Introduction
The extracellular matrix (ECM) is a dynamic three-dimensional scaffold composed of fibrillar macromolecules that provide structure for tissue cells to reside in. Specific proteolytic enzymes of the ECM mediate its continuous and controlled remodeling, a feature that is of vital importance throughout the life of all multicellular organisms []. The physiology of some major ECM proteins is regulated by metzincin proteases, including the disintegrin metalloproteinases with thrombospondin motifs (ADAMTS) []. This family belongs to the metzincin protease superfamily that has one conserved Met residue in the metalloproteinase domain (MPD) and a catalytic zinc ion in the active site [, ]. The ADAMTS family includes 19 secreted metalloproteinases and seven ADAMTS-like proteins that have no catalytic activity [, ]. They are largely evolutionarily conserved, reflecting their ubiquitous importance in embryogenesis and tissue homeostasis. Unsurprisingly, ADAMTS family members have recently been implicated in the pathophysiology of a growing number of vascular diseases, such as coronary artery disease (CAD) and coagulation disorders []. This review focuses on ADAMTS-4 and its implications in vascular pathology. ADAMTS-4 (also known as aggrecanase-1) is expressed by an array of tissues, most prominently in endocrine organs, lungs, and brain, but also in the cardiovascular system [, ]. Notably, the majority of ADAMTS-4 substrates are principal proteoglycans expressed physiologically in smooth muscle cells (SMCs) of blood vessels and the developing heart. A good example is versican, a large ECM proteoglycan that is targeted by ADAMTS-4, which is an important reason why this metzincin protease was originally implicated in the pathophysiology of vascular diseases with an atherosclerotic background []. Atherosclerosis is a major underlying cause of cardiovascular and cerebrovascular disease and plays a crucial role in aortic aneurysms and dissections; therefore, the necessity to scrutinize the role of ADAMTS-4 in plaque formation and progression is apparent [, , ]. Therefore, this review highlights the current state of knowledge on the specific functions and action mechanisms of ADAMTS-4 in the complex pathophysiology of major vascular diseases.
Molecular Structure and Expression of ADAMTS-4
The architecture of ADAMTS-4 is rather simple in comparison to other ADAMTS family members: it consists of a proteinase domain and ancillary domains that is rather simple in comparison to other ADAMTS family members. This is a basic template that is then, didactically speaking, “upgraded” by protein and glycan structures in other ADAMTS molecules. Changing structure reflects various steps the protein passes as it traverses discrete cellular compartments on its way towards secretion and activation in the extracellular space. The ADAMTS-4 sequence starts with a signal peptide that targets the protein for secretion, followed by a prodomain whose cleavage activates the coming latent MPD (Fig. 1). The activity of MPD is likely strongly influenced by the neighboring disintegrin-like domain. This domain interacts with the substrates of ADAMTS-4 through a rather peculiar active site that can have an open or closed conformation, depending on whether a Ca2+ ion is bound to the domain []. This rather uncommon metzincin feature might serve the function of binding to accessory proteins, substrates, or even to ADAMTS-domains. The structure ends with an ancillary domain that likely also modulates ADAMTS-4 activity through ECM binding, regulation of proteinase activity, and substrate specificity fine-tuning []. This domain consists of a thrombospondin type I motif (TSR), a cysteine-rich domain, and a spacer domain, and it varies in other ADAMTS family members in the number of TSR and other specialized domains.
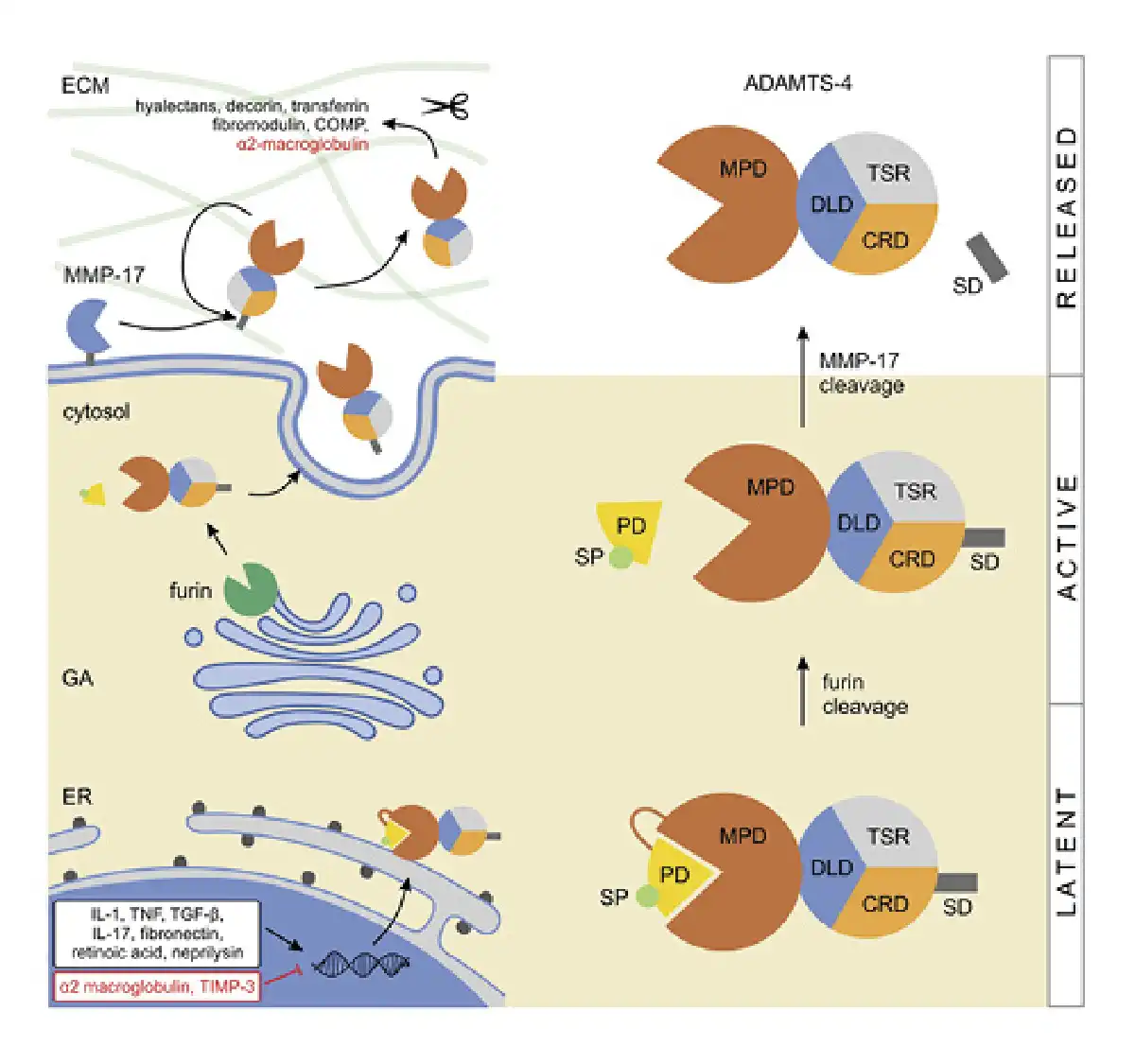
Fig. 1
Expression and activation of ADAMTS-4. Expression of ADAMTS-4 is enhanced by IL-1 and IL-17, TNF, TGF-β, fibronectin, retinoic acid, and neprilysin, and it is inhibited by α2 macroglobulin and TIMP-3 (the enhancers/inhibitors do not localize in the nucleus, that is, are depicted there for practicality). In the trans-GA, the latent ADAMTS-4 is cleaved/activated by furin, releasing its SP and PD. The protein is then secreted and bound to the ECM where the C-terminal SD is cleaved by MMP-17 or autocatalytically, effectively releasing ADAMTS-4 from the ECM. This final proteolytic step enhances the protease activity of ADAMTS-4 which cleaves multiple substrates in the ECM. COMP, cartilage oligomeric matrix protein; CRD, cystein-rich domain; DLD, disintegrin-like domain; ER, endoplasmatic reticulum; SP, single peptide; PD, prodomain; TNF, tumor necrosis factor; SD, spacer domain; IL, interleukins; GA, Golgi apparatus; ECM, extracellular matrix.
