Introduction
Alzheimer’s disease (AD) is the most common cause of dementia in the elderly. This pathology is characterized by a progressive impairment of cognitive functions and the presence of senile plaques and neurofibrillary tangles throughout the brain, including areas particularly involved in memory formation and emotional regulation. Plaques are composed of insoluble extracellular aggregates consisting mainly of amyloid-β (Aβ) peptides, while neurofibrillary tangles result from the aggregation of hyper- and abnormal phosphorylation of the microtubule-stabilizing protein Tau [, ]. Familial forms with known mutations of specific genes represent less than 5% of cases, while 95% of patients develop sporadic forms, for which several risk factors have been identified. Besides aging, there is growing evidence that environmental and lifestyle aspects may increase the probability to develop AD [, ]. Clinical observations suggest that stressful life events can reduce the age of onset of AD [], while stress-related disorders like depression or anxiety can promote AD symptoms and neuropathology [, ]. This view is particularly supported by the fact that AD patients demonstrated an early deregulation of the hypothalamic-pituitary-adrenal (HPA) axis (stress axis), as well as elevated levels of glucocorticoids (GC; stress hormone) in plasma and CSF, and GC receptors (GR) impairment [-].
The HPA axis, pivotal for the stress response, triggers the adrenal cortex to release GC. These steroid hormones readily cross the BBB and bind to low affinity GR and high affinity mineralocorticoid receptors (MR) []. GC are necessary for normal cellular activity and fundamental for many CNS functions, including learning and memory []. While MR are localized mainly in the hippocampus, GR are more ubiquitous and highly expressed in the limbic system (prefrontal cortex [PFC], hippocampus, and amygdala). These structures are strongly involved in cognitive and psychological functions and are also important components of the neural circuitry modulating HPA axis activity []. GC act synergistically with excitatory amino acids (like glutamate) and can induce neurotoxicity. Hence, a deregulation of the HPA axis activity and/or modification of GR functioning could be extremely toxic, especially in limbic structures [], and thus could contribute to the cognitive decline and psychological symptoms that occur in AD. In transgenic (Tg) animal models of AD, chronic stress accelerates the onset of cognitive deficits, triggers amyloid precursor protein (APP) misprocessing, enhances plaque pathology, reduces Aβ clearance, increases Aβ levels, and stimulates Tau hyperphosphorylation and its neuronal accumulation [-]. In an acute pathomimetic model of AD obtained after a single intracerebroventricular (icv) injection of an oligomeric solution of Aβ25–35 (oAβ25–35) [-], we demonstrated a strong, long-lasting activation of the HPA axis. This was associated with a modification of GR and MR expression in brain regions involved in the control of GC secretion (i.e., hippocampus, amygdala, and hypothalamus) [], supporting its involvement in the etiology of AD [, -]. We also observed that an antagonist and 2 selective GR modulators (sGRm) could potently counteract the effects of oAβ25–35 icv injection in the hippocampus and in the PFC, strongly arguing in favor of a therapeutic potential of modulating GR activity and a plausible pharmacological entry points to modify the progression of AD [, , ].
In the present study, we aimed to determine in vivo the physiological impact of a chronic GC supplementation in an AD context. For this purpose, we first established and validated a rat model of experimental chronic GC supplementation (chronic corticosterone [CORT] consumption in drinking water for 4 weeks). Such long-term exogenous GC exposure in rodents has been widely described to induce anxiety/depressive-like behaviors [-], HPA axis impairment [], memory and neurogenesis deficits, and hippocampal damages [, -]. Then, to evaluate the consequences of chronic GC consumption and to decipher the impact of HPA axis dysregulation on the onset of Aβ toxicity, animals chronically treated with GC were icv injected with oAβ25–35 (CCoAβ group). We evaluated AD-related cognitive deficits and pathogenic mechanisms, with a special emphasis on neuroinflammatory markers.
Materials and Methods
Animals
Adult male Sprague-Dawley rats (Janvier Lab., Le Genest-Saint-Isle, France) weighing 260–280 g (8 weeks) at the beginning of the experiments were housed 1 week before experiments in a standard animal facility of the University of Montpellier (CECEMA, registration No. D34-172-23) (12H/12H light/dark cycle with lights on at 07H00; 21 ± 1°C, food and water ad libitum). All experiments, including sacrifices, were performed in conscious rats between 09H00 and 12H00, during the diurnal trough of the HPA axis circadian rhythm. Male has been preferred to female because the latter may present a confounding factor of the estrous cycle, a parameter that needs to be systematically controlled. Although unlikely, a possibility still exists that mixing males and females in each experimental group may introduce a variable difficult to account for. Furthermore, doubling the number of groups and rats goes beyond the 3R rule. We do recognize the prospective importance of using both genders, and results will need to be verified in female.
Amyloid-β Peptide
In patients, soluble Aβ oligomers contain mainly the sequences Aβ1–40 and Aβ1–42 []. Nevertheless, they also contain peptides with shorter sequences such as Aβ25–35 or Aβ25–35/40 [-], with no difference between human and rodent []. These shorter peptides can be produced in AD patients by enzymatic cleavage of Aβ1–40 [, ]. They contain extracellular and transmembrane residues that have been reported to be a biologically active region of Aβ [-] and to contain the highly hydrophobic region forming stable aggregates []. Interest in this undecapeptide, which itself shows a β-sheet structure [, ], has grown over the last 15 years, mainly because it induces neurite atrophy, neuronal cell death, synaptic loss, as well as synaptic plasticity and memory deficits in a similar way to Aβ1–40 and Aβ1–42 [], but with better solubility and efficiency []. Aβ25–35 and scrambled Aβ25–35 peptides (Eurogentec, Angers, France) were dissolved in sterile water (1 µg/µL) and stored at −20°C. Since soluble Aβ oligomers correlate better with the progression of the disease [], Aβ25–35 and scrambled peptides were pre-aggregated by an in vitro incubation at 37°C (4 days) to obtain a solution mainly composed (more than 95%) of a mixture of soluble oligomer species (oAβ25–35), as previously characterized [].
