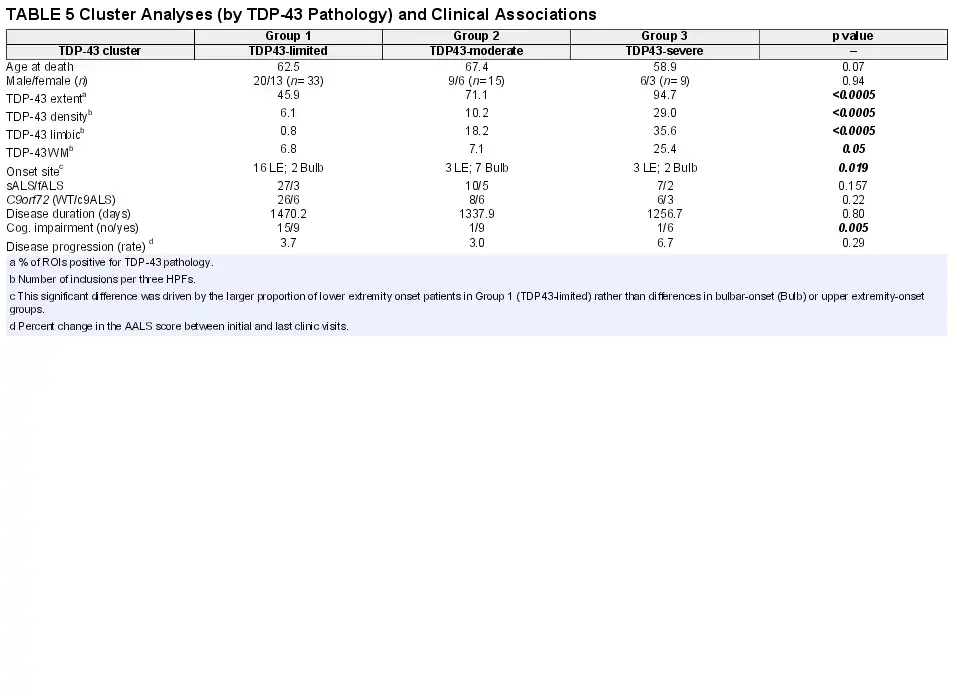
Among clinical measures, these 3 clusters significantly differed with respect to cognitive impairment (p = 0.005) with the highest frequencies of cognitive impairment being in the TDP43-severe and TDP43-moderate subgroups and lowest in the TDP43-limited subgroup. These groups also significantly differed with respect to onset site (p = 0.019) as the TDP43-limited subgroup (group 1) had a high number of lower extremity onset patients, representing 50% of the subgroup and 72% of all lower extremity onset patients. These subgroups did not significantly differ with respect to rate of clinical progression or disease duration. These subgroups, derived from cluster analysis, did not significantly differ with respect to age at death, sex, c9orf72 status, or fALS versus sALS (please also see Table 5). A Supplementary Data Table S1 is also available, which shows the results of a cluster analysis that limits the TDP43-moderate group to the right-most 6 patients in Figure 3.
DISCUSSION
The aim of this study was to assess the clinical correlates of TDP-43 pathology, particularly with respect to the duration of disease, the rate of disease progression, cognitive status, and site of symptom onset. A number of prior studies have reported on the heterogeneous spectrum of TDP-43 pathology in ALS (, ) although fewer studies have examined the specific clinical associations of that pathology ().
We identified several significant associations of TDP-43 pathology by regression and cluster-based analyses, several non-significant trends, and clinical variables that did not significantly associate with TDP-43 pathology. For instance, a non-significant trend toward greater TDP-43 pathology in c9ALS was identified; this group also had significantly shorter disease duration and significantly more severe striatal pathology. Likewise, regression analysis revealed a trend toward cognitive impairment in patients with greater TDP-43 pathologic burden. This finding was subsequently confirmed by cluster-based analyses, which showed more cognitively impaired patients in the “TDP43-moderate” and “TDP43-severe” subgroups and the lowest frequency of cognitive impairment in the “TDP43-limited” subgroup. Hierarchical clustering analysis further revealed a potentially unique pathologic subgroup—a subgroup of “TDP43-limited” patients with lower extremity onset and no cognitive impairment. Disease duration and rate of clinical progression, calculated as the per month change in AALS score, did not significantly associate with the burden of TDP-43 pathology.