Although the expression regulation of ADAMTS-4 is not fully understood, it is tightly controlled on multiple levels, including transcription, translation and through physiologic enhancers or inhibitors. Several enhancers like interleukin 1 (IL-1), tumor necrosis factor, transforming growth factor beta (TGF-β), IL-17, fibronectin, retinoic acid, and neprilysin have been identified. Conversely, alpha-2-macroglobulin and tissue inhibitor of metalloproteinases-3 (TIMP-3) are known to be its physiologic inhibitors [, ]. Additionally, TIMP-3 inhibition appears to be dependent upon ADAMTS-4 TSR and SR interaction with its aggrecan glycosaminoglycan chains [, ].
The path toward synthesizing a fully functional ADAMTS-4 molecule starts by intracellular cleavage of the prodomain, after which the protein is transported to the ECM and further activated by discrete proteases. In contrast to other ADAMTS family members which are processed by furin in the extracellular space (e.g., pro-ADAMTS-5 and -9), ADAMTS-4 is initially processed within the cell []. Latent ADAMTS-4 is cleaved in the trans-Golgi network at multiple N-terminal sites by furin proprotein convertase (Fig. 1) []. This cleavage releases the signal peptide and prodomain, yielding three forms (68, 53, and 40 kDa) that are secreted into the extracellular space []. Once outside the cell, ADAMTS-4 binds to the ECM, where the processing continues at the C-terminal part, mediated by matrix metalloproteinase (MMP)-17 (MT4-MMP) or autocatalytically [-] (Fig. 1). The cleavage of the C-terminal spacer domain is a physiologic event that seems to release ADAMTS-4 from the ECM. It also activates the proteolytic function and likely increases its substrate range, thus regulating the processing of ECM proteoglycans []. Once the protein gets trafficked to the ECM, its biologic functions (as with other aggrecanases) are dependent upon the interplay between the specificity of its exosites that bind potential substrates, and the high specificity of the actual protease cleavage site. One of the main targets of ADAMTS-4 is the proteoglycan aggrecan (accordingly its initial label was aggrecanase-1); however, its family member, ADAMTS-5, was shown to be 20 times more active in degrading aggrecan []. Additionally, ADAMTS-4 cleaves other members of the hyalectan family including versican 1, -2, and brevican [, ], hence the protein was also named as “hyalectanase” []. Other substrates of ADAMTS-4 include a known MMP inhibitor alpha-2-macroglobulin and ECM modulators like decorin and fibromodulin and also cartilage oligomeric matrix protein and transferrin [, , ]. Obika et al. [] showed that ADAMTS-4 might also play a role in the degradation of biglycan, an ECM proteoglycan.
The Role of ADAMTS-4 in Atherosclerosis and Resulting Vascular Diseases
Atherosclerosis is the result of fatty and/or fibrous material accumulation in the inner wall of arteries []. Plaques mostly consist of lipids, cholesterol, inflammatory cells, and apoptosis debris; they reduce blood flow and increase the risk of blood clot formation and embolization. A hallmark of progressive lesion formation is inflammation that favors cell adhesion to the vascular endothelium through specific glycoproteins. The lesions initiate on damaged endothelial cells which raise their expression of adhesive molecules, decrease secretion of nitric oxide, and other compounds. The net result is enhanced adhesion of pro-inflammatory low-density lipoproteins (LDLs), monocytes/macrophages, and thrombocytes on vessel walls (Fig. 2). The LDL is oxidized which in turn drives macrophage phagocytosis and pro-inflammatory cytokine secretion, and over time lipid-laden macrophages transform to foamy cells that accumulate to clumps (Fig. 2). Inflammatory cytokines secreted by the immune cells foster the migration of SMCs which contribute to LDL uptake and migrate to the forming plaque. Chronic inflammation triggers fibrosis which leads to the accumulation of dense, irregular connective tissue that hardens blood vessels [, , , , -].