Experimental Procedures
First, to validate the chronic CORT consumption model, animals received drinking water containing 1, 5, or 10 mg/kg CORT (Sigma-Aldrich, Lyon, France) dissolved in 0.45% β-cyclodextrin (Sigma-Aldrich, Lyon, France) [] for 4 weeks (online suppl. Fig. 1a; for all online suppl. material, see http://www.karger.com/doi/10.1159/000521559). A control cohort received the vehicle alone (0.45% β-cyclodextrin) in drinking water. All animals were weighed once per week. The general behavior or anxiety state of the different groups of rats was tested at the end of the treatment (after 4 weeks) in an open field or in an elevated plus maze (EPM) test, respectively. Animals were sacrificed by decapitation. Pituitary and adrenals were rapidly harvested to be weighed and blood samples were collected for CORT assay.
Then, a new cohort of rats received drinking water containing the effective dose of CORT (10 mg/kg) or the vehicle (β-cyclodextrin) for 5 weeks. At week 4, animals were divided into 3 groups. The first groups had no surgery (control groups), the second received an icv injection of incubated scrambled peptide (10 µg/rat; scrambled groups), and the third received an icv injection of oAβ25–35 (10 µg/rat; oAβ groups) (Fig. 1a). The animals were anesthetized with an intraperitoneal (ip) injection of 1 mL of a mixture of ketamine and xylazine (80 and 10 mg/kg b.w., respectively). As previously described [, ], oAβ25–35 was injected directly into the lateral ventricles using a David-Kopf stereotaxic apparatus (Phymep, Paris, France) (coordinates: AP −1 mm, L ±1.5 mm, DV −3.5 mm) []. All animals were weighed before, 4 weeks after the treatments, and 1 week after the icv injection of oAβ (week 5) (Fig. 1a). Short-term memory was tested in a T-maze 1 week after the icv injection of oAβ (Fig. 1a). Then, animals were sacrificed by decapitation. Blood samples and hippocampi were rapidly collected during the diurnal part of the HPA axis rhythm (between 09H00 and 12H00) for CORT assay and WB analysis. Naive rats received no treatment but were manipulated in the same manner as treated rats.
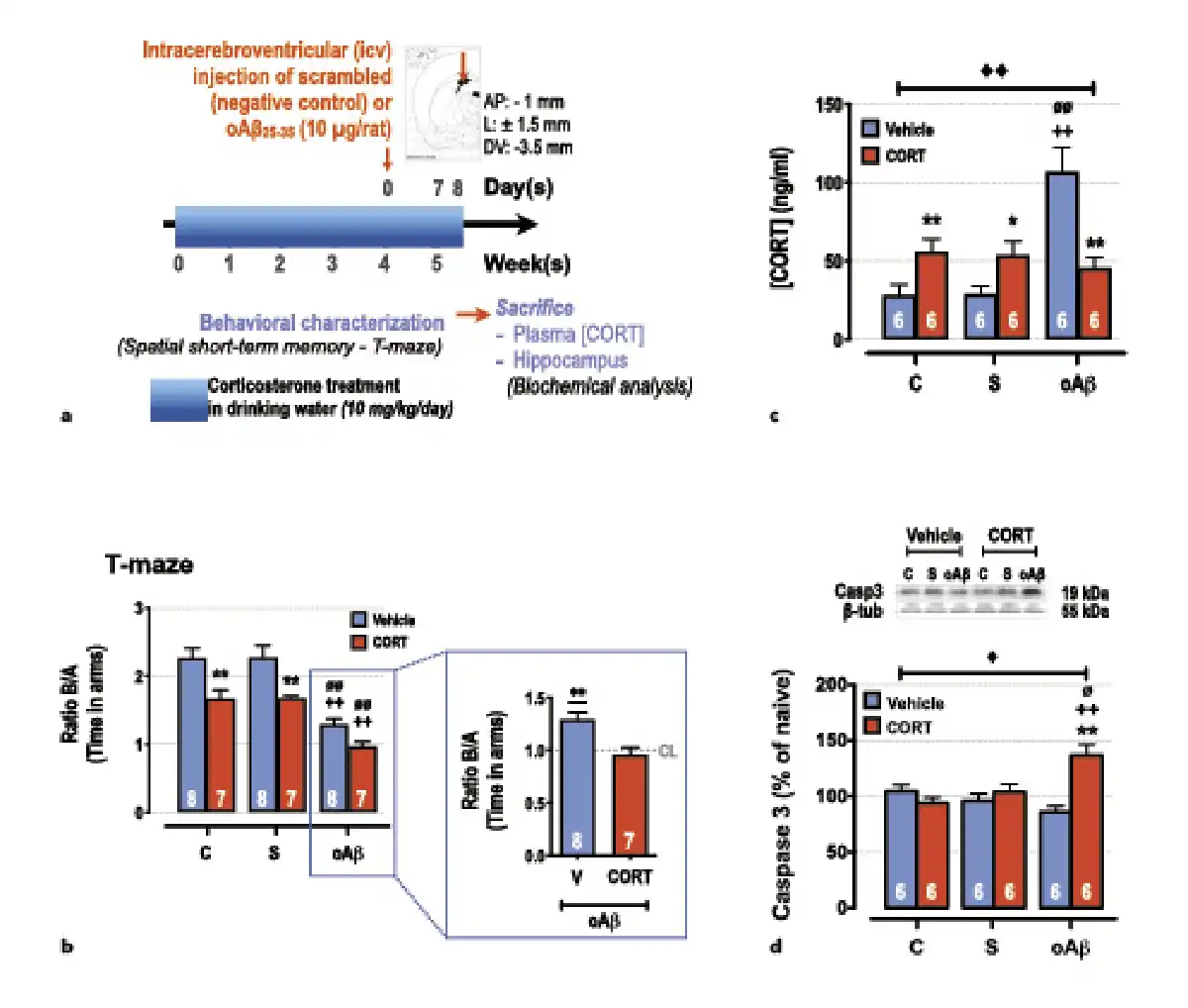
Fig. 1
Impact of chronic corticosterone (CORT) consumption on oAβ25-35-induced toxicity. a Experimental protocol – At T0, adult male rats (Sprague-Dawley) were treated with CORT in drinking water (10 mg/kg/day, dissolved in 0.45% β-cyclodextrine) for 5 weeks (CORT animals, red columns) and compared to a second group treated only with β-cyclodextrine (vehicle animals, blue columns). At week 4, animals received any icv injection (control [C] group), an icv injection of oAβ25–35 (10 µg/rat, “oAβ” group), or an icv injection of scrambled control (“S” group). The last day before sacrifice (day 34) the spatial short-term memory of each rat was tested in a T-maze. The following day (day 35), the animals were sacrificed between 9H00 and 12H00 (during the diurnal trough of the HPA axis circadian rhythm), blood samples, and hippocampi were rapidly collected for CORT assay and Western blot analysis, respectively. b Spatial short-term memory performance was determined in a T-maze test and was expressed as the ratio of the time spent in the initially closed arm (B) over the time spent in the previous arm (A). Two-way ANOVA: F2, 39 = 44.7 for group, p < 0.0001; F1, 39 = 36.4 for treatment, p < 0.0001; and F2, 39 = 1.09 for interaction, ns. In addition, all data were analyzed in comparison to chance level (CL), i.e., maximal deficit, using a one-sample t test. All data are different to the CL, except rats co-treated with CORT and oAβ. c Plasma concentrations of CORT were determined by using ELISA and expressed as ng/mL. Two-way ANOVA: F2, 30 = 9.48 for group, p < 0.001; F1, 30 = 0.15 for treatment, ns; and F2, 30 = 15.3 for interaction, p < 0.0001. d Variations of apoptosis hallmark (caspase-3, 19 kDa) were evaluated in each group and normalized with the variations of β-tubulin (β-tub, 55 kDa) and compared with non-injected rats (control group: C). Two-way ANOVA: F2, 30 = 1.99 for group, ns; F1, 30 = 8.45 for treatment, p < 0.01; and F2, 30 = 11.0 for interaction, p < 0.001. Data are expressed as means ± SEM in percent of naive rats. *p < 0.05 and **p < 0.01 versus respective group treated with vehicle. ++p < 0.01 versus respective C group. ∅p < 0.05 and ∅∅p < 0.01 versus respective scrambled (S) group, ◆p < 0.05 and ◆◆p < 0.01 versus selected group, **p < 0.01 versus CL.
General Behavior
