Introduction
Pulmonary hypertension is a severe disease characterized by vasoconstriction and remodeling of pulmonary arteries, leading to increased pulmonary vascular resistance (PVR) and increased afterload on the right ventricle. This can result in right ventricular hypertrophy and heart failure that is closely related to a poor prognosis [, ].
Transglutaminases are enzymes that have calcium-dependent transamidase activity and consists of a family of several members including TG1-TG7 (genes tgm1-tgm7) and the blood coagulation factor XIIIa []. Transglutaminase 2 (TG2) also called tissue transglutaminase catalyzes cross-linking of matrix proteins by inducing a N-[Latin Small Letter Open E](γ-glutamyl)lysine bond, and recent studies suggest that TG2 is involved in serotonylation of proteins, such as fibronectin and Rho kinase, which are thought to play a role in pulmonary hypertension [, ]. TG2 in the lung parenchyma also plays a role in lung inflammation and fibrosis in bleomycin-treated mice [, ]. Animal studies have suggested that TG2 plays a role in remodeling of systemic resistance arteries in systemic hypertension [-] contributing to large artery stiffness [], and it is closely related to development of left ventricular hypertrophy [-]. TG2 mRNA is increased in the right ventricle of chronic hypoxic rats and rats with pulmonary artery banding [, ]. Moreover, TG2 is involved in endothelin-induced hypertrophy of neonatal cardiomyocytes [] and hypoxia-induced smooth muscle proliferation in vitro []. Recent studies have also suggested that TG2 activity is increased in the lungs of mice and rats with, respectively, hypoxia- and monocrotaline-induced pulmonary hypertension [, ]. Therefore, several studies suggest that TG2 plays a role in pulmonary hypertension, but it is unclear whether inhibition of TG2 will prevent the development of pulmonary hypertension.
The small organic disulphide cystamine (NH2–(CH2)2–S–S–(CH2)2–NH2) has been suggested to inhibit TG2 competitively by binding to the active site and acting as an alternative substrate [], although more recent research suggest an irreversible mechanism of action via oxidation of cysteine residues in TG2 []. Inhibition of caspase-3 activity and elevation of antioxidants are also thought to contribute to the pharmacodynamics effects of cystamine []. Remodeling of systemic small arteries depends on TG2 [] and administration of cystamine has been shown to inhibit eutrophic inward remodeling in phenylephrine-induced hypertensive rats [] and to reduce blood pressure in spontaneously hypertensive rats []. Thus, it is suggested that inhibition of TG2 prevents the formation of cross-links in the extracellular matrix and hence inhibit vascular and cardiac remodeling.
Although it is unclear whether TG2 is a cause or consequence of pulmonary hypertension, our hypothesis was that TG2 is upregulated in the lung and the right ventricle and involved in the development of right ventricular hypertrophy during chronic hypoxia, and that treatment with cystamine would inhibit the development of pulmonary arterial muscularization, fibrosis, pulmonary hypertension, and right ventricular hypertrophy. Furthermore, we expected that the effect of cystamine on right ventricular hypertrophy would be additive in combination with the pulmonary vasodilator sildenafil. First, we investigated the effect of cystamine on the transamidase activity of lung and liver homogenates from normoxic rats as well as intact lungs from normoxic and hypoxic mice. Second, we measured the effect of cystamine on right ventricular systolic pressure (RVSP), lung fibrosis, remodeling of pulmonary arteries, and TG2 expression in normoxic and hypoxic rats. The phosphodiesterase type 5 inhibitor, sildenafil, is first-line treatment for pulmonary hypertension [], and was therefore included as a positive control treatment in the present study.
Material and Methods
Effect of Cystamine on Transamidase Activity in vitro and ex vivo
To measure transamidase activity in vitro, the Specific Tissue Transglutaminase Colometric Microassay Kit TG2-CovtTest (Zedira, Darmstadt, Germany) was used. This test is based on the incorporation of a biotinylated substrate specific for TG2 (Biotin-pepT26) into an amine donor (spermine)-coated strip [, ]. Protein extracts from normoxic rat liver containing 250 μg of total protein (measured by a modified Lowry method) were incubated for 20 min with different doses of cystamine (25–2,500 μmol/L), and the assay was then performed according to the manufacturer’s indications. Assays were also carried out as described above in protein extracts from normoxic rat lungs containing 5 μg of total protein, but due to reduced sample amount, the Tissue Transglutaminase Colometric Picoassay Kit (Zedira, Darmstadt, Germany) was used instead, which is less specific as it also detects the activity of TG3 and factor XIII but presents higher sensitivity. This picoassay kit is similar to the TG2-Covtest but it uses biotinyl-cadaverine as a substrate and HA-HMS as the amino donor coating the strips.
To measure transglutaminase activity in more physiologically relevant conditions, we used an assay based on the incorporation of the tranglutaminase substrate 5-(biotinamido)pentylamine (BP) to structural proteins was used as previously described []. Sixteen-week-old female C57BL/6 mice were exposed to hypobaric hypoxia while others were left as the normoxic control group. After 2 weeks of exposure to normoxia or hypoxia, the animals were weighed and euthanized by cervical dislocation. Immediately after the mice were sacrificed, heart, lungs, and liver were removed and kept in cold (5°C) physiological saline solution (PSS). The heart was isolated, and both atria were removed. The free wall of the right ventricle (RV) was separated from left ventricle and septum (LV + S). Each part was weighed and the ratios of right ventricle to left ventricle plus septum (RV/(LV + S)) were calculated and used to assess right ventricular hypertrophy. Lung tissue contains a large amount of vessels with lumen diameters of 20–80 μm, where the remodeling contributing to increased pulmonary pressure takes place in chronic hypoxic mice and rats [, ]. Left lung was then divided and incubated in PSS-containing BP (1 mmol/L) and an increased concentration of Ca2+ (2.5 mmol/L) at 37°C for 4 h, in presence and absence of cystamine (1.5 and 15 μmol/L). During incubation, PSS was continuously bubbled with 5% CO2-75% N2-20% O2 to maintain pH (7.4). Then, unreacted BP was washed by rinsing the arteries with PBS before tissue homogenization and protein extraction. Then 10 μg of total proteins were subjected to Western blot using a stain-free gel (Bio-Rad). After transfer to the membrane, total protein was visualized using stain-free technology by exposing the membranes to UV light and the images were captured using a PXi 4 Touch image analysis system (Syngene). The membrane was blocked in 5% bovine serum albumin (BSA) for 2 h and then incubated with HRP-conjugated streptavidin (Amersham Bioscience; dilution 1:10,000 in 0.5% BSA) for 2 h to detect BP incorporation. The membrane was then developed using an ECL-Plus kit (General Electric [GE] Health care, Copenhagen, Denmark). The images were captured by a luminescence camera using a PXi 4 Touch image analysis system (Syngene). Blots were quantified using GeneTools 4 software (Syngene) and normalized to the total protein. For each experimental group, the average activity in control conditions was set to 100% and the results are expressed as percentage of transamidase activity normalized to control conditions.
Chronic Hypoxic Rat Model
All animal experiments followed the revised NIH publication no. 86–23, entitled ”Principle of laboratory animal care,” and they were approved by the National Danish Animals Experiments Inspectorate (Permission 2006/561-1160 and Permission 2011/561-2011). Nine-week-old male Wistar rats (n = 80) were divided into a normoxic group and 4 groups exposed to hypobaric hypoxia and the following treatments: (1) hypoxic group treated with vehicle (H); (2) hypoxic group treated with cystamine (HC); (3) hypoxic group treated with sildenafil (HS); (4) hypoxic group treated with cystamine and sildenafil (HCS).
Sildenafil was dissolved in the drinking water with dosage of 25 mg/kg/day adjusted to water intake that was similar in sildenafil and vehicle-administered animals. Cystamine was administered subcutaneously in mini-osmotic pumps (Model 2002; Alzet, Cupertino, CA, USA) while vehicle-treated animals were sham operated with pumps filled with distilled water. We have previously found that a dosage of 80 mg/kg/day resulted in plasma cystamine concentrations of ∼2.5 μM and lowering of the systemic blood pressure [], while 40 mg/kg/day inhibited small arterial remodeling without changing the systemic blood pressure []. Therefore, the animals were treated with a dosage of 40 mg/kg/day.
The mini-osmotic pumps were implanted in the neck of the rats under subcutaneously administrated anesthesia with midazolam 0.4125 mg/100 g and fentanyl citrate 0.0381 mg/100 g plus fluanisone 0.825 mg/100 g (rodent mixture). The rats were given postoperational analgesia with Temgesic 0.13 mL. After 2 days, they were exposed to hypobaric hypoxia or normoxia. They were stored in a 12-h light:dark cycle at 20°C ventilated room with free access to water and chow.
In the hypobaric hypoxic chamber, the pressure was decreased to 560 mbar corresponding to an oxygen tension of 10–12%. The chamber was opened 3 times a week for about 30 min for cleaning the cages and renewing of water and chow.
After 2 weeks of exposure to normoxia or hypoxia, the animals were weighed and anesthetized by inhalation of isoflurane followed by subcutaneous injection of rodent mixture. Anesthesia was supplied during the procedure, and it was assessed by checking deep reflexes.
Hemodynamic Measurements
The animal was deposited on its back on a heat plate and with a rectal probe to measure the core temperature (Physitemp Instruments Inc., Clifton, NJ, USA). A pressure catheter (2F MicroTip Pressure Catheter; Millar Instruments Inc., Houston, TX, USA) was inserted into the RV through the right jugular vein. The catheter was connected to a QUAD Bridge connected to Powerlab 4/20 (AD Instruments, Oxfordshire, UK), and the pressure profile was registered by Chart 5.5 software (AD Instruments). After measurement of the RVSP and heart rate, the catheter was inserted into the left carotid artery for measurement of systemic blood pressure. Pressure profiles were recorded for 3 min. Right ventricular end-diastolic pressure (RVEDP), changes in the pressure over time during isovolumetric contraction and relaxation (dP/dtmax and dP/dtmin, respectively) were calculated directly from the pressure curves.