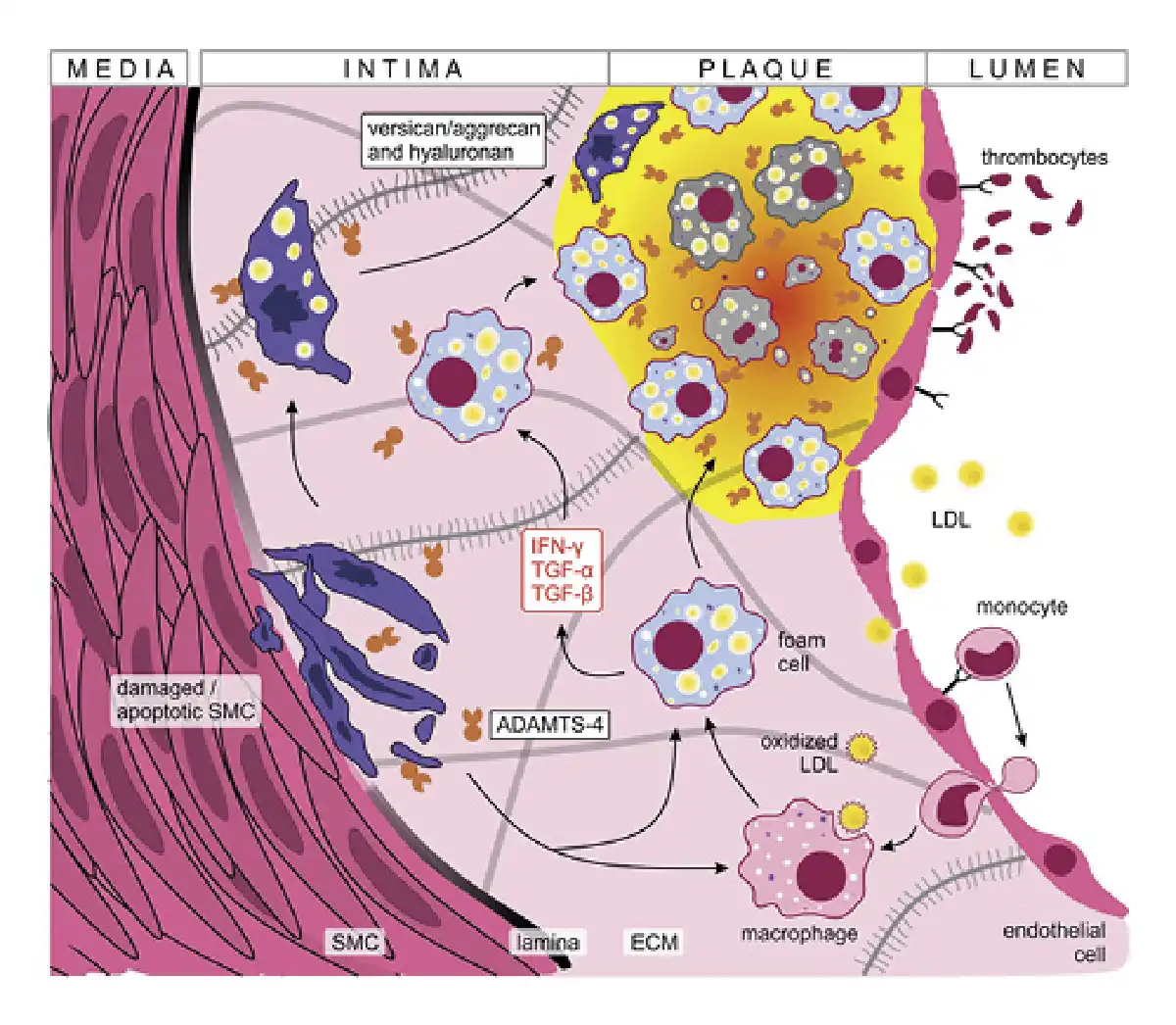
Fig. 2
ADAMTS-4 in atherosclerotic plaque formation. Damaged endothelial cells express adhesion molecules promoting LDL uptake, monocyte extravasation and thrombocyte adhesion. The pro-inflammatory environment (supported in part by macrophage activation) favors the transformation of lipid-laden macrophages to foamy cells that accumulate to clumps forming the atherosclerotic plaque. SMCs migrate under the pro-inflammatory conditions and contribute to oxidized LDL uptake, plaque formation, and ECM thickening. Damaged and apoptotic SMCs overexpress versican and aggrecan which are cleaved by ADAMTS-4, further promoting vessel atrophy. Finally, chronic inflammation triggers fibrosis leading to the accumulation of connective tissue that hardens blood vessels. IFN-γ, interferon γ; ECM, extracellular matrix.
ADAMTS family members are recognized to play significant roles in the process of plaque formation [, ]. Their importance lies partly in the control of expression levels of versican, one of the most abundant proteoglycans produced by SMCs []. Versican has numerous roles that promote vascular disease progression through cell proliferation, migration, adhesion, and ECM remodeling []. In stress conditions, like increased blood flow and shear stress, the damaged intimal SMCs overexpress versican and aggrecan molecules which are then cleaved by ADAMTS-4. This in turn promotes SMC apoptosis and worsens blood vessel atrophy, further driving ADAMTS-4 production, which was shown to be synthesized by endothelial cells and localized to endosomes on the cell surface [, , -]. Moreover, the pro-inflammatory microenvironment present in the plaque drives the expression of ADAMTS-4 enzymes by macrophages and macrophage-like cells derived from vascular SMCs [, , , ]. In brief, the pro-inflammatory plaque microenvironment supports a positive feedback loop where versican and aggrecan enhance plaque formation and corrode vessel walls (Fig. 2). Cleavage of these proteoglycans by ADAMTS-4 further destabilizes the atherosclerotic plaques leading to unfavorable patient outcomes [, , , ].