The open-field test was used to characterize general behavior of rats, as previously reported []. The apparatus consists of a squared open-field (1 m2 × 0.6 m high) made of white PVC with an infrared light emitting floor. An infrared sensitive CCD camera was placed above the field and connected to a video tracking system (Noldus EthoVisonXT, France). Animals were allowed to freely explore the open-field during 10 min. This test evaluates locomotion-related parameters, such as the total distance travelled or walking speed; exploratory-related parameters, including locomotion, immobility, and number of rearings; anxiety-related indexes, such as latency to start and thigmotaxis (locomotion along the walls); and stereotyped responses, like rearing or grooming.
Spatial Short-Term Memory
As previously reported, the T-maze test was used to rapidly assess cognitive ability in rats, especially the short-term memory deficits when performed in two successive sessions [-, , ]. The T-maze consisted of two short arms (A and B), extending from a longer alley (C) and enclosed with high walls. The test involved two trials separated by 1 h. During the training session, one short arm (B) was closed. Rats were placed at the end of the long alley, allowed to visit the maze for 10 min, and then returned into their home cage. During the test session, which was video-tracked (Noldus EthoVisonXT, France), animals were placed in the maze for 2 min, with free access to all arms. The number of visits and time spent in each arm were measured. The results were expressed as ratio of the time spent in the initially closed novel arm, over the time spent in the previous arm and as a ratio of the number of entries into the novel arm over the familiar one. The apparatus was cleaned with diluted ethanol (50%) between animals.
Anxiety Behavior
The anxiety state of rats was measured using their ability to explore open and enclosed arms of an EPM, as previously detailed []. The clear plexiglass apparatus consisted of 2 open arms (50 × 10 cm) and 2 enclosed arms (50 × 10 × 45 cm high), extending from a central platform and placed 60 cm above the floor. Each rat was placed at the center of the EPM facing the closed arm and its exploration behavior was recorded for 10 min (Noldus EthoVisonXT, France). The results were expressed as total time spent in the open arms and the total number of entries was counted to verify general motor activity. An entry into an arm was recorded if the animal crossed the line that connected that arm with the central platform with all 4 legs. The apparatus was cleaned with diluted ethanol (50%) between animals.
CORT Assay
Blood samples were collected at the time of sacrifice on 1 mg/mL EDTA (Sigma-Aldrich, France), centrifuged at 4°C, and plasma stored at −20°C until assayed for CORT []. Plasma CORT concentrations were assayed using a conventional ELISA kit (Enzo-Life Sciences, Farmingdale, NY, USA) in a 10 µL plasma sample diluted (1:40) with the assay buffer. The assay sensitivity was 27 pg/mL. The intra- and inter-assay coefficients were 6.6 and 7.8%, respectively.
WB Analysis
WB was performed as previously described [] in the whole hippocampus. All antibodies used are detailed in the online Supplementary Table 2. Briefly, after sacrifice, hippocampi were microdissected, weighed, immediately frozen in liquid nitrogen and stored at −20°C. Tissues were sonicated (VibraCell; Sonics & Materials, Newtown, CT, USA) in a lysis buffer [] and centrifuged (4°C). Supernatants were collected and the protein concentration was measured using a BCA kit (ThermoFisher Scientific, Illkirch-Graffenstaden, France). Sixty microgram from each sample was taken for WB analysis. Samples were separated in SDS-polyacrylamide gel (12%) and transferred to a PVDF membrane (Merck-Millipore, Molsheim, France). The membrane was incubated overnight (4°C) with the primary antibody, washed and then incubated for 2 h with the appropriate horseradish peroxidase-conjugated secondary antibody. Peroxidase activity was revealed by using enhanced-chemiluminescence reagents (Luminata-Crescendo, Merck-Millipore, Burlington, MA, USA). The intensity of peroxidase activity was quantified using Image-J software (NIH, Bethesda, MA, USA). β-tubulin was used as a loading control for all immunoblotting experiments.
Statistical Analysis
Data are presented as mean ± SEM and analyzed using one-way or 2-way ANOVA followed by a Tukey’s multiple comparisons test (GraphPad-Prism 9.0). In addition, T-maze data are analyzed in comparison to chance level, i.e., maximal deficit, using a one-sample t test (GraphPad-Prism 9.0). p < 0.05 was considered significant. The number of animals in each group is indicated within the columns and was determined by a statistical power analysis based on our previous studies (G*Power software). Before each analysis of variance, the Gaussian distribution was evaluated and validated by a Kolmogorov-Smirnov test (GraphPad-Prism 9.0).