Cardiac output (CO) was measured in rats not used for RVSP measurements using the thermodilution technique in anaesthetized animals as described previously []. Briefly, the rat was anesthetized as described previously and a catheter for delivery of 0.2 mL saline, 22–24°C, inserted in the right jugular vein. The thermodilution curve was recorded with a T-type ultrafast thermocouple (AD Instruments, Oxfordshire, England) placed in the aorta, and CO was calculated as earlier described [] using Chart 7 Cardiac Output Software (AD Instruments, Oxfordshire, England). Systemic vascular resistance and PVR were calculated as mean pressures divided by CO.
Organ Weights and Muscularization of Pulmonary Arteries
The animals were sacrificed by decapitation. The heart, lungs, and liver were removed and stored in cold PSS. Right ventricular hypertrophy was evaluated as (RV/LV + S). Liver and lung weights were obtained as well.
For evaluation of pulmonary arterial muscularization, the left lung was isolated; a catheter inserted into the pulmonary artery, and 0.1 mL of heparin 100 IE/mL was injected. The lung was perfused with PSS-containing albumin (2%) followed by PSS with papaverine (10−4M) and finally with formalin 4% at a perfusion pressure of 20 mm Hg. Sections of the lung were stored in formalin buffer 4%, embedded in paraffin and stained with antibody against human smooth muscle actin (1:200; Dako, Glostrup, Denmark) and human von Willebrand factor (1:4,000; Dako, Denmark). At ×400 magnification small pulmonary vessels of each animal ranging from 20 to 80 μm in internal diameter were counted by a blinded observer and noted as muscular, partially muscular, or nonmuscular. The numbers are expressed as percentage of the total number of small arteries.
Sirius Red Staining for Collagen
Collagen content was assessed using picrosirius red staining []. The sections of the left lung were fixated with formaldehyde and imbedded with paraffin and cut in 10 μm transverse sections. After deparaffinization, the sections were incubated with Picro-Sirius Red solution (Abcam, UK). The sections were rinsed twice in a 0.5% acetic acid solution, followed by dehydration in absolute alcohol.
In a blinded manner, a minimum of 6 images per animal were randomly obtained and analyzed under circularly polarized white light. The images were processed in ImageJ (image analysis software, US National Institutes of Health, Bethesda, MA, USA). The red-stained collagen was identified in grayscale images, using adjusted threshold color. The resulting data on collagen content in lung tissue are given as percent collagen area of the tissue area.
Localization and Immunoblotting of TG2
Pieces from the RV from normoxic and hypoxic rats were stored in formalin 4% until they were embedded into paraffin. Slices of approximately 3 μm were made from each piece. Removing paraffin from the sections was done by washing in different solutions each for 5 min following the schedule: 2x xylene (xylene: mixture of isomers [AppliChem, Germany]), 2 × 99% ethanol, 96% ethanol, 50% ethanol, and water. The slices were incubated in 3% H2O2 for 10 min and afterward washed 2 × 5 min in Coon’s buffer (Na2H2PO4 [2H2O], NaH2PO4 [H2O], NaCl – ad. 10,000 mL H2O). The sections were treated with either citrat buffer (10 mM [Tri-sodium citrate dihydrate 5 mM, dinatriumhydrogencitrat 5 mM, pH adjusted to 6.0]) or TEG buffer (Tris 10 mM, EGTA 0.5 mM, pH adjusted to 9.0), then for 2 × 5 min in a microwave at 650 W and afterward washed in Coon’s buffer. The serum was removed and the primary antibody to TG2 (1:1,000) diluted in 1% BSA was added and incubated overnight at 4°C in a moisture chamber.
The sections were washed in Coon’s buffer and then incubated with secondary antibody (Bionylated Link – Universal LSABTM 2 Kit/HRP, Rabbit/Mouse; Dako, CA, USA) for 20 min at 20°C in a moisture chamber, then the samples were washed with Coon’s buffer and incubated with streptavidin (Streptavidin; Dako) for 20 min at 20°C in a moisture chamber. After washing with Coon’s buffer, DAB (DAB Chromogen tablets dissolved in Coon’s buffer; Dako) was added for 5 min. The sections were incubated with Mayer’s hematoxylin for 1 min, washed in H2O and incubated for 5 min in each solution: 50% ethanol, 96% ethanol, and 99% ethanol. Negative controls were made exactly identical but without incubation with primary antibody.
The lungs for immunoblotting were perfused with heparin 100 IE/mL and then homogenized in 600 μL of lysis buffer in a homogenizer (Precellys 24; Bertin Technologies, France). Heart tissue was homogenized in 300 μL lysis buffer using the same technique. Western blotting was performed as previously described []. Samples containing an equal amount of protein were loaded on 4–12% polyacrylamide Bis-Tris criterion gel. Proteins were blotted onto a polyvinylidine difluoride (PVDF membrane, which was incubated with primary antibodies (TG 2 Ab-2) overnight and with secondary antibody (HRP-conjugated goat anti-mouse IgG) for 2 h. The membranes were developed using an enhanced chemiluminescence detection system (ECL-Plus; Amersham Biosciences, Buckinghamshire, UK) and expressed on X-ray films. Therefore, the blots were analyzed by optical density measurements (Image Quant TL; Amersham Biosciences) and the densitometric analysis of TG2 and N-[Latin Small Letter Open E](γ-glutamyl)lysine was given in relation to protein content and also normalized to panactin. Hypoxia regulates expression of glyceraldehyde 3-phosphate dehydrogenase [], and therefore we chose to normalize protein expression to panactin, despite it was increased to the same level in all hypoxic groups compared to tissue from normoxic control animals.
Drugs and Solutions
Cystamine dihydrochloride (Sigma-Aldrich, St. Louis, MO, USA), sildenafil citrate (Pfizer Inc., Groton, CT, USA), Dormicum (midazolam 5 mg/mL; F. Hoffmann-La Roche AG, Basel, Switzerland), Hypnorm (fentanyl citrate 0.315 mg/mL, fluanisone 10 mg/mL; VetaPharma Ltd, Leeds, UK), Temgesic (buprenorphinum 0.3 mg/mL; Schering-Plough, Belgium), isofluran (4.5%, oxygen flow 2 L/min, FORENE®; Abbott Scandinavia AB, Solna, Sweden), and bovine albumin (Sigma-Aldrich) were used. PSS consisted of the following (mM): NaCl 119 mM, KCl 4.7 mM, MgSO4 (7H2O) 1.17 mM, NaHCO3 25 mM, KH2PO4 1.18 mM, EDTA 0.026 mM), glucose 5.5 mM). Formalin 4% (Bie & Berntsen, Herlev, Denmark), papaverine (Sigma-Aldrich), U46619 (Sigma-Aldrich), acetylcholine (Sigma-Aldrich), lysis buffer (20 mM Tris/HCL, 5 mM EGTA, 150 mM NaCl, 20 mM glycorophosphate, 10 mM NaF, 1% triton X-100, 0.1% tween-20, adjusted to pH 7.5 and added 10 μL orthovanadate, 4 μL PMSF, 100 μL PIM per mL of lysis buffer), human smooth muscle actin (Clone 1A4) (Dako, Denmark), human Von Willebrand factor (Dako), TG 2 Ab-2 (clone TG100) (Thermo Scientific, CA, USA), and HRP-conjugated goat anti-mouse IgG (Zymed, San Francisco, CA, USA) were used.
Statistical Analysis
N indicates the number of animals used for each experiment. For the statistical analysis, the computer software GraphPad Prism 7.0 (San Diego, CA, USA) was used. The animal data were analyzed by use of one-way analysis of variance, and in case of significance, Bonferroni’s posttest was made to compare it to the vehicle-treated hypoxic group (H). The assumptions of the models were investigated by inspecting Q-Q plots, and data were logarithmically transformed when necessary in order to generate a Gaussian-distributed data set. In case of clustered data, the data were analyzed by use of a nonparametric rank test. All values are presented as mean ± SD and considered significant when p < 0.05.
Results
Effect of Cystamine on TG2 Activity
First, we examined the presence of TG1-TG7 mRNA by qualitative RT-PCR and sequence analysis. In rat pulmonary arteries, we found expression of TG1 and TG2. Lung tissue samples showed expression of TG1, TG2, and TG5, while in rat liver there was expression of TG1, TG2, and TG5 (see online suppl. Fig. 1; for all online suppl. material, see http://www.karger.com/doi/10.1159/000515511).
To investigate the effect of cystamine on transamidase activity, a test based on the incorporation of a specific TG2 substrate (Biotin-pepT26) was performed, and incubation with the irreversible selective TG2 inhibitor, VA5, inhibited transamidase activity in the liver suggesting that the transamidase activity can be attributed to TG2 activity. The effect of increasing concentrations of cystamine was examined in rat liver and showed that relatively high cystamine concentrations (>30 μM) inhibited transamidase activity in the liver (Fig. 1a) and even higher concentrations were required in the lung (Fig. 1b). However, in this assay the homogenized tissue is treated with DTT leading to full tissue activation of transamidase activity. Therefore, we examined the effect of cystamine on transamidase activity in lung tissue by measuring 5-BP incorporation (Fig. 1c). Lung tissue from control and chronic hypoxic mice was used. The chronic hypoxic mice had right ventricular hypertrophy (Fig. 1d), and there was a tendency to increased transamidase activity (Fig. 1e). Cystamine (1.5 and 15 μM) significantly inhibited the 5-BP incorporation, while there was no effect of vehicle (Fig. 1f).