Over time, the ECM degrades which can lead to breaks in the formed atherosclerotic plaques. This process can result in thrombosis with life-threatening complications, such as myocardial infarction or ischemic stroke. ADAMTS-1, -4, -5, and -7 also regulate the long-term stability and/or lipid deposition in the atherosclerotic plaque [, ]. However, it is noteworthy that Lee et al. [] indicated that ADAMTS-1 is more likely to be involved in plaque destabilization than -4 or -5. Furthermore, genome-wide association studies revealed functionally relevant single nucleotide polymorphisms in the ADAMTS-7 locus which were associated with CAD []. However, to the best of our knowledge, no such associations have been suggested for ADAMTS-4 in relation to CAD. Interestingly, in ADAMTS-4 knockout (KO) mice, a decrease in high fat diet-induced atherosclerosis and increased plaque stability was observed []. The impact of ADAMTS-4 on the development and progression of atherosclerosis is clearly present; however, its potential therapeutic use through direct targeting is questionable due to its diverse roles in ECM homeostasis and the seemingly overlapping functions of ADAMTS family members.
Coronary Artery Disease
Atherosclerotic plaques can occur across the arterial system, but the consequences of blood vessel occlusion are striking when lesions form in the coronary arteries. This pathologic process causes CAD, the leading cause of death worldwide, which includes a spectrum of diagnoses from angina pectoris, myocardial infarction, silent myocardial ischemia, to sudden cardiac death []. Several studies have suggested that serum expression levels of ADAMTS-4 could be used for diagnosis and prediction of severity of CAD and to distinguish patients with CAD from patients without CAD, with 76% sensitivity and 69% specificity. Furthermore, serum levels of ADAMTS-4 in patients with atheroma plaques were significantly higher compared to controls and could predict CAD with 76.2% sensitivity and 67.7% specificity [, ]. The severity of the disease was also correlated to serum ADAMTS-4 levels, as patients with increased number of diseased vessels and severe stenosis had higher expression levels []. However, as atherosclerosis is not limited to the coronary milieu, the elevated ADAMTS-4 levels could be a sign of a more generalized vascular disease.
The correlation of serum expression levels of ADAMTS-4 to the risk of developing CAD was also investigated in other patient groups with different underlying diseases. The studied conditions could all be indirectly linked to atherosclerotic changes in blood vessels, suggesting a possible common role for ADAMTS-4 in their pathophysiology. ADAMTS-4 expression levels were, thus, positively correlated with higher cardiovascular risk in patients with idiopathic hypogonadotropic hypogonadism and primary hyperparathyroidism [, ]. Moreover, significantly higher levels of ADAMTS-4 in diabetic than nondiabetic CAD patients was observed, which indicates that ADAMTS-4 might also contribute to diabetes-associated atherosclerosis []. Elevated levels of ADAMTS-4 were also implicated in renal vascular changes and loss of parenchyma in chronic kidney disease, an independent risk factor for all-cause mortality in cardiovascular diseases in the general population []. Taken together these results suggest that the serum expression levels of ADAMTS-4 are elevated in multiple unrelated pathologic conditions, warranting caution in considering this metalloproteinase as a specific disease biomarker.
Cerebrovascular Disease
Ischemic stroke is another major ramification of atherosclerosis – it is the leading cause of long-term disability and the second leading cause of death worldwide []. After vessel occlusion, necrotic brain tissue is infiltrated by macrophages which support inflammation and cause secondary damages to the ischemia-affected and surrounding brain tissues []. Necrotic tissue and leukocytes that are present in the infiltrate support a pro-inflammatory microenvironment. The secreted interferon-γ, TGF-β, and tumor necrosis factor-α increase the level of protein and mRNA expression of ADAMTS-4 on the infiltrated macrophages [, , ]. This is supported by a postmortem study of ischemia-affected areas of 4 people who died of stroke that found elevated ADAMTS-4 levels. This finding was further confirmed in a mouse model of ischemic stroke that revealed elevated ADAMTS-4 levels in the affected areas, which might be the result of “damage control,” since ADAMTS-4 metalloprotease catalyzes the degradation of chondroitin sulfate-rich proteoglycans, which inhibit neuroregeneration []. As previously mentioned, it also alleviates the detrimental effects of versican overexpression that is related to atherosclerotic plaque formation. This hypothesis is also supported by mice ischemic stroke models, which revealed that recombinant human ADAMTS-4, while having no effect on infarction area, greatly reduced leukocyte infiltration of necrotic tissue relative to the untreated mice [].
ADAMTS-4 in Pathogenesis of Vascular Wall Disease
ECM degradation and the ensuing atherosclerosis, among others, are crucial factors in the development of aortic aneurysms – abnormal bulges occurring at weak spots in the aortic wall [, -]. An aneurysm may also increase the chance of tearing of the aorta lining – leading to aortic dissection and, possibly, a highly fatal aortic rupture. Diagnosis of these diseases currently relies on radiological imaging, which makes systematic screening fairly inaccessible for the entire population and presents the need for a noninvasive way of timely diagnosis and prevention [, ]. As ECM degradation is a key event in the pathogenesis of aortic dissection and aneurysm formation, the involvement of metalloproteinases in the pathophysiology is expected []. ECM degradation also leads to the release of morphogens such as VEGF or TGF-β that are implicated in dysregulation of the inflammatory response in blood vessels [].
Vorkapic et al. [] found ADAMTS-4 levels to be decreased in patients with aortic disease; however, these results should be interpreted with caution due to the ubiquitous link hinted between vessel wall damage and ADAMTS-4 expression. Furthermore, several other researchers found ADAMTS-4 expression to be increased in the aortas of patients with aneurysms and chronic dissection in comparison to controls []. To add, a small study found that patients with Stanford type A aortic dissection had significantly higher plasma concentrations of ADAMTS-4 than controls []. As ADAMTS family members have significant structural similarities, there is a strong possibility of overlapping functions, as evidenced by the ADAMTS-4−/− KO mice that suffer no major changes in growth and physiology [].
Although the role of ADAMTS-4 in the pathogenesis of these diseases is far from clear, additional insight is provided from the mentioned animal models. ADAMTS-4−/− KO mice had lower incidence of aortic aneurysm and dissection formation, as well as lower incidence of aortic rupture; importantly, their aortas had reduced degradation of versican and elastic fibers, as well as reduced macrophage infiltration and apoptosis []. Ren et al. [] observed that ADAMTS-4 translocates to the nucleus of SMCs where it cleaves PARP-1, one of the key molecules responsible for DNA repair and cell survival, thus directly implicating it in the SMC apoptotic process.