Results
Validation of the Chronic CORT Consumption Model
To validate this model (online suppl. Fig. 1a), we assessed relevant parameters associated with chronic CORT treatment in drinking water [, , ]. The daily dose of 10 mg/kg of CORT was the sole dose that significantly increased the plasma concentrations of CORT, while the vehicle (0.45% β-cyclodextrin) and the doses of 1 and 5 mg/kg induced no modification in comparison to control rats (online suppl. Fig. 1b). The dose of 10 mg/kg/day was also the most efficient to atrophy adrenal glands (online suppl. Table 1) and to induce anxiety-like behavior. Indeed, in the open-field test (online suppl. Fig. 1c), we identified several characteristic features of anxiety, such as the increase of the latency to start moving and of the total immobility time. We also observed a decrease of visits in the arena center and the number of grooming and rearing, while locomotion was not affected by the different doses. To confirm these general observations, we performed a well-defined anxiety test, the EPM. Animals treated with the two highest doses of CORT (5 and 10 mg/kg/day) displayed a decrease of the time spent in the open arms, characteristic of an anxious state (online suppl. Fig. 1d).
Impact of Chronic CORT on oAβ Toxicity
We next evaluated the impact of chronic CORT consumption (10 mg/kg/day) on the Aβ toxicity 1 week after the icv injection of oAβ25–35 (CCoAβ group) (Fig. 1a). Five weeks of chronic CORT consumption in drinking water induced short-term memory deficits (T-maze test) and potentiated significantly the oAβ25–35-induced alterations of memory performances (Fig. 1b). No effects of scrambled peptide nor vehicle treatment were evidenced (Fig. 1-4). One week after the icv injection of oAβ25–35, an increase of plasma CORT concentrations was observed, which was partially blunted by the upstream chronic CORT consumption (Fig. 1c). In addition, an additive effect of CCoAβ treatment was evidenced on body weight (Table 1) and on the hippocampal expression of cleaved-caspase 3, while the two treatments alone did not impact the later parameter (Fig. 1d).
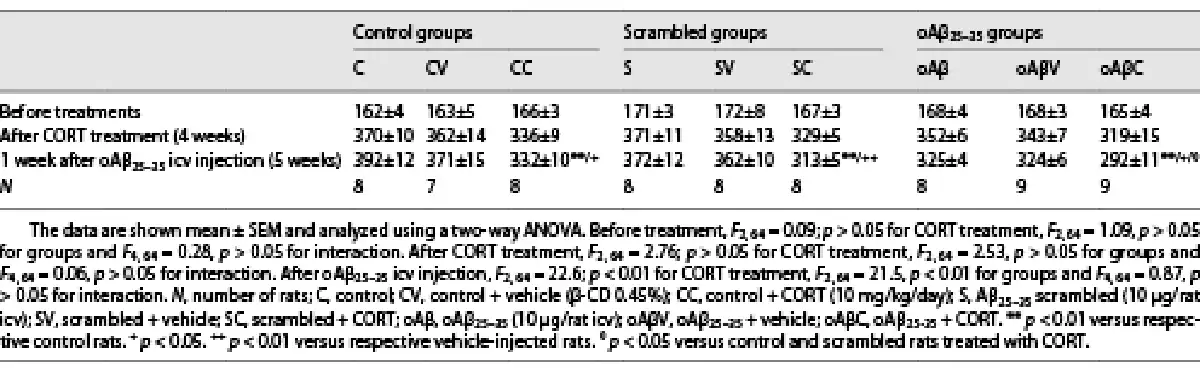
Fig. 2
The cumulative effects in the hippocampus of chronic CORT consumption and oAβ25–35 injection on GR signaling pathways (a), were evaluated by Western blot. Variations of the expression of GR (95 kDa) (b, c), the phosphorylation of GR (p[Ser211]GR, 95 kDA) (b, d), the expression ratio of HSP90/HSP70 (90 kDa/70 kDa) (b, e), ANNEXIN A1 (35 kDA) (b, f), Fyn (59 kDa) (b, g), the activation of GSK-3β (ratio of p[Tyr216]GSK-3β/GSK-3β total, 46 kDa) (h, i), the Cdk5 (30 kDa) pathways (h, j), and the expression ratio of p25/p35 (25 and 35 kDa) (h, k) were evaluated in each group and normalized with the variations of β-tubulin (β-tub, 55 kDa). For experimental protocol see Fig. 1a. Two-way ANOVA: GR: F2, 30 = 3.71 for group, p < 0.05; F1, 30 = 20.8 for treatment, p < 0.001; and F2, 30 = 2.63 for interaction, ns; pGR: F2, 30 = 4.48 for group, p < 0.05; F1, 30 = 21.5 for treatment, p < 0.0001; and F2, 30 = 3.98 for interaction, p < 0.05; HSP90/HSP70: F2, 30 = 10.7 for group, p < 0.001; F1, 30 = 7.23 for treatment, p < 0.001; and F2, 30 = 0.53 for interaction, ns; annexin A1: F2, 30 = 17.4 for group, p < 0.0001; F1, 30 = 43.8 for treatment, p < 0.0001; and F2, 30 = 1.06 for interaction, ns; Fyn: F2, 30 = 11.1 for group, p < 0.001; F1, 30 = 15.9 for treatment, p < 0.001; and F2, 30 = 0.83 for interaction, ns; p[Tyr216]GSK-3β/GSK-3β: F2, 30 = 18.4 for group, p < 0.0001; F1, 30 = 13.8 for treatment, p < 0.001; and F2, 30 = 1.95 for interaction, ns; Cdk5: F2, 30 = 3.38 for group, p < 0.05; F1, 30 = 40.3 for treatment, p < 0.0001; and F2, 30 = 2.75 for interaction, ns; p25/p35: F2, 30 = 19.5 for group, p < 0.0001; F1, 30 = 56.5 for treatment, p < 0.0001; and F2, 30 = 1.21 for interaction, ns. Data are expressed as means ± SEM in % of naive rats. *p < 0.05 and **p < 0.01 versus respective group treated with vehicle. +p < 0.05 and ++p < 0.01 versus respective control (C) group. ∅p < 0.05 and ∅∅p < 0.01 versus respective scrambled (S) group, ◆◆p < 0.01 versus selected group.