Individuals with c9orf72 expansion constitute a unique clinical subgroup of sALS and fALS patients with more rapid cognitive decline and poor survival (, ) (significantly shorter disease duration also being found in our study). In addition to sequencing data, these patients have distinct morphologic hallmarks on pathologic examination of brain tissue that allows them to be identified (, ). In this study, the population of patients with c9orf72 expansion had variably severe TDP-43 neuropathology despite the fact that they had uniformly poor survival. Within the striatal region, c9ALS patients in this study did have a significantly greater burden of TDP-43 pathology (p = 0.0029), including an interesting “thread-like” pattern of white matter inclusions (Fig. 2d). Regarding whole brain TDP-43 and c9ALS, prior studies have identified more extensive TDP-43 pathology in these patients (, ) and by regression analysis in our study a similar trend was identified. However, cluster-based analyses did not identify c9ALS patients as a distinct TDP-43 pathology subgroup and c9ALS patients were spread out among the TDP43-limited, TDP-43-moderate, and TDP43-severe subgroups (indicated by the red arrows in Figure 3). Unexpectedly, c9ALS patients with rapid progression and cognitive impairment but comparatively sparse TDP-43 pathology were identified in this study. Similarly, a recent report () described 2 c9ALS patients with severe cognitive impairment and/or ALS yet minimal TDP-43 pathology. This study and the present findings suggest that clinical impairments and shorter disease durations in patients with c9orf72 expansion may not necessarily be linked to TDP-43 neuropathology directly in all cases (). In these patients an early pathologic event is the accumulation of RNA foci and non-AUG-mediated translation of expansion transcripts () and RNA sequencing studies have suggested these patients have widespread and extensive transcriptional dysregulation (). The process by which the accumulation of RNA foci, dipeptide repeats, and/or transcriptional dysregulation leads to subsequent accumulation of TDP-43 inclusions remains under investigation.
The finding that TDP-43 neuropathology in ALS associates with some though not all clinical aspects of the disease suggests an important role of TDP-43 pathologic burden, but also a role for other pathologies not investigated here. TDP-43 pathology is common to almost all forms of sALS and fALS, with the notable exception of patients with SOD1 mutations. TDP-43 pathology is not exclusive to ALS among human neurodegenerative diseases, co-occurring in many age-related dementing illnesses in particular (). This finding, and the knowledge of the proximal-most pathogenetic event in some ALS patients (e.g. c9ALS), suggests that pathologic deposition of TDP-43 may be a downstream event of the initiating pathology, at least in some cases. This view is counterbalanced by the rare occurrence of fALS cases with TARDBP mutations (, ), the putative role of DNA and RNA-binding proteins in ALS disease pathogenesis (), and the fairly specific occurrence of TDP-43 pathology in motor neuron groups in ALS and FTD/ALS. Nonetheless, both patient and animal studies suggests that cellular dysfunction in ALS is multifactorial, potentially including mitochondrial dysfunction (), activation of pro-apoptotic pathways (), neuronal hyperexcitability () and cytoskeletal instability, dysregulation of glutamate transport (, ), and a neurocytotoxic pro-inflammatory state lead by activated microglia (). The absence of an association between duration of disease and rate of progression and TDP-43 burden may require one or more of these alternate mechanisms of cell injury to be examined (e.g. neuroinflammatory response). For instance, the reduction of regulatory T-lymphocytes peripherally and their associated transcription factor FoxP3 has been shown to lead to more rapid clinical progression and reduced survival (). Additionally, the absence of an association between duration and TDP-43 proteinopathy in this study and in earlier studies (, ) raises the question as to whether some ALS patients without FTD have extra-motor TDP-43 neuropathology at the onset of motor symptoms (and thus the date of clinical diagnosis). This question cannot be answered in a cross-sectional autopsy study and the incidence of ALS is too low in the population to confidently identify presymptomatic pathology. However, a subset of FTLD patients have been shown to have TDP-43 pathology in motor neurons (), which suggests the possibility that non-FTLD patients developing ALS might also have extramotor TDP-43 pathology at the time of clinical presentation. Along similar lines, it has been suggested that ALS is a “multifactorial and multisystem” disorder, at least in some patients, unified by motor neuron involvement (). This concept remains intriguing although difficult to test in cross-sectional autopsy studies such as this one.
Our study also confirms and extends earlier observations that TDP-43 neuropathology in ALS frequently affects regions outside the motor neuron system (, , ). Staging schemes have been proposed to capture the spectrum of TDP-43 neuropathology in ALS (, ) and our findings concur with a prior study that duration of disease has no relationship to the extent of TDP-43 pathology (). As described in those and earlier studies (), we found many of the same brain regions involved (motor nuclei and motor cortex, red nuclei and substantia nigra, striatum and thalamus, extramotor cortex, and mesial temporal lobe), particularly in patients with more extensive whole brain pathology. However, we did not find a stereotypic pattern of pathology across patients, found heterogeneous morphology of inclusions when present, and not a single patient had motor system-limited TDP-43 neuropathology. The identification of TDP43-limited, TDP43-moderate, and TDP43-severe subgroups suggests that further investigation may yield additional clinicopathologic subgroups beyond those identified here. The present study and an earlier study of ours () have also identified TDP-43 pathology in unexpected regions such as basal forebrain (Fig. 2f), cerebellar deep nuclei (Fig. 2e), hypothalamus, claustrum and insula, presubiculum and entorhinal cortex, and rarely in other structures, such as neurons of the pontine nuclei and in occipital cortex (Table 3).