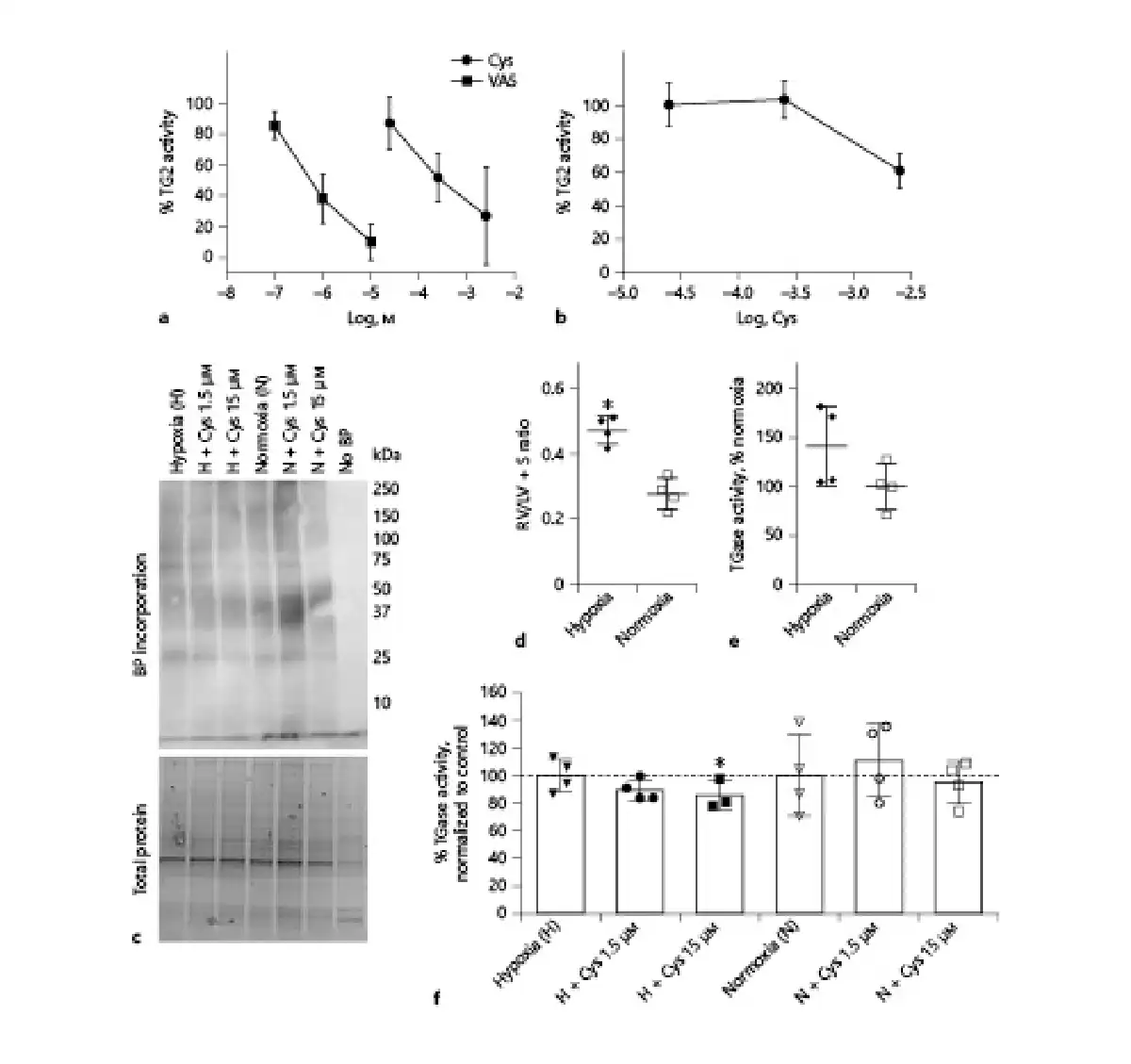
Fig. 1
Transglutaminase inhibition by Cys. a, b In vitro transglutaminase activity normalized to control in response to concentration-response curves of Cys and selective irreversible TG2 inhibitor VA5 in tissue extract from liver (a) and lungs (b) from n = 4 normoxic rats. c Ex vivo transamidase activity measured through BP incorporation in the lung tissue of normoxic and hypoxic mice in absence or presence of Cys. In the negative control, the incubation period was carried out in absence of BP (no BP). d RV/LV + S of the hypoxic and normoxic mice used in (c), *p < 0.05 versus normoxic mice. e Average transamidase activity of the animals used in (c) presented as percentage of activity in normoxic mice. f Average transamidase activity in response to Cys of the mice used in (c) presented as percentage of their respective controls. Values are means ± SD. One-way analysis of variance with Bonferroni posttest: *p < 0.05 versus vehicle. Cys, cystamine; RV/LV + S, right ventricular weight/left ventricular weight + septum weight ratio; BP, biotin-pentylamine; H, hypoxia; N, normoxia.
Effect of Cystamine and Sildenafil in Chronic Hypoxic Rats
Table 1 shows the organ and body weights of the animals by the end of the study. Fourteen days after exposure to chronic hypoxia, the body weight was decreased by 14%, and the heart to body weight, and lung to body weight ratios were markedly increased by 16 and 48%, respectively, while liver weights were unaltered. Treatment with cystamine, sildenafil, or the combination of cystamine and sildenafil did not change the organ or body weights in the hypoxic rats (Table 1).
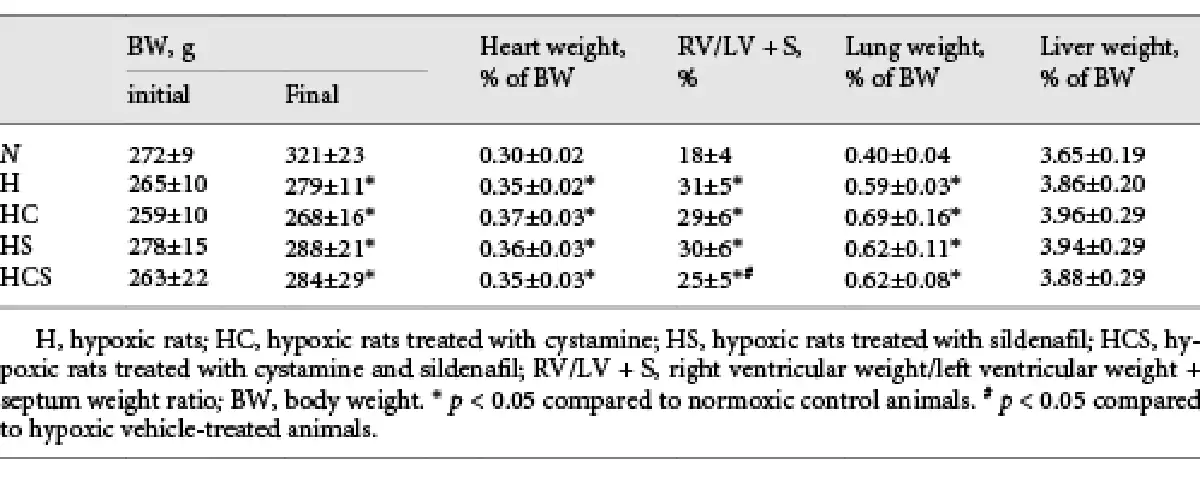
The degree of right ventricular hypertrophy was determined as the weight of the right ventricular wall to the weight of RV/LV + S. RV hypertrophy increased by 72% in the chronic hypoxic vehicle-treated rats and was unaltered by treatment with cystamine or sildenafil but significantly decreased in the group treated with the combination of cystamine and sildenafil (Table 1).
Hemodynamic Measurements and Cardiac Hypertrophy
In vehicle-treated chronic hypoxic rats, RVSP markedly increased compared to normoxic rats (Fig. 2). Treatment with cystamine failed to prevent the increase in RVSP in chronic hypoxic rats. RVSP was decreased in chronic hypoxic rats treated with sildenafil compared to vehicle-treated hypoxic rats and decreased in the rats treated with the combination of sildenafil and cystamine compared to the chronic hypoxic rats treated with cystamine (Fig. 2a). In chronic hypoxic rats, the CO was not changed by the treatments with cystamine or sildenafil (Fig. 2b). The estimated PVR was increased in vehicle-treated hypoxic compared to normoxic animals. Within the hypoxic groups, PVR was unaltered in the cystamine-treated chronic hypoxic rats, while sildenafil lowered PVR compared to the cystamine-treated chronic hypoxic rats (Fig. 1c).