Second, Wang et al. [] suggested that a central molecule of the canonical TGF-β pathway called mothers against decapentaplegic homolog-4 is connected to an increased risk of thoracic aortic aneurysms and dissections. Low expression levels of a functional mothers against decapentaplegic homolog-4 variant were linked to increased versican cleavage via ADAMTS-4, leading to increased SMC apoptosis and proteoglycan degradation []. Versican fragments formed by ADAMTS-4 accumulate in the human aorta and stimulate vascular SMC migration into the intima, which then contribute foam cell formation by lipoprotein uptake, proliferation, and ECM synthesis which is required for fibrous cap production [, , ].
Finally, Li et al. [] found that miRNA (MiR-126a-5p) reduced ADAMTS-4 expression and limited aneurysm formation in mouse angiotensin II-induced aortic aneurysm models. They further demonstrated that ADAMTS-4-directed neutralizing antibody blockade had protective effects. Moreover, further research uncovered that ADAMTS-4 antibody administration led to a decrease in IL-1 signaling transduction in chondrocytes [-]. When taking these findings into account, it is possible that ADAMTS-4 exerts its effects on the aorta via inflammatory response/immune dysregulation which might lead to indirect collagen I and elastin degradation via ADAMTS-4 immunomodulation []. These processes probably work synergistically in the progression of aortic disease pathogenesis and contribute towards creating a vicious circle involving ECM degradation and immune dysregulation. However, ADAMTS-4 presumably presents a small thread in the greater aneurysmatic/dissecting proteome pattern.
Apart from its involvement in the pathogenesis of the arterial wall, ADAMTS-4 seems to play a role in the diseases of the venous component of the vascular system. An example is chronic venous disease, a highly prevalent condition affecting up to 77% of the population above 70 years of age [-]. A proposed mechanism for chronic venous disease development is an imbalance in the regulation of metzincins and their TIMPs that can either lead to overgrowth or atrophy of vessel walls. In some vessel regions, the disease presents as SMC hypertrophy where ECM accumulates, and in other regions, the venous wall is atrophic due to SMC apoptosis and ECM degradation []. ADAMTS-4 likely plays a role in the balancing of these processes, as it is an important modulator of ECM and SMC physiology. Serra et al. [, ] showed that serum levels of ADAMTS-4 positively correlate with increasing levels of chronic venous insufficiency. Moreover, varicosity development is accompanied by downregulation of both aggrecan and its associated aggrecanases, ADAMTS-1 and -4 []. Additionally, ADAMTS-4 could be indirectly involved in the pathologic changes of vein grafts used for coronary artery bypass surgery. The stenosis (narrowing) of venous grafts is presumed to be, at least to some extent, a consequence of versican hyperproduction coupled to its decreased degradation. As degradation of versican is mediated by ADAMTS-4, its expression regulation could be a factor in intimal hyperplasia leading to unfavorable patient outcomes [, ]. Furthermore, the production of aggrecan is induced on vascular injury, which is important in autologous vein grafts. Aggrecan production in the vessel wall is increased when a graft is transplanted from low-pressure to arterial high-pressure and high-stress environment []. It is known that loss and degradation of aggrecan is an important factor in vein stiffness, and metzincins could play a role in these changes []. However, although alluring, the regulatory roles of ADAMTS-4 in these processes need to be elucidated by further studies in order to utilize these findings.
Conclusion
ADAMTS proteases are a highly conserved family of proteins that are expressed throughout different tissues and play important roles in differentiation and homeostasis. Their dysregulation is thus expectedly implicated in the pathogenesis and progression of numerous diseases. Here, we highlighted the emerging role ADAMTS-4 might play in vascular pathology and appreciated its importance in atherosclerosis and vessel wall abnormalities, as well as in diseases resulting from these two pathophysiologic processes. However, it is important not to oversimplify the role of ADAMTS-4 and not to consider it to be a sole culprit for the progression of these conditions: ADAMTS-4 is likely only a small piece of the complex puzzle that are the pathogeneses of atherosclerosis and vessel wall abnormalities. ADAMTS-4 exerts its effects in concert with a myriad of other molecules, probably in a time and tissue-context dependent matter. Furthermore, ADAMTS-4 is a highly conserved and pleiotropic molecule that exerts a plethora of functions, not limited to vasculature. For these reasons and due to the lack of such evidence, we believe that the possibility of therapeutic potential of ADAMTS-4 inhibition in the context of halting vascular disease progression at this point is somewhat premature, although the presented evidence indicates possible merits of this approach. For the near future, it appears to be more likely, that ADAMTS-4 might find its clinical purpose as a biomarker for vascular disease, perhaps as one of the indicators in a larger cytokine panel. However, further clinical and preclinical research is needed to clearly elucidate the role of this molecule and to expound the findings that were thus far made.
Conflict of Interest Statement
The authors do not have any conflicts of interest to disclose.
Funding Sources
This work was supported by the Scientific Center of Excellence for Reproductive and Regenerative Medicine (project “Reproductive and regenerative medicine – exploration of new platforms and potentials,”) Grant Agreement KK.01.1.1.01.0008 which is funded by the European Union through the European Regional Development Fund.
Author Contributions
L.G., R.N., S.H., G.S., J.B., and L.M. all made substantial contributions to the manuscript or conception and design of the work. L.G. contributed to conceptualization ideas, manuscript writing, and supervision; R.N. contributed to conceptualization ideas, manuscript writing, and design of figures; S.H., G.S., J.B. contributed to conceptualization ideas and manuscript writing; L.M. contributed to manuscript writing and design of figures. All authors read and approved the final manuscript.
References
- 1. Huxley-Jones J, Robertson DL, Boot-Handford RP. On the origins of the extracellular matrix in vertebrates. Matrix Biol. 2007;26:2–11.