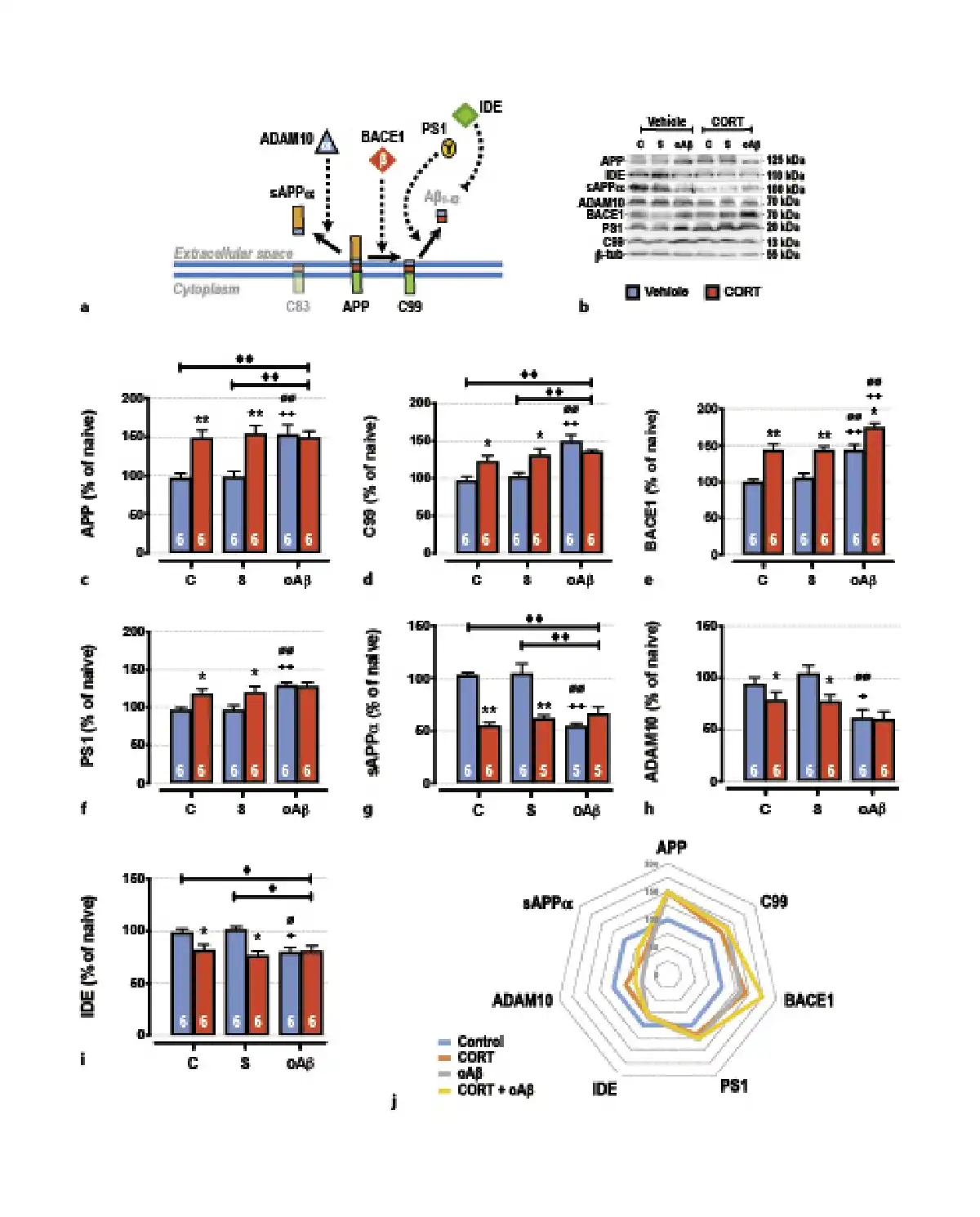
Fig. 3
The cumulative effects in the hippocampus of chronic CORT consumption and oAβ25–35 injection on APP processing (a), were evaluated by Western blot. Variations of the expression of APP (120 kDa) (b, c), C99 (13 kDa) (b, d), BACE1 (β-secretase, 70 kDa) (b, e), PS1 (γ-secretase, 20 kDa) (b, f), sAPPα (100 kDa) (b, g), Adam10 (α-secretase, 70 kDa) (b–h), IDE (110 kDa) (b, i) were evaluated in each group and normalized with the variations of β-tubulin (β-tub, 55 kDa). For experimental protocol, see Fig. 1a. Two-way ANOVA: APP: F2, 30 = 5.14 for group, p < 0.01; F1, 30 = 19.5 for treatment, p < 0.0001; and F2, 30 = 5.98 for interaction, p < 0.01; C99: F2, 30 = 11.6 for group, p < 0.001; F1, 30 5.34 for treatment, p < 0.05; and F2, 30 = 5.48 for interaction, p < 0.01; BACE1: F2, 30 = 16.7 for group, p < 0.0001; F1, 30 = 41.3 for treatment, p < 0.0001; and F2, 30 = 0.34 for interaction, ns; PS1: F2, 30 = 7.85 for group, p < 0.01; F1, 30 = 8.75 for treatment, p < 0.01; and F2, 30 = 2.63 for interaction, ns; sAPPα: F2, 27 = 9.56 for group, p < 0.001; F1, 27 = 35.8 for treatment, p < 0.0001; and F2, 27 = 18.5 for interaction, p < 0.0001; ADAM10: F2, 30 = 9.52 for group, p < 0.001; F1, 30 = 5.81 for treatment, p < 0.05; and F2, 30 = 1.38 for interaction, ns; IDE: F2, 30 = 3.14 for group, p < 0.05; F1, 30 = 14.3 for treatment, p < 0.001; and F2, 30 = 4.54 for interaction, p < 0.05. j Radar chart displaying the profile of APP processing (non-amyloidogenic pathway; sAPPα, ADAM10, and IDE, vs. amyloidogenic pathway; C99, BACE1, and PS1) occurring in each experimental condition (control, treated with CORT, treated with oAβ, or treated with CORT and oAβ). Data are expressed as means ± SEM in % of naive rats. *p < 0.05 and **p < 0.01 versus respective group treated with vehicle. +p < 0.05 and ++p < 0.01 versus respective control (C) group. ∅p < 0.05 and ∅∅p < 0.01 versus respective scrambled (S) group, ◆p < 0.05 and ◆◆p < 0.01 versus selected group.