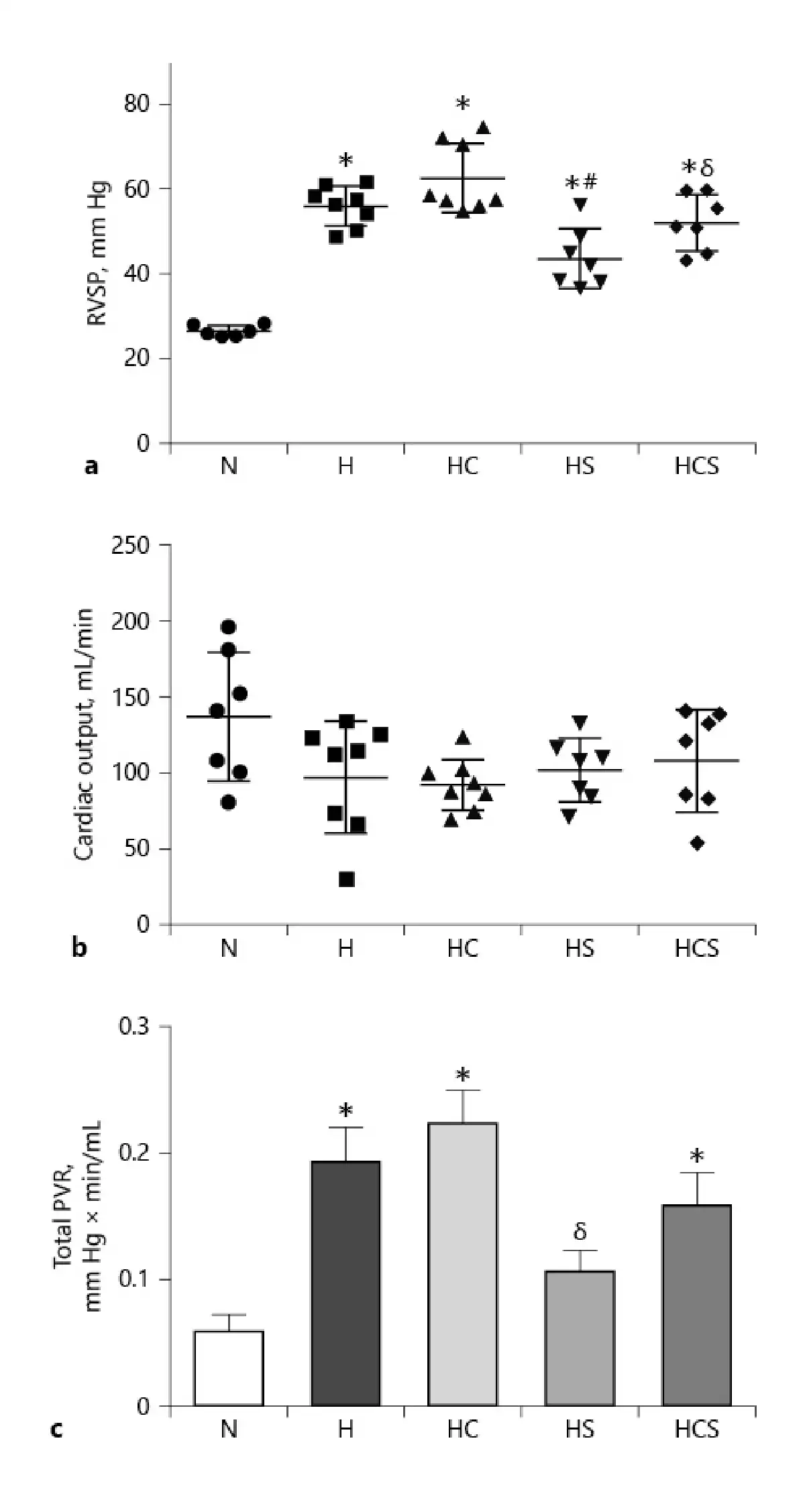
Fig. 2
Effect of cystamine and sildenafil on hemodynamic measurements in the pulmonary circulation. a RVSP. b CO. c Total PVR. n = 6–8 in each group. Values are means ± SEM. One-way analysis of variance with Bonferroni posttest: *p < 0.05 versus normoxic rats, #p < 0.05 versus vehicle-treated hypoxic rats, δp < 0.05 versus cystamine-treated hypoxic rats. H, hypoxic rats; HC, hypoxic rats treated with cystamine; HS, hypoxic rats treated with sildenafil; HCS, hypoxic rats treated with cystamine and sildenafil; RVSP, right ventricular systolic pressure; CO, cardiac output; PVR, pulmonary vascular resistance.
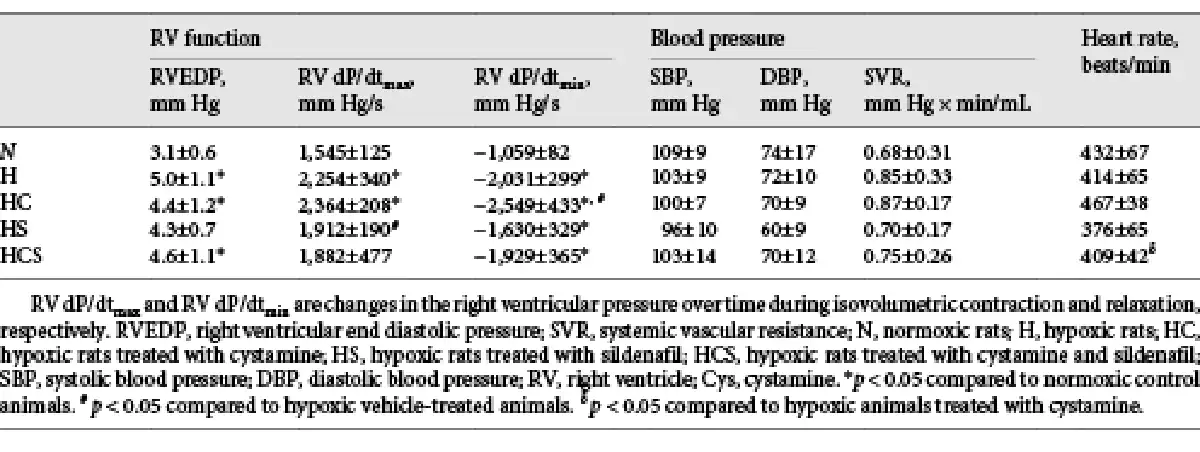
In addition to RVSP being higher in the chronic hypoxic rats, RVEDP was increased in the chronic hypoxic rats with exception of the sildenafil-treated animals (Table 2). Chronic hypoxic-induced pulmonary hypertensive rats exhibited a marked elevation in RV dP/dtmax and dP/dtmin. The increased RV dP/dtmax indicates RV systolic dysfunction, whereas the increased RV dP/dtmin indicates RV diastolic dysfunction. In the hypoxic rats treated with cystamine, the diastolic function was significantly worse, while treatment with sildenafil improved the systolic heart function in the hypoxic rats. The heart rate was similar in the normoxic and vehicle-treated hypoxic animals, and there were no differences in systemic systolic and diastolic blood pressure or systemic vascular resistance in any of the 5 groups (Table 2).
Effects of Cystamine and Sildenafil on Muscularization and Fibrosis in Pulmonary Arteries in Chronic Hypoxic Rats
As observed in previous studies [], lung sections stained for Von Willebrand factor showed only positive immunoreaction in the endothelial cell layer (Fig. 3a), while α-actin stained the smooth muscle layer (Fig. 3b). In lung sections from the chronic hypoxic compared to normoxic rats, the number of nonmuscularized arterioles (20–80 μm in diameter) was significantly decreased, and this was also the case in hypoxic rats treated with cystamine (Fig. 3c). Only the combination of cystamine and sildenafil partially prevented the decrease in nonmuscularized arterioles (Fig. 3c). The number of partially muscularized arteries was not significantly changed in hypoxic versus normoxic lung sections (Fig. 3d), while the number of fully muscularized arteries was significantly increased (Fig. 3e). Monotherapy with cystamine failed to change the muscularization of the pulmonary arteries in chronic hypoxic rats, and only the combined treatment with cystamine and sildenafil significantly reduced the muscularization of the pulmonary arterioles in chronic hypoxic rats (Fig. 3e).
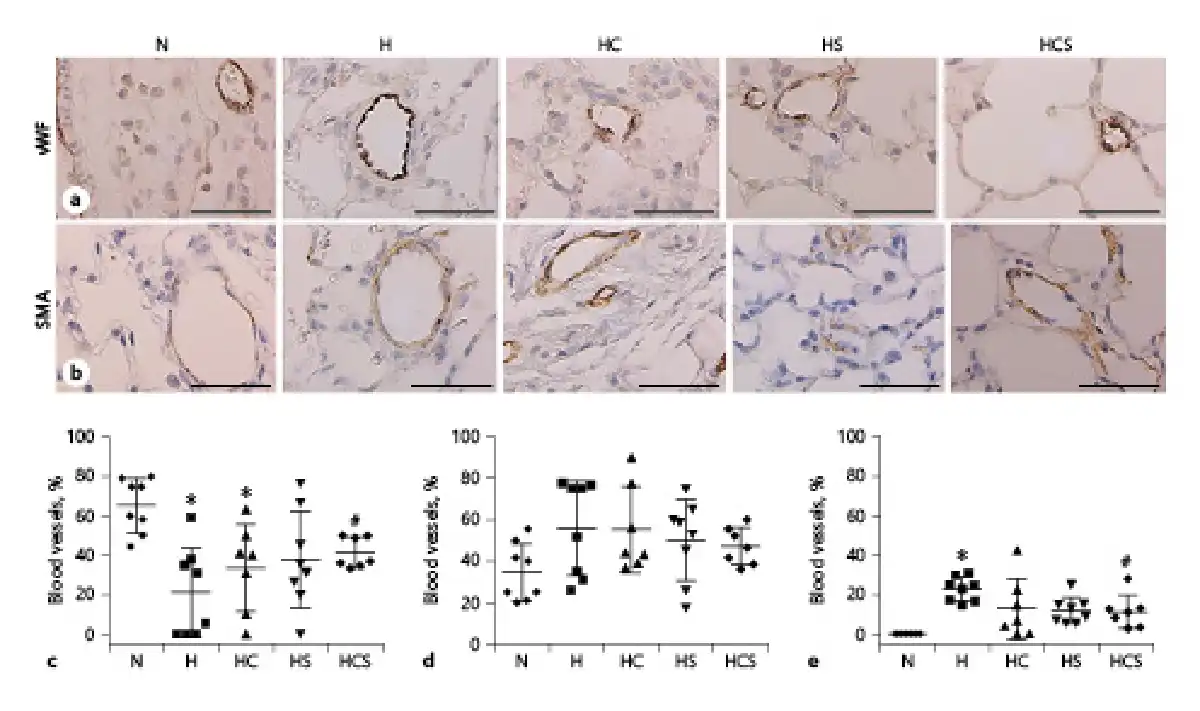
Fig. 3
Effect of cystamine and sildenafil on pulmonary arterial muscularization. Examples of lung sections showing positive immunoreaction of pulmonary arteries to vWF (a) and SMA (b). The black bars corresponds to 50 μm. Numbers of pulmonary arteries with diameters of 20–80 μm, which are nonmuscularized arteries (c), partially muscularized (d), and fully muscularized (e) expressed as percentage of total numbers of arteries. n = 6–8 in each group. Values are means ± SD. One-way analysis of variance with Bonferroni posttest: *p < 0.05 versus normoxic rats, #p < 0.05 versus vehicle-treated hypoxic rats. RVSP, right ventricular systolic pressure; CO, cardiac output; PVR, pulmonary vascular resistance; H, hypoxic rats; HC, hypoxic rats treated with cystamine; HS, hypoxic rats treated with sildenafil; HCS, hypoxic rats treated with cystamine and sildenafil; vWF, von Willebrand factor; SMA, smooth muscle actin.