- 2. Wayne GJ, Deng S-J, Amour A, Borman S, Matico R, Carter HL, et al. TIMP-3 inhibition of ADAMTS-4 (aggrecanase-1) is modulated by interactions between aggrecan and the C-terminal domain of ADAMTS-4. J Biol Chem. 2007;282:20991–8.
- 3. Wang P, Tortorella M, England K, Malfait A-M, Thomas G, Arner EC, et al. Proprotein convertase furin interacts with and cleaves pro-ADAMTS4 (aggrecanase-1) in the trans-Golgi network. J Biol Chem. 2004;279:15434–40.
- 4. Rivera S, Khrestchatisky M, Kaczmarek L, Rosenberg GA, Jaworski DM. Metzincin proteases and their inhibitors: foes or friends in nervous system physiology?J Neurosci. 2010;30:15337–57.
- 5. Santamaria S, de Groot R. ADAMTS proteases in cardiovascular physiology and disease: ADAMTS, cardiovascular roles. Open Biol. 2020;10:200333.
- 6. Mead TJ, Apte SS. ADAMTS proteins in human disorders. Matrix Biol. 2018;71–72:225–39. http://dx.doi.org/10.1016/j.matbio.2018.06.002.
- 7. Kelwick R, Desanlis I, Wheeler GN, Edwards DR. The ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) family. Genome Biol. 2015;16:113.
- 8. The Human Protein Atlas. ADAMTS4. Protein atlas version 20.1. 2021. Available from: https://www.proteinatlas.org/ENSG00000158859-ADAMTS4 Accessed 2021 Sep 2.
- 9. Salter RC, Ashlin TG, Kwan APL, Ramji DP. ADAMTS proteases: key roles in atherosclerosis?J Mol Med. 2010;88:1203–11.
- 10. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Prim. 2019;5:56.
- 11. Golledge J, Norman PE. Atherosclerosis and abdominal aortic aneurysm: cause, response, or common risk factors?Arterioscler Thromb Vasc Biol. 2010;30(6):1075–7. http://dx.doi.org/10.1161/ATVBAHA.110.206573.
- 12. Mosyak L, Georgiadis K, Shane T, Svenson K, Hebert T, McDonagh T, et al. Crystal structures of the two major aggrecan degrading enzymes, ADAMTS4 and ADAMTS5. Protein Sci. 2008;17:16–21.
- 13. Kelwick R, Desanlis I, Wheeler GN, Edwards DR. The ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) family. Genome Biol. 2015;16:113.
- 14. Porter S, Clark IM, Kevorkian L, Edwards DR. The ADAMTS metalloproteinases. Biochem J. 2005;386:15–27.
- 15. Tortorella MD, Arner EC, Hills R, Gormley J, Fok K, Pegg L, et al. ADAMTS-4 (aggrecanase-1): N-terminal activation mechanisms. Arch Biochem Biophys. 2005;444:34–44.
- 16. Troeberg L, Fushimi K, Scilabra SD, Nakamura H, Dive V, Thøgersen IB, et al. The C-terminal domains of ADAMTS-4 and ADAMTS-5 promote association with N-TIMP-3. Matrix Biol. 2009;28:463–9.
- 17. Miwa HE, Gerken TA, Huynh TD, Duesler LR, Cotter M, Hering TM. Conserved sequence in the aggrecan interglobular domain modulates cleavage by ADAMTS-4 and ADAMTS-5. Biochim Biophys Acta. 2009;1790:161–72.
- 18. Gendron C, Kashiwagi M, Lim NH, Enghild JJ, Thøgersen IB, Hughes C, et al. Proteolytic activities of human ADAMTS-5. J Biol Chem. 2007;282:18294–306.
- 19. Flannery CR, Zeng W, Corcoran C, Collins-Racie LA, Chockalingam PS, Hebert T, et al. Autocatalytic cleavage of ADAMTS-4 (aggrecanase-1) reveals multiple glycosaminoglycan-binding sites. J Biol Chem. 2002;277:42775–80.
- 20. Kashiwagi M, Enghild JJ, Gendron C, Hughes C, Caterson B, Itoh Y, et al. Altered proteolytic activities of ADAMTS-4 expressed by C-terminal processing. J Biol Chem. 2004;279:10109–19.
- 21. Gao G, Plaas A, Thompson VP, Jin S, Zuo F, Sandy JD. ADAMTS4 (aggrecanase-1) activation on the cell surface involves C-terminal cleavage by glycosylphosphatidyl inositol-anchored membrane type 4-matrix metalloproteinase and binding of the activated proteinase to chondroitin sulfate and heparan sulfate on Sy. J Biol Chem. 2004;279:10042–51.
- 22. Gao G, Westling J, Thompson VP, Howell TD, Gottschall PE, Sandy JD. Activation of the proteolytic activity of ADAMTS4 (aggrecanase-1) by C-terminal truncation. J Biol Chem. 2002;277:11034–41.
- 23. Fushimi K, Troeberg L, Nakamura H, Lim NH, Nagase H. Functional differences of the catalytic and non-catalytic domains in human ADAMTS-4 and ADAMTS-5 in aggrecanolytic activity. J Biol Chem. 2008;283:6706–16.
- 24. Santamaria S, Yamamoto K, Teraz-Orosz A, Koch C, Apte SS, de Groot R, et al. Exosites in hypervariable loops of ADAMTS spacer domains control substrate recognition and proteolysis. Sci Rep. 2019;9:10914.
- 25. Tortorella MD, Arner EC, Hills R, Easton A, Korte-Sarfaty J, Fok K, et al. α2-Macroglobulin is a novel substrate for ADAMTS-4 and ADAMTS-5 and represents an endogenous inhibitor of these enzymes. J Biol Chem. 2004;279:17554–61.
- 26. Acharya C, Yik JHN, Kishore A, Van Dinh V, Di Cesare PE, Haudenschild DR. Cartilage oligomeric matrix protein and its binding partners in the cartilage extracellular matrix: interaction, regulation and role in chondrogenesis. Matrix Biol. 2014;37:102–11.
- 27. Obika M, Vernon RB, Gooden MD, Braun KR, Chan CK, Wight TN. ADAMTS-4 and biglycan are expressed at high levels and co-localize to podosomes during endothelial cell tubulogenesis in vitro. J Histochem Cytochem. 2014;62:34–49.