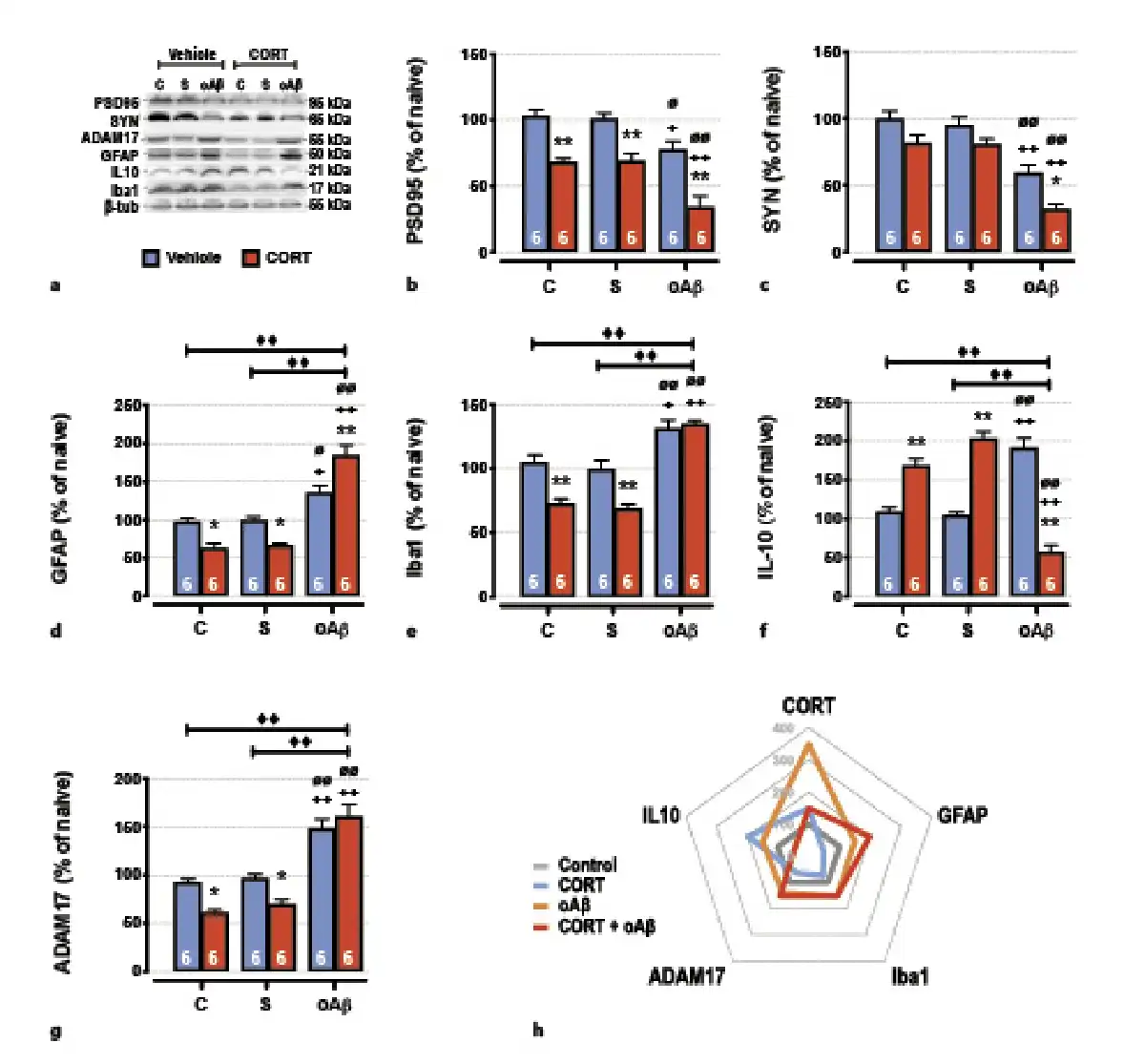
Fig. 4
The cumulative effects in the hippocampus of chronic CORT consumption and oAβ25–35 injection on synaptic deficits and neuroinflammation were evaluated by Western blot. Variations of the expression of PSD95 (95 kDa) (a, b), synaptotagmine (SYN, 65 kDA) (a, c), GFAP (50 kDa) (a, d), Iba1 (21 kDa) (a, e), IL-10 (25 kDa) (a, f), and ADAM17 (55 kDa) (a, g) were evaluated in each group and normalized with the variations of β-tubulin (β-tub, 55 kDa). For experimental protocol see Fig. 1a. Two-way ANOVA: PSD95: F2, 30 = 18.8 for group, p < 0.0001; F1, 30 = 66.7 for treatment, p < 0.0001; and F2, 30 = 0.60 for interaction, ns; SYN: F2, 30 37.8 for group, p < 0.0001; F1, 30 16.7 for treatment, p < 0.001; and F2, 30 = 0.37 for interaction, ns; GFAP: F2, 30 = 69.5 for group, p < 0.0001; F1, 30 = 1.99 for treatment, ns; and F2, 30 = 19.0 for interaction, p < 0.0001; Iba1: F2, 30 = 55.9 for group, p < 0.0001; F1, 30 = 22.9 for treatment, p < 0.0001; and F2, 30 = 7.80 for interaction, p < 0.01; IL-10: F2, 30 = 5.84 for group, p < 0.01; F1, 30 = 1.20 for treatment, ns; and F2, 30 = 100 for interaction, p < 0.0001; ADAM17: F2, 30 = 67.4 for group, p < 0.0001; F1, 30 = 6.49 for treatment, p < 0.05; and F2, 30 = 5.43 for interaction, p < 0.01. h Radar chart displaying the inflammatory profile (anti-inflammatory mediators; IL-10 and ADAM-17 vs. pro-inflammatory mediators; GFAP, Iba1) occurring in each experimental conditions (control, treated with CORT, treated with oAβ or treated with CORT, and oAβ) and in relation with plasma CORT levels. Data are expressed as means ± SEM in % of naive rats. *p < 0.05 and **p < 0.01 versus respective group treated with vehicle. +p < 0.05 and ++p < 0.01 versus respective control (C) group. ∅p < 0.05 and ∅∅p < 0.01 versus respective scrambled (S) group, ◆◆p < 0.01 versus selected group.