Picrosirius red staining was used to evaluate lung fibrosis. Examining the sections in polarized light revealed that the staining tended to be decreased in lung sections of all drug-treated rats (Fig. 4a–g), but the decrease was only significant in lung sections of cystamine-treated hypoxic rats (Fig. 4f).
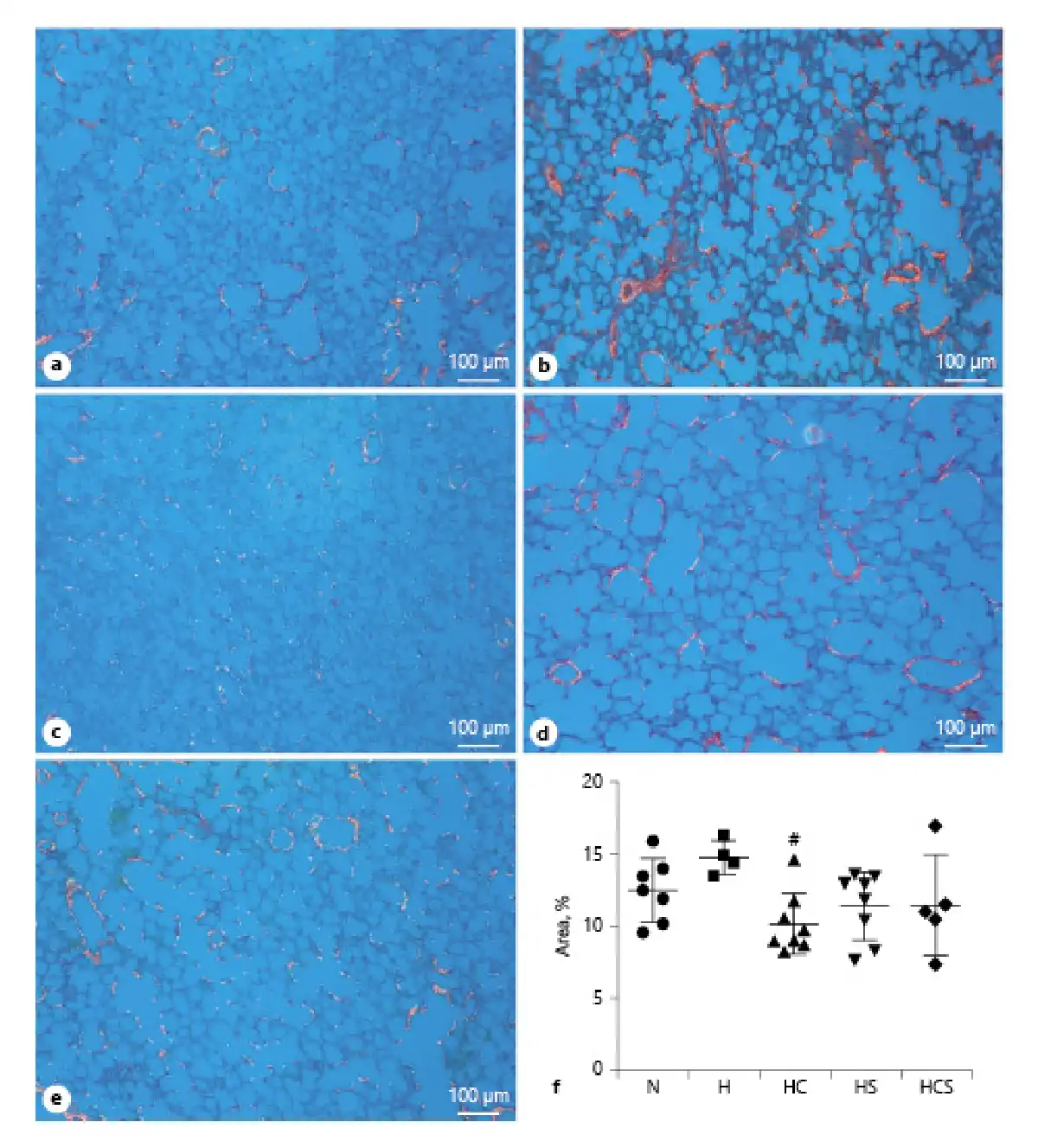
Fig. 4
Effect of pharmacological treatments on lung fibrosis in hypoxic rats. Representative images of fibrosis in lungs, observed under circularly polarized white light, from N rat (a), H rats (b), HC (c), HS (d), and HCS (e). f Quantification of interstitial lung fibrosis as measured by picrosirius red staining under circularly polarized white light. The bar in the pictures corresponds to 100 μm. Values are means ± SD. One-way analysis of variance with Bonferroni posttest: #p < 0.05 versus H rats. H, hypoxic rats; HC, hypoxic rats treated with cystamine; HS, hypoxic rats treated with sildenafil; HCS, hypoxic rats treated with cystamine and sildenafil.
Effect of Cystamine and Sildenafil on TG2 Immunoreaction in Heart and Lung Tissue
Expression of TG2 in sections was visualized in the RV and lungs by immunostaining showing that TG2 is localized in the cardiomyocytes of the RV (Fig. 5a, b), and in the lungs TG2 was localized to the endothelium and the smooth muscle layer of the arteries (Fig. 5c, d). As reported previously, immunoblotting for human TG2 full-length protein gave a band at 82 kDa []. The content of TG2 evaluated by immunoblotting was increased several hundred percent in the RV by hypoxia [] (Fig. 5e). Although less pronounced, TG2 expression was also significantly increased in the lung (p < 0.05, Students t test) from hypoxic animals (Fig. 5f). In the RV of the chronic hypoxic rats, the loading control panactin was also markedly increased compared to the RV from normoxic rats, but there was no difference in panactin among the hypoxic groups (online suppl. Fig. 2). Expressing the TG2 immunoreaction relative to panactin showed treatment with cystamine, sildenafil, or the combination did not decrease TG2 immunoreaction in the RV or lung of the chronic hypoxic rats (Fig. 5g, h).
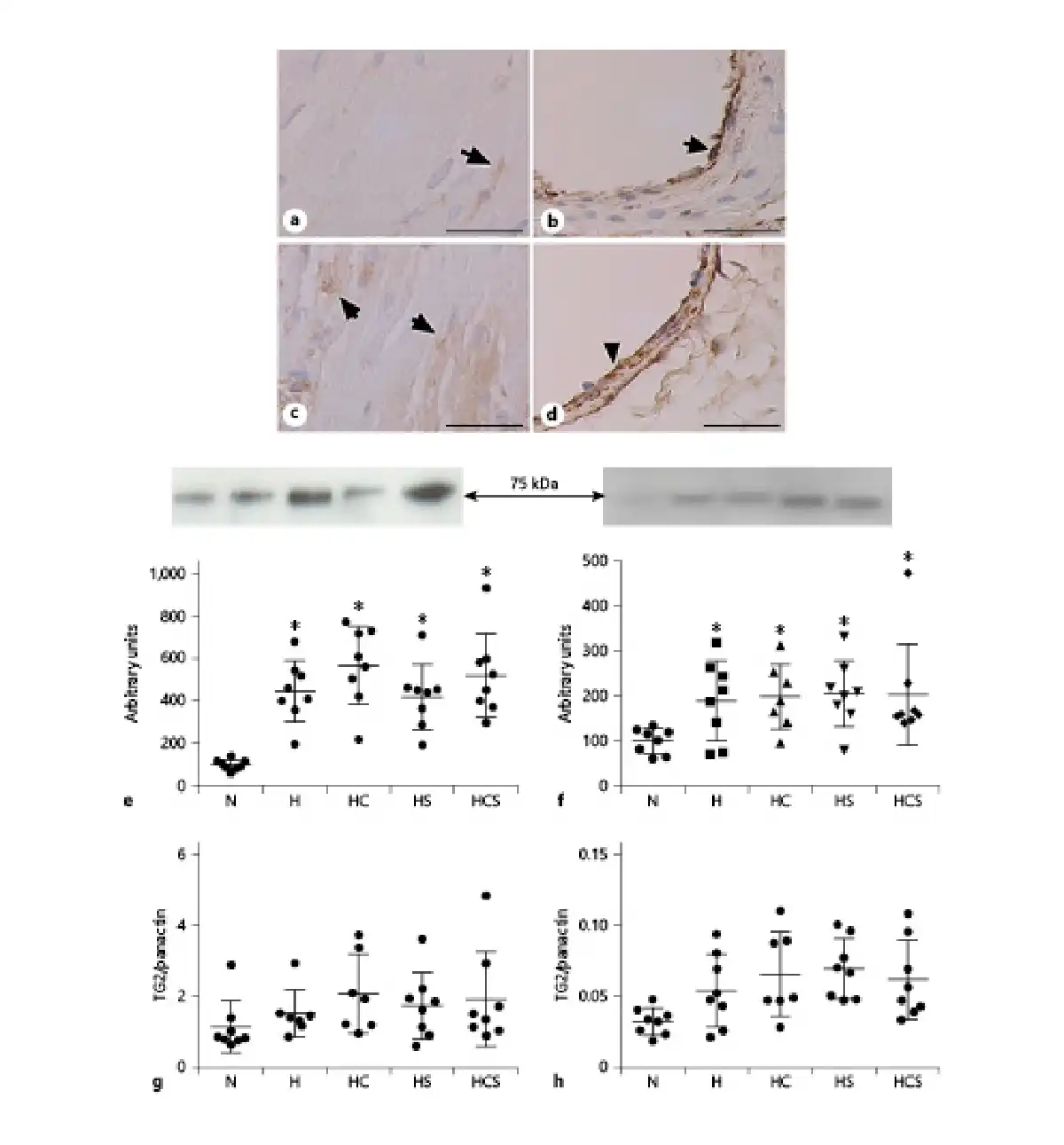
Fig. 5
Effect of cystamine and sildenafil treatment on tissue TG2 expression. TG2 immunoreaction in RV (a) and pulmonary artery (b) from normoxic rat, and RV (c) and pulmonary artery (d) from hypoxic rats. The bar corresponds to 50 μm. Quantification of immunoblots for TG2 expression in relation to total protein in RV (e) and lung (f). Expressed relative to panactin immunoblotting in RV (g) and lung (h) from N rats, H rats, and HC, HS, and HCS. Representative immunoblots are inserted above the graphs for TG2 (e, f). n = 6–8 in each group. Values are means ± SD. One-way analysis of variance with Bonferroni posttest: *p < 0.05 versus normoxic rats. TG2, transglutaminase 2; H, hypoxic rats; HC, hypoxic rats treated with cystamine; HS, hypoxic rats treated with sildenafil; HCS, hypoxic rats treated with cystamine and sildenafil; RV, right ventricle.