- 28. Rienks M, Barallobre-Barreiro J, Mayr M. The emerging role of the ADAMTS family in vascular diseases. Circ Res. 2018;123:1279–81. http://dx.doi.org/10.1161/CIRCRESAHA.118.313737.
- 29. Merrilees MJ, Beaumont B, Scott LJ. Comparison of deposits of versican, biglycan and decorin in saphenous vein and internal thoracic, radial and coronary arteries: correlation to patency. Coron Artery Dis. 2001;12:7–16. http://dx.doi.org/10.1097/00019501-200102000-00002.
- 30. Ashlin TG, Kwan AP, Ramji DP. Regulation of ADAMTS-1, -4 and -5 expression in human macrophages: differential regulation by key cytokines implicated in atherosclerosis and novel synergism between TL1A and IL-17. Cytokine. 2013;64:234–42. http://dx.doi.org/10.1016/j.cyto.2013.06.315.
- 31. Uluçay S, Çam FS, Batır MB, Sütçü R, Bayturan Ö, Demircan K. Novel association between TGFb1 and ADAMTS4 in coronary artery disease: a new potential mechanism in the progression of atherosclerosis and diabetes. Anatol J Cardiol. 2015;15:823–9.
- 32. Yang SN, Osman N, Burch ML, Little P. Factors affecting proteoglycan synthesis and structure that modify the interaction with lipoproteins. Clin Lipidol. 2009;4:479–92.
- 33. Wight TN, Kinsella MG, Evanko SP, Potter-Perigo S, Merrilees MJ. Versican and the regulation of cell phenotype in disease. Biochim Biophys Acta. 2014;1840:2441–51.
- 34. Kenagy RD, Min S-K, Clowes AW, Sandy JD. Cell death-associated ADAMTS4 and versican degradation in vascular tissue. J Histochem Cytochem. 2009;57:889–97.
- 35. Kim S-M, Huh J-W, Kim E-Y, Shin M-K, Park J-E, Kim S-W, et al. Endothelial dysfunction induces atherosclerosis: increased aggrecan expression promotes apoptosis in vascular smooth muscle cells. BMB Rep. 2019;52:145–50.
- 36. Koch CD, Lee CM, Apte SS. Aggrecan in cardiovascular development and disease. J Histochem Cytochem. 2020;68:777–95. http://dx.doi.org/10.1369/0022155420952902.
- 37. Karakose M, Caliskan M, Arslan MS, Demirci T, Karakose S, Tutal E, et al. Association of ADAMTS4 and ADAMTS9 levels with cardiovascular risk in patients with primary hyperparathyroidism. Endocr Res. 2018;43:15–20.
- 38. Sandy JD, Westling J, Kenagy RD, Iruela-Arispe ML, Verscharen C, Rodriguez-Mazaneque JC, et al. Versican V1 proteolysis in human aorta in vivo occurs at the Glu441-Ala442 bond, a site that is cleaved by recombinant ADAMTS-1 and ADAMTS-4. J Biol Chem. 2001;276:13372–8.
- 39. Lee CW, Hwang I, Park CS, Lee H, Park DW, Kang SJ. Comparison of ADAMTS-1, -4 and -5 expression in culprit plaques between acute myocardial infarction and stable angina. J Clin Pathol. 2011;64:399–404.
- 40. Kumar S, Chen M, Li Y, Wong FHS, Thiam CW, Hossain MZ, et al. Loss of ADAMTS4 reduces high fat diet-induced atherosclerosis and enhances plaque stability in ApoE−/− mice. Sci Rep. 2016;6:31130.
- 41. Hanson MA, Fareed MT, Argenio SL, Agunwamba AO, Hanson TR. Coronary artery disease. Prim Care. 2013;40:1–16.
- 42. Chen L, Yang L, Zha Y, Cui L. Association of serum a disintegrin and metalloproteinase with thrombospodin motif 4 levels with the presence and severity of coronary artery disease. Coron Artery Dis. 2011;22:570–6.
- 43. Ozler S, Isci Bostanci E, Oztas E, Kuru Pekcan M, Gumus Guler B, Yilmaz N. The role of ADAMTS4 and ADAMTS9 in cardiovascular disease in premature ovarian insufficiency and idiopathic hypogonadotropic hypogonadism. J Endocrinol Invest. 2018;41:1477–83.
- 44. Weiner DE, Tighiouart H, Amin MG, Stark PC, MacLeod B, Griffith JL, et al. Chronic kidney disease as a risk factor for cardiovascular disease and all-cause mortality: a pooled analysis of community-based studies. J Am Soc Nephrol. 2004;15(5):1307–15. http://dx.doi.org/10.1097/01.asn.0000123691.46138.e2.
- 45. Donkor ES. Stroke in the 21st century: a snapshot of the burden, epidemiology, and quality of life. Stroke Res Treat. 2018;2018:3238165. http://dx.doi.org/10.1155/2018/3238165.
- 46. Kuriakose D, Xiao Z. Pathophysiology and treatment of stroke: present status and future perspectives. Int J Mol Sci. 2020;21:7609. http://dx.doi.org/10.3390/ijms21207609.
- 47. Ren P, Zhang L, Xu G, Palmero LC, Albini PT, Coselli JS, et al. ADAMTS-1 and ADAMTS-4 levels are elevated in thoracic aortic aneurysms and dissections. Ann Thorac Surg. 2013;95:570–7.
- 48. Wågsäter D, Björk H, Zhu C, Björkegren J, Valen G, Hamsten A, et al. ADAMTS-4 and -8 are inflammatory regulated enzymes expressed in macrophage-rich areas of human atherosclerotic plaques. Atherosclerosis. 2008;196:514–22.
- 49. Lemarchant S, Wojciechowski S, Vivien D, Koistinaho J. ADAMTS-4 in central nervous system pathologies. J Neurosci Res. 2017;95:1703–11.
- 50. Lemarchant S, Dunghana H, Pomeshchik Y, Leinonen H, Kolosowska N, Korhonen P, et al. Anti-inflammatory effects of ADAMTS-4 in a mouse model of ischemic stroke. Glia. 2016;64:1492–507.