Impact on GR Signaling Pathways
As previously reported in the PFC [], the icv injection of oAβ25–35 altered GR-associated signaling pathways in the hippocampus (Fig. 2a). Indeed, 1 week after oAβ25–35, the expression of total and activated GR (phosphorylated on Ser211) was increased [] (Fig. 2b–d) and was associated with an increase of heat-shock protein 90/70 (HSP90/70) ratio (Fig. 2b, e), two GR chaperones reflecting the activity of GR and highly involved in AD pathophysiology [-] and an increase of annexin A1 (a potent endogenous anti-inflammatory effector functioning in synergy with GR) [, ] (Fig. 2b, f). We also observed the increased expression of the Src-kinase Fyn (Fig. 2b, g), the active form of GSK3β (phosphorylated on Tyr216) (Fig. 2h, i), the total form of Cdk5 (Fig. 2h, j), and the p25/p35 ratio (associated with the activation of Cdk5) [] (Fig. 2h, k). These three enzymes are particularly relevant since they are involved in GR activation but also in the hyperphosphorylation of Tau [-]. Chronic CORT consumption induced a similar effect as oAβ25–35 on all of these markers, except on annexin A1 (Fig. 2b, f) and the p25/p35 ratio where we evidenced an additive effect of CCoAβ treatments (Fig. 2h, k).
Impact on APP Metabolism
Then, we characterized by Western blot the different pathways of APP maturation, through the assessment of the main cellular elements involved in the amyloidogenic and non-amyloidogenic pathways (Fig. 3a). One week after oAβ25–35 injection and as previously reported [, ], we evidenced the induction of the amyloidogenic pathway in the hippocampus (Fig. 3j), consistent with an Aβ1–42 increase, as previously reported by our group [, , ]. We observed an increased expression of both full-length APP (precursor of amyloid proteins) (Fig. 3b, c), C99 (precursor of Aβ peptides) (Fig. 3b, d), BACE1 (β-APP cleaving enzyme, β-secretase) (Fig. 3b, e), and PS1 (presenilin-1, γ-secretase subunit) (Fig. 3b, f). This induction is associated with a concomitant inhibition of non-amyloidogenic pathways (Fig. 3j), characterized by a decrease of sAPPα (α-secretase-cleaved soluble APP ecto-domain) (Fig. 3b, g), α-secretase ADAM10 (A disintegrin and metalloprotease domain-containing protein 10) (Fig. 3b, f), and IDE (insulin-degrading enzyme, involved in the clearance of Aβ) (Fig. 3b, g). Interestingly, chronic CORT consumption induced the same modifications as those observed 1 week after the icv injection of oAβ25–35 (Fig. 3), and CCoAβ treatment induced no supplementary effects, except for BACE1 where we showed a significant additive effect (Fig. 3b, e).
Impact on Synaptic Deficits and Neuroinflammation
Decreased memory performances and GR signaling pathway alterations observed 1 week after the icv injection of oAβ25–35 were associated in the hippocampus with pre- (synaptotagmine) and postsynaptic deficits (Fig. 4a–c). These deficits were potentiated by the prior overexposure to exogenous CORT in animals injected with oAβ25–35 (Fig. 4a–c). Moreover, and as previously reported [, ], synaptic deficits induced by the Aβ toxicity were associated with important modifications of hippocampal inflammatory processes. Indeed, after 1 week, the icv injection of oAβ25–35 provoked both the induction of pro-inflammatory mechanisms (activation of astrocytes [GFAP] and recruitment of microglia [Iba1]) (Fig. 4a, d, e) and the induction of anti-inflammatory components, as interleukin-10 (IL-10) (Fig. 4a, e, f). By contrast, while chronic CORT favored anti-inflammatory pathways (Fig. 4g) by inhibiting astrocytes activation (Fig. 4a, d), microglia recruitment (Fig. 4a, e), and IL-10 induction (Fig. 4a, f), the CCoAβ group displayed exacerbated neuroinflammation (Fig. 4g). Indeed, co-treatment increased the activation of astrocytes and the recruitment of microglia (Fig. 4a, d, e) and inhibited IL-10 (Fig. 4a, f). In addition, ADAM17, another α-secretase (also known as tumor necrosis factor-α converting enzyme or TACE), highly activated in neuroinflammatory processes and promoting pro-inflammatory cytokines maturation in turn (for review, see []), was upregulated after the icv injection of oAβ25–35 (Fig. 4a, g). And although chronic CORT inhibited the expression of this metalloprotease, as previously observed with ADAM10 (Fig. 3b, f), the CCoAβ treatment re-established the hippocampal increase of ADAM17 (Fig. 4a, g).
Discussion
In previous studies, we provided evidence for a vicious cycle between AD and the HPA axis. We showed that the pathology, and especially the Aβ toxicity, rapidly increases GC secretion which, in turn, modulates APP processing, Aβ1–42 synthesis, and Tau phosphorylation [-]. Here, in order to characterize the repercussion of chronic GC consumption on AD pathophysiology, we treated animals with CORT in drinking water upstream of the icv injection of oAβ25–35. We reported that chronic CORT consumption in the oAβ25–35 model exacerbated cognitive decline, amyloid pathology (Fig. 3j), and neuroinflammation (Fig. 4h), notably in close association with an enhanced activity of GR signaling pathways. Many biochemical AD hallmarks impaired by oAβ25–35 injection were equivalently impaired by chronic GC treatment. We could hypothesize that in AD, the deregulation of these intracellular pathways could be due to GC overexposure and HPA axis impairment induced by Aβ toxicity, more than Aβ toxicity itself. This not only implies a specific susceptibility to stress in AD but also brings new information on how chronic GC consumption could be an important risk factor for AD.