Discussion
In the present study, we found that TG2 expression in the lung and the RV from hypoxic rats was unaltered by the treatment with cystamine and sildenafil. Cystamine inhibited transamidase activity in the liver and lungs of hypoxic animals at high concentrations and BP incorporation at lower concentrations relevant for the in vivo effect of cystamine on TG2. In contrast to the phosphodiesterase type 5 inhibitor, sildenafil, cystamine failed to have effect on the development of pulmonary hypertension, RV hypertrophy, and pulmonary arterial muscularization in the chronic hypoxic rat. Moreover, there was no additive effect of the combination of cystamine and sildenafil compared to only treatment with sildenafil. However, cystamine reduced fibrosis measured using picrosirius staining in the lung of the chronic hypoxic rats, indirectly supporting cystamine reached effective concentrations despite lack of effect on pulmonary hypertension in the chronic hypoxic rats.
Effect of Cystamine and Sildenafil on the RV in Pulmonary Hypertension
In previous microarray studies confirmed by qPCR and immunoblotting of the RV, we observed upregulation of TG2 in relation to the increase in RVSP in chronic hypoxic rats []. In agreement with these studies and those by others [], TG2 protein was expressed in the myocardium and was markedly increased in the RV of hypoxic rats in the present study. Overexpression of TG2 has been shown to result in left ventricular hypertrophy and interstitial fibrosis []. However, in the present study cystamine treatment by itself did not inhibit RV hypertrophy in the hypoxic rats. Moreover, endothelin-1 expression and plasma levels are increased by hypoxia [, ], and both endothelin-1 and aldosterone have been shown to be associated with elevated TG2 expression and/or activity in cardiomyocytes []. Therefore, a likely explanation is that high-pressure load in the RV [] and/or increased circulating levels of endothelin-1 [] leads to upregulation of TG2 which may contribute to the adaptation rather than the development of right ventricular hypertrophy in the hypoxic rats.
Cystamine was reported to inhibit the development of right ventricular hypertrophy in chronic hypoxic rats and monocrotaline-induced pulmonary arterial hypertension [, ]. In contrast, in the present study, treatment with cystamine failed to prevent the development of RV hypertrophy measured as RV/LV + S. One of the main differences is the drug administration. In previous studies, cystamine was either administrated twice daily by intraperitoneal administration [] or by oral gavage [], while in the present study cystamine was administered in mini-osmotic pumps. In contrast to intraperitoneal and oral administration yielding high plasma peak concentrations, administration by mini-osmotic pumps results in lower steady plasma concentrations. In previous studies in our laboratory, where the plasma concentration of cystamine was measured in rats treated with twice the dose and the same administration route as used in the present study, a concentration of 1–2 μM cystamine was detected [].
In platform assays, 73 μM cystamine inhibits TG2; cystamine concentrations above 100–200 μM may also inhibit TG1 and caspase-3, and in isolated cells up to 500 μM only reduced transamidase activity with 70% []. In agreement with these observations, we observed that millimolar cystamine concentrations were required to inhibit the transamidase activity in liver and lung tissue from hypoxic rats, but in this assay TG2 is maximally activated by DTT. Another approach used to measure in vivo activity of TG2 is the incorporation of BP, and this activity is inhibited in the presence of a competitive irreversible inhibitor of the open state of the enzyme [, ]. Incubation of lung sections from hypoxic mice revealed in the present study that 1.5–15 μM cystamine significantly inhibits 5-BP incorporation. Based on previous results of plasma-cystamine concentrations [], these results support that cystamine in vivo may inhibit TG2, while it is unlikely with the dose applied in the present study that it will inhibit other potential targets. Therefore, the relation of plasma cystamine to pharmacodynamic effect on right ventricular hypertrophy suggest that TG2 inhibition is insufficient to prevent RV hypertrophy in chronic hypoxic rats. Thus, inhibition of other signal pathways by higher concentrations of cystamine may contribute in inhibition of RV hypertrophy in studies where cystamine was administered by intraperitoneal administration or oral gavage [, ]. In support of our observations, a selective irreversible TG2 and TG1 inhibitor ERW1041E failed to prevent the development of RV hypertrophy in mice with pulmonary hypertension induced by a combination of hypoxia plus SU-5416 [].
In the same mouse pulmonary hypertension model, however, ERW104E significantly reduced RVSP []. In contrast, in the present study, cystamine failed to alter RVSP. However, in agreement with our previous studies showing that sildenafil reduces RVSP in hypoxic rats [, ], sildenafil alone or in combination with cystamine also significantly reduced RVSP in the hypoxic rats in the present study. Moreover, systolic ventricular function, measured as dP/dtmax, decreased by treatment with sildenafil compared to the vehicle-treated hypoxic rats, whereas cystamine failed to improve systolic ventricular function, and actually appeared to accelerate the deterioration in RV diastolic function, measured as an increase in dP/dtmin, compared to vehicle-treated rats. In small systemic arteries, cystamine was found to inhibit both inward and outward remodeling []. Moreover, TG2 null mice had increased mortality and enhanced ventricular dilation following left ventricular pressure overload suggesting maladaptation to high pressure []. Although further investigation will be required, it cannot be excluded that cystamine actually inhibits the adaptation of the RV to high RVSP and RVEDP. This is supported in the present study by the observation that RV weight expressed relative to RVSP is significantly less pronounced in cystamine-treated animals compared to hypoxic controls (online suppl. Fig. 3). Although other approaches are required to confirm our observations, our results may suggest that cystamine delays the remodeling process in the RV in response to an increased afterload.
Effects of Cystamine and Sildenafil on Muscularization in Pulmonary Arteries from Chronic Hypoxic Rats
TG2 is expressed in the lung endothelium and epithelium in mice []. In the present study, we found expression of TG1 and TG2 in small pulmonary arteries, while in the lung TG1, TG2, and TG5 expression was evident. Using the same primers, we have previously detected TG4 expression, confirmed by sequencing, in the rat ear and testes [], but this variant was not detected in the peripheral rat lung in the present study. TG4 is expressed in larger arteries [], and therefore we cannot exclude that it will also be expressed in samples including proximal large pulmonary arteries. In agreement with previous studies [], we found that chronic hypoxia upregulates TG2 expression in the lung, and the expression remained high in hypoxic rats treated with cystamine and sildenafil.
Chronic hypoxia markedly upregulates the PVR due to increased vascular tone, hematocrite, muscularization, fibrosis [], and rarefaction often reflected by pulmonary arterial occlusive lesions []. Sildenafil is a potent vasodilator in the pulmonary circulation [] and decreased the PVR. In contrast to sildenafil, cystamine is a less potent vasodilator in mesenteric small arteries [, ], and very high concentrations (>100 μM) of cystamine were required to relax pulmonary small arteries (online suppl. Fig. 4), and in agreement with these findings cystamine treatment did not decrease the PVR in the chronic hypoxic rats.
Cystamine was found to reduce arterial wall thickness in pulmonary arteries from chronic hypoxic rats [], and recently TG2 was found to play an important role in stimulation of pulmonary vascular smooth muscle proliferation when lowering oxygen from 21 to 3% in cell cultures []. We found that chronic hypoxia increased the muscularization of the pulmonary arteries, while the treatment with cystamine alone failed to reduce pulmonary arterial muscularization. In agreement with previous studies [, ], treatment with sildenafil decreased the number of fully muscularized arterioles in the lung of chronic hypoxic rats. However, the most remarkable effect of the treatments was that the number of nonmuscularized arterioles decreased in vehicle and cystamine-treated animals, and this effect was prevented in rats treated with sildenafil. These findings agree with our recent observations that the pulmonary vasodilator, levosimendan, is also associated with less pulmonary arterial occlusive lesions []. Thus, our findings suggest that in contrast to cystamine, the vasodilator effect of sildenafil maintains an open lumen in pulmonary small arteries in chronic hypoxic rats, and this contributes to the lowering of the PVR and pressure.
Collagen accumulates in lung tissue and pulmonary arteries, and it is important for the stiffening in the large proximal pulmonary arteries [-], and in distal pulmonary arteries it plays an important role for right ventricular afterload in chronic hypoxic rats [, ]. In the hypoxic rats, fibrosis measured by picrosirius red staining was increased in the lung tissue and was decreased in the lung tissue of rats treated with cystamine. The mechanism involved in cystamine-induced decrease in collagen content is unclear. However, increased TG2 activity has also been suggested to lead to platelet-derived growth factor and transforming growth factor activation [, ] and hence induction of collagen formation. There was no additive effect of cystamine and sildenafil treatment on lung fibrosis suggesting that cystamine treatment would not provide additional effect compared to monotherapy with sildenafil.