- 51. Golledge J, Tsao PS, Dalman RL, Norman PE. Circulating markers of abdominal aortic aneurysm presence and progression. Circulation. 2008;118:2382–92.
- 52. Hellenthal FAMVI, Buurman WA, Wodzig WKWH, Schurink GWH. Biomarkers of AAA progression. Part 1: extracellular matrix degeneration. Nat Rev Cardiol. 2009;6:464–74.
- 53. Jana S, Hu M, Shen M, Kassiri Z. Extracellula’r matrix, regional heterogeneity of the aorta, and aortic aneurysm. Exp Mol Med. 2019;51:1–15.
- 54. Sweeting MJ, Masconi KL, Jones E, Ulug P, Glover MJ, Michaels JA, et al. Analysis of clinical benefit, harms, and cost-effectiveness of screening women for abdominal aortic aneurysm. Lancet. 2018;392:487–95.
- 55. Ying AJ, Affan ET. Abdominal aortic aneurysm screening: a systematic review and meta-analysis of efficacy and cost. Ann Vasc Surg. 2019;54:298–303.e3. http://dx.doi.org/10.1016/j.avsg.2018.05.044.
- 56. Zhang X, Shen YH, Lemaire SA. Thoracic aortic dissection: are matrix metalloproteinases involved?Vascular. 2010;17:147–57.
- 57. Pearce WH, Shively VP. Abdominal aortic aneurysm as a complex multifactorial disease: interactions of polymorphisms of inflammatory genes, features of autoimmunity, and current status of MMPs. Ann N Y Acad Sci. 2006;1085:117–32. http://dx.doi.org/10.1196/annals.1383.025.
- 58. Vorkapic E, Folkesson M, Magnell K, Bohlooly-Y M, Länne T, Wågsäter D. ADAMTS-1 in abdominal aortic aneurysm. PLoS One. 2017;12:e0178729.
- 59. Li K, Wang ZW, Hu Z, Ren Z, Hu X, Li L, et al. Assessing serum levels of ADAMTS1 and ADAMTS4 as new biomarkers for patients with type A acute aortic dissection. Med Sci Monit. 2017;23:3913–22.
- 60. Glasson SS, Askew R, Sheppard B, Carito BA, Blanchet T, Ma H-L, et al. Characterization of and osteoarthritis susceptibility in ADAMTS-4-knockout mice. Arthritis Rheum. 2004;50:2547–58.
- 61. Ren P, Hughes M, Krishnamoorthy S, Zou S, Zhang L, Wu D, et al. Critical role of ADAMTS-4 in the development of sporadic aortic aneurysm and dissection in mice. Sci Rep. 2017;7:12351.
- 62. Wang Y, Huang H-Y, Bian G-L, Yu Y-S, Ye W-X, Hua F, et al. A functional variant of SMAD4 enhances thoracic aortic aneurysm and dissection risk through promoting smooth muscle cell apoptosis and proteoglycan degradation. EBioMedicine. 2017;21:197–205.
- 63. Newby AC, Zaltsman AB. Fibrous cap formation or destruction: the critical importance of vascular smooth muscle cell proliferation, migration and matrix formation. Cardiovasc Res. 1999;41:345–60. http://dx.doi.org/10.1016/s0008-6363(98)00286-7.
- 64. Li L, Ma W, Pan S, Li Y, Wang H, Wang B, et al. MiR-126a-5p limits the formation of abdominal aortic aneurysm in mice and decreases ADAMTS-4 expression. J Cell Mol Med. 2020;24:7896–906.
- 65. Majumdar MK, Chockalingam PS, Bhat RA, Sheldon R, Keohan C, Blanchet T, et al. Immortalized mouse articular cartilage cell lines retain chondrocyte phenotype and respond to both anabolic factor BMP-2 and pro-inflammatory factor IL-1. J Cell Physiol. 2008;215:68–76.
- 66. Shiraishi A, Mochizuki S, Miyakoshi A, Kojoh K, Okada Y. Development of human neutralizing antibody to ADAMTS4 (aggrecanase-1) and ADAMTS5 (aggrecanase-2). Biochem Biophys Res Commun. 2016;469:62–9.
- 67. Tortorella MD, Liu R-Q, Burn T, Newton RC, Arner E. Characterization of human aggrecanase 2 (ADAM-TS5): substrate specificity studies and comparison with aggrecanase 1 (ADAM-TS4). Matrix Biol. 2002;21:499–511.
- 68. Serra R, Gallelli L, Butrico L, Buffone G, Caliò FG, De Caridi G, et al. From varices to venous ulceration: the story of chronic venous disease described by metalloproteinases. Int Wound J. 2017;14:233–40.
- 69. Serra R, Andreucci M, De Caridi G, Massara M, Mastroroberto P, de Franciscis S. Functional chronic venous disease: a systematic review. Phlebology. 2017;32:588–92.
- 70. Pugsley MK, Tabrizchi R. The vascular system. An overview of structure and function. J Pharmacol Toxicol Methods. 2000;44:333–40. http://dx.doi.org/10.1016/s1056-8719(00)00125-8.
- 71. Chen Y, Peng W, Raffetto JD, Khalil RA. Matrix metalloproteinases in remodeling of lower extremity veins and chronic venous disease. Prog Mol Biol Transl Sci. 2017;147:267–99.
- 72. Barallobre-Barreiro J, Oklu R, Lynch M, Fava M, Baig F, Yin X, et al. Extracellular matrix remodelling in response to venous hypertension: proteomics of human varicose veins. Cardiovasc Res. 2016;110:419–30.
- 73. Kenagy RD, Kikuchi S, Evanko SP, Ruiter MS, Piola M, Longchamp A, et al. Versican is differentially regulated in the adventitial and medial layers of human vein grafts. PLoS One. 2018;13:e0204045.
- 74. Yasmin, Maskari RA, McEniery CM, Cleary SE, Li Y, Siew K, et al. The matrix proteins aggrecan and fibulin-1 play a key role in determining aortic stiffness. Sci Rep. 2018;8:8550.