TDP-43 contributes to the pathology of ALS (, ) and has the potential to be a useful biomarker of the disease (). Here, we identified several clinical correlates of whole brain TDP-43 pathology in ALS, as well as 3 pathologic subgroups that may be predictive of onset site and cognitive status, if not all clinical variables studied. Our findings also suggest that TDP-43 pathology in ALS is heterogeneous and that much remains to be explained in terms of the pathologic correlates of disease duration and progression in particular. Refinement and extension of clinico-pathologic subgroups in ALS, or identification of novel clinico-pathologic associations using other approaches or pathologic measures of interest, may yield greater patient-specific insights into this clinically, genetically, and pathologically heterogeneous disease.
ACKNOWLEDGMENTS
We are grateful for the excellent assistance provided by the technical personnel of the autopsy, histology and immunohistochemistry sections in the Department of Pathology and Genomic Medicine (Houston Methodist Hospital), as well as the assistance of Sandra Steptoe and Yuelan Ren in the Research Pathology Core. We are also grateful to Dr. Kathryn Stockbauer, PhD and Dr. Helen Chifotides, PhD, both of the Department of Pathology and Genomic Medicine at Houston Methodist Hospital, for their careful reading of the manuscript and edits. We are grateful to the ALS patients and their family members for the donations that made this work possible.
REFERENCES
- 1. Ravits J. Focality, stochasticity and neuroanatomic propagation in ALS pathogenesis. Exp Neurol2014;262:121–6
- 2. Ravits J, Appel S, Baloh RH, Deciphering amyotrophic lateral sclerosis: what phenotype, neuropathology and genetics are telling us about pathogenesis. Amyotroph Lateral Scler Frontotemporal Degener2013;14(Suppl. 1):5–18
- 3. Moloney EB, de Winter F, Verhaagen J. ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front Neurosci2014;8:252
- 4. Killian JM, Wilfong AA, Burnett L, Decremental motor responses to repetitive nerve stimulation in ALS. Muscle Nerve1994;17:747–54
- 5. Ringholz GM, Appel SH, Bradshaw M, Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology2005;65:586–90
- 6. Wheaton MW, Salamone AR, Mosnik DM, Cognitive impairment in familial ALS. Neurology2007;69:1411–7
- 7. Jawaid A, Murthy SB, Wilson AM, A decrease in body mass index is associated with faster progression of motor symptoms and shorter survival in ALS. Amyotroph Lateral Scler2010;11:542–8
- 8. Baltadzhieva R, Gurevich T, Korczyn AD. Autonomic impairment in amyotrophic lateral sclerosis. Curr Opin Neurol2005;18:487–93
- 9. Gilbert RM, Fahn S, Mitsumoto H, Parkinsonism and motor neuron diseases: twenty-seven patients with diverse overlap syndromes. Mov Disord2010;25:1868–75
- 10. McCluskey LF, Elman LB, Martinez-Lage M, Amyotrophic lateral sclerosis-plus syndrome with TAR DNA-binding protein-43 pathology. Arch Neurol2009;66:121–4
- 11. Mehta P, Kaye W, Bryan L, Prevalence of amyotrophic lateral sclerosis—United States, 2012–2013. MMWR Surveill Summ2016;65:1–12
- 12. Chio A, Logroscino G, Hardiman O, Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler2009;10:310–23
- 13. Irwin DJ, McMillan CT, Brettschneider J, Cognitive decline and reduced survival in C9orf72 expansion frontotemporal degeneration and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry2013;84:163–9
- 14. Zu T, Liu Y, Banez-Coronel M, RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A2013;110:E4968–77
- 15. Peters OM, Ghasemi M, Brown RH Jr. Emerging mechanisms of molecular pathology in ALS. J Clin Invest2015;125:2548
- 16. Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol2010;9:995–1007
- 17. Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature2016;539:197–206
- 18. Israelson A, Arbel N, Da Cruz S, Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron2010;67:575–87
- 19. Guegan C, Vila M, Rosoklija G, Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis. J Neurosci2001;21:6569–76
- 20. Gonatas NK, Stieber A, Mourelatos Z, Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis. Am J Pathol1992;140:731–7
- 21. Hirano A, Donnenfeld H, Sasaki S, Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol1984;43:461–70
- 22. Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med1992;326:1464–8