Limitations
Cystamine is considered a pan transglutaminase inhibitor as half-maximal inhibition (IC50) for human TG2 and TG1 is, respectively, 73 and 264 μM, and above 100 μM for inhibition of caspase-3 []. Therefore, it could perhaps have been an advantage to the test the hypothesis of the present study in TG2 knockout mice. However, the global TG2 knockout mouse phenotype resembles that of the maturity onset of diabetes []. Due to the less pronounced development of pulmonary hypertension in chronic hypoxic mice compared to rats [, , ], the presence of diabetes may have larger influence on the pulmonary circulation and heart than pulmonary hypertension. Therefore, as in our previous studies, the hypobaric hypoxic rat was chosen and found to have increased pulmonary pressure, PVR, muscularization of the pulmonary arteries, and RV hypertrophy [, , ].
Conclusion and Perspectives
In summary, TG2 is upregulated in the heart and lung of chronic hypoxic rats and the transglutaminase inhibitor cystamine reduces lung tissue fibrosis. Otherwise, cystamine failed to prevent pulmonary hypertension, RV hypertrophy, and pulmonary arterial muscularization in the chronic hypoxic rat. Although the selective TG2 inhibitor ERW104E failed to inhibit development of RV hypertrophy in a mouse model of pulmonary arterial hypertension [], different series of inhibitors with high selectivity for TG2 have been developed mainly for treatment of celiac disease and fibrotic kidney disease [, ]. Due to different binding kinetic and conformational change in the TG2 induced by these inhibitors, it cannot be excluded that one of these inhibitors may have larger impact than cystamine on pulmonary hypertension.
Acknowledgements
We thank Heidi Knudsen for technical assistance and Prof. Emeritus Carl Christian Danielsen for kindly letting us use his polarized light microscope. Pfizer kindly donated sildenafil citrate powder.
Statement of Ethics
All animal experiments followed the revised NIH publication No. 86–23, entitled ”Principle of laboratory animal care,” and they were approved by the National Danish Animals Experiments Inspectorate (Permission 2006/561-1160 and Permission 2011/561-2011).
Conflict of Interest Statement
The authors have no conflicts of interest to disclose.
Funding Sources
Ulf Simonsen was supported by grants from the Danish Heart Foundation (A4012-22702), the Novo Nordisk Foundation (NNF190C0055688, NNF20OC0065767), and the Danish Research Council.
Author Contributions
Conception or design of the work: Lars K. Markvardsen, Charlotte U. Andersen, Ulf Simonsen. Acquisition and analysis of data: (1) PCR studies (Susie Mogensen), (2) hemodynamic in vivo studies (Lars K. Markvardsen and Lene Sønderskov), (3) immunohistochemistry, immunoblotting, morphology and fibrosis studies, TG2 activity studies (Lars K. Markvardsen, Christine Wandall-Frostholm, Estefano Pinilla, and Judit Prat-Duran), and (4) small-vessel pharmacology experiments (Mathilde Aalling). Interpretation of data for the work: all authors. Drafting and revising the manuscript critically for important intellectual content: all authors.
References
- 1. Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest. 2009;135(3):794–804.http://dx.doi.org/10.1378/chest.08-0492.
- 2. Vonk Noordegraaf A, Chin KM, Haddad F, Hassoun PM, Hemnes AR, Hopkins SR, et al. Pathophysiology of the right ventricle and of the pulmonary circulation in pulmonary hypertension: an update. Eur Respir J. 2019;53(1):1801900.http://dx.doi.org/10.1183/13993003.01900-2018.
- 3. Griffin M, Casadio R, Bergamini CM. Transglutaminases: nature’s biological glues. Biochem J. 2002;368(2):377–96.
- 4. Guilluy C, Rolli-Derkinderen M, Tharaux PL, Melino G, Pacaud P, Loirand G. Transglutaminase-dependent RhoA activation and depletion by serotonin in vascular smooth muscle cells. J Biol Chem. 2007;282(5):2918–28.http://dx.doi.org/10.1074/jbc.M604195200.
- 5. Wei L, Warburton RR, Preston IR, Roberts KE, Comhair SA, Erzurum SC, et al. Serotonylated fibronectin is elevated in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2012;302(12):L1273–9.http://dx.doi.org/10.1152/ajplung.00082.2012.
- 6. Oh K, Park HB, Byoun OJ, Shin DM, Jeong EM, Kim YW, et al. Epithelial transglutaminase 2 is needed for T cell interleukin-17 production and subsequent pulmonary inflammation and fibrosis in bleomycin-treated mice. J Exp Med. 2011;208(8):1707–19.http://dx.doi.org/10.1084/jem.20101457.
- 7. Olsen KC, Sapinoro RE, Kottmann RM, Kulkarni AA, Iismaa SE, Johnson GV, et al. Transglutaminase 2 and its role in pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(6):699–707.http://dx.doi.org/10.1164/rccm.201101-0013OC.
- 8. Bakker EN, Buus CL, Spaan JA, Perree J, Ganga A, Rolf TM, et al. Small artery remodeling depends on tissue-type transglutaminase. Circ Res. 2005;96(1):119–26.http://dx.doi.org/10.1161/01.RES.0000151333.56089.66.
- 9. Eftekhari A, Rahman A, Schaebel LH, Chen H, Rasmussen CV, Aalkjaer C, et al. Chronic cystamine treatment inhibits small artery remodelling in rats. J Vasc Res. 2007;44(6):471–82.http://dx.doi.org/10.1159/000106465.
- 10. Engholm M, Eftekhari A, Chwatko G, Bald E, Mulvany MJ. Effect of cystamine on blood pressure and vascular characteristics in spontaneously hypertensive rats. J Vasc Res. 2011;48(6):476–84.http://dx.doi.org/10.1159/000327773.
- 11. Huelsz-Prince G, Belkin AM, Vanbavel E, Bakker EN. Activation of extracellular transglutaminase 2 by mechanical force in the arterial wall. J Vasc Res. 2013;50(5):383–95.http://dx.doi.org/10.1159/000354222.
- 12. Jung SM, Jandu S, Steppan J, Belkin A, An SS, Pak A, et al. Increased tissue transglutaminase activity contributes to central vascular stiffness in eNOS knockout mice. Am J Physiol Heart Circ Physiol. 2013;305(6):H803–10.http://dx.doi.org/10.1152/ajpheart.00103.2013.
- 13. Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, et al. Enhanced Gαq signaling: a common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc Natl Acad Sci U S A. 1998;95(17):10140–5.
- 14. Shinde AV, Frangogiannis NG. Tissue transglutaminase in the pathogenesis of heart failure. Cell Death Differ. 2018;25(3):453–6.http://dx.doi.org/10.1038/s41418-017-0028-9.
- 15. D’Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, et al. Transgenic Gαq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci U S A. 1997;94(15):8121–6.
- 16. Small K, Feng JF, Lorenz J, Donnelly ET, Yu A, Im MJ, et al. Cardiac specific overexpression of transglutaminase II (G(h)) results in a unique hypertrophy phenotype independent of phospholipase C activation. J Biol Chem. 1999;274(30):21291–6.http://dx.doi.org/10.1074/jbc.274.30.21291.
- 17. Baandrup JD, Markvardsen LH, Peters CD, Schou UK, Jensen JL, Magnusson NE, et al. Pressure load: the main factor for altered gene expression in right ventricular hypertrophy in chronic hypoxic rats. PLoS One. 2011;6(1):e15859.http://dx.doi.org/10.1371/journal.pone.0015859.
- 18. Li X, Wei XL, Meng LL, Chi MG, Yan JQ, Ma XY, et al. Involvement of tissue transglutaminase in endothelin 1-induced hypertrophy in cultured neonatal rat cardiomyocytes. Hypertension. 2009;54(4):839–44.http://dx.doi.org/10.1161/HYPERTENSIONAHA.109.130161.
- 19. Penumatsa KC, Toksoz D, Warburton RR, Hilmer AJ, Liu T, Khosla C, et al. Role of hypoxia-induced transglutaminase 2 in pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol. 2014;307(7):L576–85.http://dx.doi.org/10.1152/ajplung.00162.2014.
- 20. DiRaimondo TR, Klöck C, Warburton R, Herrera Z, Penumatsa K, Toksoz D, et al. Elevated transglutaminase 2 activity is associated with hypoxia-induced experimental pulmonary hypertension in mice. ACS Chem Biol. 2014;9(1):266–75.http://dx.doi.org/10.1021/cb4006408.
- 21. Wang HM, Liu WZ, Tang FT, Sui HJ, Zhan XJ, Wang HX. Cystamine slows but not inverses the progression of monocrotaline-induced pulmonary arterial hypertension in rats. Can J Physiol Pharmacol. 2018;96(8):783–9.http://dx.doi.org/10.1139/cjpp-2017-0720.
- 22. Jeitner TM, Delikatny EJ, Ahlqvist J, Capper H, Cooper AJ. Mechanism for the inhibition of transglutaminase 2 by cystamine. Biochem Pharmacol. 2005;69(6):961–70.http://dx.doi.org/10.1016/j.bcp.2004.12.011.
- 23. Palanski BA, Khosla C. Cystamine and disulfiram inhibit human transglutaminase 2 via an oxidative mechanism. Biochemistry. 2018;57(24):3359–63.http://dx.doi.org/10.1021/acs.biochem.8b00204.
- 24. Lesort M, Lee M, Tucholski J, Johnson GVW. Cystamine inhibits caspase activity: implications for the treatment of polyglutamine disorders. J Biol Chem. 2003;278(6):3825–30.
- 25. Galiè N, Channick RN, Frantz RP, Grünig E, Jing ZC, Moiseeva O, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J. 2019;53(1):1801889.
- 26. Perez Alea M, Kitamura M, Martin G, Thomas V, Hitomi K, El Alaoui S. Development of an isoenzyme-specific colorimetric assay for tissue transglutaminase 2 cross-linking activity. Anal Biochem. 2009;389(2):150–6.http://dx.doi.org/10.1016/j.ab.2009.03.029.
- 27. Sugimura Y, Hosono M, Wada F, Yoshimura T, Maki M, Hitomi K. Screening for the preferred substrate sequence of transglutaminase using a phage-displayed peptide library: identification of peptide substrates for TGase 2 and factor XIIIa. J Biol Chem. 2006;281(26):17699–706.http://dx.doi.org/10.1074/jbc.M513538200.
- 28. Wandall-Frostholm C, Skaarup LM, Sadda V, Nielsen G, Hedegaard ER, Mogensen S, et al. Pulmonary hypertension in wild type mice and animals with genetic deficit in KCa2.3 and KCa3.1 channels. PLoS One. 2014;9(5):e97687.
- 29. Thorsen LB, Eskildsen-Helmond Y, Zibrandtsen H, Stasch JP, Simonsen U, Laursen BE. BAY 41-2272 inhibits the development of chronic hypoxic pulmonary hypertension in rats. Eur J Pharmacol. 2010;647(1–3):147–54.http://dx.doi.org/10.1016/j.ejphar.2010.08.032.
- 30. Pozeg ZI, Michelakis ED, McMurtry MS, Thébaud B, Wu XC, Dyck JR, et al. In vivo gene transfer of the O2-sensitive potassium channel Kv1.5 reduces pulmonary hypertension and restores hypoxic pulmonary vasoconstriction in chronically hypoxic rats. Circulation. 2003;107(15):2037–44.http://dx.doi.org/10.1161/01.CIR.0000062688.76508.B3.
- 31. Rittié L. Method for picrosirius red-polarization detection of collagen fibers in tissue sections. Methods Mol Biol. 2017;1627:395–407.
- 32. Østergaard L, Honoré B, Thorsen LB, Baandrup J, Eskildsen-Helmond Y, Laursen BE, et al. Pulmonary pressure reduction attenuates expression of proteins identified by lung proteomic profiling in pulmonary hypertensive rats. Proteomics. 2011;11(23):4492–502.http://dx.doi.org/10.1002/pmic.201100171.
- 33. Yamaji R, Fujita K, Takahashi S, Yoneda H, Nagao K, Masuda W, et al. Hypoxia up-regulates glyceraldehyde-3-phosphate dehydrogenase in mouse brain capillary endothelial cells: involvement of Na+/Ca2+ exchanger. Biochim Biophys Acta. 2003;1593(2–3):269–76.http://dx.doi.org/10.1016/s0167-4889(02)00397-x.
- 34. Engholm M, Pinilla E, Mogensen S, Matchkov V, Hedegaard ER, Chen H, et al. Involvement of transglutaminase 2 and voltage-gated potassium channels in cystamine vasodilatation in rat mesenteric small arteries. Br J Pharmacol. 2016;173(5):839–55.http://dx.doi.org/10.1111/bph.13393.
- 35. Kourembanas S, Marsden PA, McQuillan LP, Faller DV. Hypoxia induces endothelin gene expression and secretion in cultured human endothelium. J Clin Invest. 1991;88(3):1054–7.http://dx.doi.org/10.1172/JCI115367.
- 36. Pisarcik S, Maylor J, Lu W, Yun X, Undem C, Sylvester JT, et al. Activation of hypoxia-inducible factor-1 in pulmonary arterial smooth muscle cells by endothelin-1. Am J Physiol Lung Cell Mol Physiol. 2013;304(8):L549–61.http://dx.doi.org/10.1152/ajplung.00081.2012.
- 37. Schaertl S, Prime M, Wityak J, Dominguez C, Munoz-Sanjuan I, Pacifici RE, et al. A profiling platform for the characterization of transglutaminase 2 (TG2) inhibitors. J Biomol Screen. 2010;15(5):478–87.http://dx.doi.org/10.1177/1087057110366035.
- 38. Pinilla E, Comerma-Steffensen S, Prat-Duran J, Rivera L, Matchkov VV, Buus NH, et al. Transglutaminase 2 inhibitor LDN 27219 age-dependently lowers blood pressure and improves endothelium-dependent vasodilation in resistance arteries. Hypertension. 2021 Jan;77(1):216–27.
- 39. Andersen CU, Mulvany MJ, Simonsen U. Lack of synergistic effect of molsidomine and sildenafil on development of pulmonary hypertension in chronic hypoxic rats. Eur J Pharmacol. 2005;510(1–2):87–96.http://dx.doi.org/10.1016/j.ejphar.2005.01.020.
- 40. Shinde AV, Dobaczewski M, De Haan JJ, Saxena A, Lee KK, Xia Y, et al. Tissue transglutaminase induction in the pressure-overloaded myocardium regulates matrix remodelling. Cardiovasc Res. 2017;113(8):892–905.http://dx.doi.org/10.1093/cvr/cvx053.
- 41. Soveg F, Abdala-Valencia H, Campbell J, Morales-Nebreda L, Mutlu GM, Cook-Mills JM. Regulation of allergic lung inflammation by endothelial cell transglutaminase 2. Am J Physiol Lung Cell Mol Physiol. 2015;309(6):L573–83.http://dx.doi.org/10.1152/ajplung.00199.2015.
- 42. Mandegar M, Fung YC, Huang W, Remillard CV, Rubin LJ, Yuan JX. Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvasc Res. 2004;68(2):75–103.http://dx.doi.org/10.1016/j.mvr.2004.06.001.
- 43. Hansen MS, Andersen A, Holmboe S, Schultz JG, Ringgaard S, Simonsen U, et al. Levosimendan prevents and reverts right ventricular failure in experimental pulmonary arterial hypertension. J Cardiovasc Pharmacol. 2017;70(4):232–8.http://dx.doi.org/10.1097/FJC.0000000000000508.
- 44. Laursen M, Beck L, Kehler J, Christoffersen CT, Bundgaard C, Mogensen S, et al. Novel selective PDE type 1 inhibitors cause vasodilatation and lower blood pressure in rats. Br J Pharmacol. 2017;174(15):2563–75.http://dx.doi.org/10.1111/bph.13868.
- 45. Sebkhi A, Strange JW, Phillips SC, Wharton J, Wilkins MR. Phosphodiesterase type 5 as a target for the treatment of hypoxia-induced pulmonary hypertension. Circulation. 2003;107(25):3230–5.http://dx.doi.org/10.1161/01.CIR.0000074226.20466.B1.
- 46. Drexler ES, Bischoff JE, Slifka AJ, McCowan CN, Quinn TP, Shandas R, et al. Stiffening of the extrapulmonary arteries from rats in chronic hypoxic pulmonary hypertension. J Res Natl Inst Stand Technol. 2008 Jul–Aug;113(4):239–49.http://dx.doi.org/10.6028/jres.113.018.
- 47. Ooi CY, Wang Z, Tabima DM, Eickhoff JC, Chesler NC. The role of collagen in extralobar pulmonary artery stiffening in response to hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2010;299(6):H1823–31.http://dx.doi.org/10.1152/ajpheart.00493.2009.
- 48. Su J, Logan CC, Hughes AD, Parker KH, Dhutia NM, Danielsen CC, et al. Impact of chronic hypoxia on proximal pulmonary artery wave propagation and mechanical properties in rats. Am J Physiol Heart Circ Physiol. 2018;314(6):H1264–78.http://dx.doi.org/10.1152/ajpheart.00695.2017.
- 49. Poiani GJ, Tozzi CA, Yohn SE, Pierce RA, Belsky SA, Berg RA, et al. Collagen and elastin metabolism in hypertensive pulmonary arteries of rats. Circ Res. 1990;66(4):968–78.http://dx.doi.org/10.1161/01.res.66.4.968.
- 50. Wang Z, Schreier DA, Abid H, Hacker TA, Chesler NC. Pulmonary vascular collagen content, not cross-linking, contributes to right ventricular pulsatile afterload and overload in early pulmonary hypertension. J Appl Physiol. 2017;122(2):253–63.http://dx.doi.org/10.1152/japplphysiol.00325.2016.
- 51. Nunes I, Gleizes PE, Metz CN, Rifkin DB. Latent transforming growth factor-beta binding protein domains involved in activation and transglutaminase-dependent cross-linking of latent transforming growth factor-beta. J Cell Biol. 1997;136(5):1151–63.http://dx.doi.org/10.1083/jcb.136.5.1151.
- 52. Zemskov EA, Mikhailenko I, Smith EP, Belkin AM. Tissue transglutaminase promotes PDGF/PDGFR-mediated signaling and responses in vascular smooth muscle cells. J Cell Physiol. 2012;227(5):2089–96.http://dx.doi.org/10.1002/jcp.22938.
- 53. Bernassola F, Federici M, Corazzari M, Terrinoni A, Hribal ML, DE Laurenzi V, et al. Role of transglutaminase 2 in glucose tolerance: knockout mice studies and a putative mutation in a MODY patient. FASEB J. 2002;16(11):1371–8.http://dx.doi.org/10.1096/fj.01-0689com.
- 54. Elmedal B, De Dam MY, Mulvany MJ, Simonsen U. The superoxide dismutase mimetic, tempol, blunts right ventricular hypertrophy in chronic hypoxic rats. Br J Pharmacol. 2004;141(1):105–13.http://dx.doi.org/10.1038/sj.bjp.0705580.
- 55. Zhuang R, Khosla C. Substrates, inhibitors, and probes of mammalian transglutaminase 2. Anal Biochem. 2020;591:113560. . In press.
- 56. Prat-Duran J, Pinilla E, Nørregaard R, Simonsen U, Buus NH. Transglutaminase 2 as a novel target in chronic kidney disease – Methods, mechanisms and pharmacological inhibition. Pharmacol Ther. 2021;222:107787. . In press.