- 23. Rothstein JD, Van Kammen M, Levey AI, Selective loss of glial glutamate transporter GTL-1 in amyotrophic lateral sclerosis. Ann Neurol1995;38:73–84
- 24. Peters OM, Ghasemi M, Brown RH. Emerging mechanisms of molecular pathology in ALS. J Clin Invest2015;125:1767–79
- 25. Appel SH, Zhao W, Beers DR, The microglial-motoneuron dialogue in ALS. Acta Myol2011;30:4–8
- 26. Henkel JS, Beers DR, Wen S, Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med2013;5:64–79
- 27. Hooten KG, Beers DR, Zhao W, Protective and toxic neuroinflammation in amyotrophic lateral sclerosis. Neurotherapeutics2015;12:364–75
- 28. Mackenzie IR, Bigio EH, Ince PG, Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol2007;61:427–34
- 29. Brettschneider J, Del Tredici K, Toledo JB, Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol2013;74:20–38
- 30. Braak H, Brettschneider J, Ludolph AC, Amyotrophic lateral sclerosis – a model of corticofugal axonal spread. Nat Rev Neurol2013;9:708–14
- 31. Cykowski MD, Takei H, Van Eldik LJ, Hippocampal sclerosis but not normal aging or Alzheimer disease is associated with TDP-43 pathology in the basal forebrain of aged persons. J Neuropathol Exp Neurol2016;75:397–407
- 32. Nelson PT, Trojanowski JQ, Abner EL, “New old pathologies”: AD, PART, and cerebral age-related TDP-43 with sclerosis (CARTS). J Neuropathol Exp Neurol2016;75:482–98
- 33. Josephs KA, Whitwell JL, Weigand SD, TDP-43 is a key player in the clinical features associated with Alzheimer's disease. Acta Neuropathol2014;127:811–24
- 34. Cooper-Knock J, Hewitt C, Highley JR, Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain2012;135:751–64
- 35. Appel V, Stewart SS, Smith G, A rating scale for amyotrophic lateral sclerosis: description and preliminary experience. Ann Neurol1987;22:328–33
- 36. Cykowski MD, Takei H, Schulz PE, TDP-43 pathology in the basal forebrain and hypothalamus of patients with amyotrophic lateral sclerosis. Acta Neuropathol Commun2014;2:171
- 37. Montine TJ, Phelps CH, Beach TG, National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol2012;123:1–11
- 38. McKee AC, Cairns NJ, Dickson DW, The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol2016;131:75–86
- 39. Brettschneider J, Arai K, Del Tredici K, TDP-43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol2014;128:423–37
- 40. Brettschneider J, Van Deerlin VM, Robinson JL, Pattern of ubiquilin pathology in ALS and FTLD indicates presence of C9ORF72 hexanucleotide expansion. Acta Neuropathol2012;123:825–39
- 41. Mackenzie IR, Frick P, Neumann M. The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol2014;127:347–57
- 42. Murray ME, DeJesus-Hernandez M, Rutherford NJ, Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol2011;122:673–90
- 43. Vatsavayai SC, Yoon SJ, Gardner RC, Timing and significance of pathological features in C9orf72 expansion-associated frontotemporal dementia. Brain2016;139:3202–16
- 44. Prudencio M, Belzil VV, Batra R, Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat Neurosci2015;18:1175–82
- 45. Van Deerlin VM, Leverenz JB, Bekris LM, TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol2008;7:409–16
- 46. Rutherford NJ, Zhang YJ, Baker M, Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet2008;4:e1000193
- 47. Shi P, Gal J, Kwinter DM, Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim Biophys Acta2010;1802:45–51
- 48. Wainger BJ, Kiskinis E, Mellin C, Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep2014;7:1–11
- 49. Dickson DW, Josephs KA, Amador-Ortiz C. TDP-43 in differential diagnosis of motor neuron disorders. Acta Neuropathol2007;114:71–9
- 50. Strong MJ. Progress in clinical neurosciences: the evidence for ALS as a multisystems disorder of limited phenotypic expression. Can J Neurol Sci2001;28:283–98
- 51. Geser F, Brandmeir NJ, Kwong LK, Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch Neurol2008;65:636–41
- 52. Fatima M, Tan R, Halliday GM, Spread of pathology in amyotrophic lateral sclerosis: assessment of phosphorylated TDP-43 along axonal pathways. Acta Neuropathol Commun2015;3:47
- 53. Neumann M, Sampathu DM, Kwong LK, Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science2006;314:130–3
- 54. Geser F, Prvulovic D, O'Dwyer L, On the development of markers for pathological TDP-43 in amyotrophic lateral sclerosis with and without dementia. Prog Neurobiol2011;95:649–62