Model of Chronic GC Consumption
First, we aimed to validate our chronic GC consumption model, well known to be a model of chronic anxiety/depression [, ] associated with memory deficits and hippocampal alterations [, ]. According to the literature, in our study, we focused on the anxious state displayed in animals treated for 4 weeks with CORT in drinking water. At the optimal dose determined (i.e., 10 mg/kg/day), animals developed many anxiety-like behaviors in the open field and EPM tasks. At this dose, we also evidenced an increase of GC plasma levels associated with an atrophy of adrenal glands. These results are in accordance with studies showing that chronic CORT administration suppressed the inhibitory feedback of GC on the HPA axis activity [, ], characteristic of mood disorders such as generalized anxiety or major depression [, ]. In accordance to previous works conducted by others groups, we validated this model as a chronic model of GC overexposure, displaying HPA axis impairment and anxiety-like behaviors.
Synergistic Effects
Second, we compared the effects of chronic GC consumption alone or in combination with the icv injection of oAβ25–35. CORT treatment caused short-term memory and synaptic deficits in the hippocampus, with no modification on caspase 3 levels. These alterations following GC overexposure are widely described in the literature. It was shown that chronic stress or GC administration altered synaptic terminal structure, neurogenesis, and induced neuronal atrophy, more particularly in the hippocampus [, , -]. Since CORT treatment also induced anxiety-like behaviors in the EPM, we could not totally exclude this emotional aspect in the T-maze performance. However, as reported by others, CORT treatment induced many hippocampal alterations (dendritic retraction, decreased cell proliferation, and global volume) [, ] sufficient to induce such short-term memory deficits. In addition, Han et al. [] also reported an aggravation of cognitive impairment in APP/PS1 mice submitted to chronic unpredictable stress. By contrast, it is interesting to note that CCoAβ group displayed exacerbated memory and synaptic deficits. Furthermore, we also observed a synergistic effect of this co-treatment that induced caspase 3 expression in the hippocampus, whereas the two treatments alone did not impact it. This suggested that the potentiation of synaptic and memory deficits could be due to the induction of apoptosis processes.
Additive Effects
Moreover, we evidenced some additive effects of the exogenous GC treatment and the icv injection of oAβ25–35. Indeed, we showed that the induction of BACE1 was potentiated. This enzyme was identified as the major β-secretase-like protein, highly involved in the amyloidogenic cleavage of APP, and thus in Aβ peptides production []. These findings are consistent with the study conducted by Green et al. []. The authors have shown that 1-week treatment of dexamethasone in 4-month-old 3xTg mice significantly increased BACE1 expression and higher levels of Aβ1–40 and Aβ1–42. Our result was in accordance with the literature since BACE1 production could be triggered by chronic stress as well as by Aβ. In fact, BACE1 transcription is under a direct control of GC since a glucocorticoid response element has been described in the promoter region of BACE1 []. In addition, BACE1 production could be also indirectly regulated by stress and Aβ through the activation of other intracellular mediators. (i) Both chronic GC overexposure [, ] and Aβ toxicity [, ] induce the c-jun N-terminal kinase/activator protein 1 (JNK) pathway (or stress-activated kinase), one of the main pathways favoring BACE1 production []. (ii) The p25/Cdk5 pathway, upregulated in our study, was also known to trigger BACE1 transcription []. This complex of enzymes is highly involved in AD pathology, particularly for its action on Tau hyperphosphorylation [, ], but also on GR activation via phosphorylation on Ser211 in rodent (Ser224 in human) []. Some studies demonstrated that chronic unpredictable stress [, ], as well as Aβ pathology [, ] increased p25/Cdk5 expression and activity. (iii) These mechanisms could be due to an increased activity of calpain-1, a member of cysteine proteases family showing aberrant activity in AD []. This enzyme is particularly involved in the activation of GSK-3β [] and the maturation of p35 to p25 []. Interestingly, we demonstrated recently in the oAβ25–35 model that all these enzymes (i.e., GSK3-β, p25/Cdk5, and calpain-1) were directly or indirectly regulated by GC and GR in the prefrontal cortex []. Our results suggest that calpain-1 activity may also be potentiated by chronic GC overexposure since the hippocampal expression of 2 of its principal substrates was increased. So, by these different mechanisms, it was not surprising that combined toxicity of chronic CORT consumption and oAβ25–35 injection had additive effects on the expressions and/or activities of BACE1 and p25/Cdk5. Since these 2 enzymes are highly involved in amyloid and tau pathologies, it brings new data on how chronic GC consumption is detrimental in AD development. However, further specific investigations are needed to characterize precisely tau system, in particular to assess the levels of tau hyperphosphorylation epitopes, its eventual aggregation, its localization and its staining pattern.
Effects on APP Metabolism and GR Signaling Pathways
On the principal proteins associated with GR signaling pathways (i.e., GR, p[Ser211]GR, HSP90/HSP70, GSK3-β, Cdk5, and Fyn) and APP metabolism (i.e., APP, C99, IDE, sAPPα, ADAM10, and PS1) assessed in our study, there was no additive effect of chronic CORT on the Aβ toxicity. Interestingly, in 3xTg mice, 1-week treatment with dexamethasone was able to induce an increase of APP and C99 levels []. However, the authors used 4-month-old 3xTg animals before the establishment of important AD-like symptoms. Thus, dexamethasone could potentiate the expression of these markers. In our study, the modifications induced by each treatment (chronic CORT or oAβ25–35) were closely similar. This suggests common regulatory mechanisms and supports the important role played by GR in the pathophysiology of AD (for review, see [, , , ]). Indeed, we recently demonstrated in this acute model of AD (oAβ25–35) that these pathways were impaired [] and associated with a strong and long-lasting disruption of the HPA axis activity and functionality []. Then, to evaluate the contribution of GR in the Aβ toxicity, we tried to restore their functionality using new selective GR modulators (sGRm) [, ]. This family of compounds has the advantage to selectively abrogate pathological effects of GR overactivation, while retaining their physiological function [, ]. By restoring their functionality with sGRm, we counteracted all parameters measured and induced by the Aβ toxicity, highlighting the central role played by GR in the development of AD []. Taken together, all of these results highly suggest that a part of Aβ toxicity could be a consequence of the GC overexposure and GR signaling impairment.
However, the 2 α-secretases ADAM10 and ADAM17 are differentially regulated in the hippocampus by chronic CORT consumption and oAβ25–35. We found that Aβ toxicity increased ADAM17 levels while inhibiting ADAM10 expression. By contrast, chronic CORT decreased the two α-secretases. These proteases are neuroprotective by triggering the non-amyloidogenic pathway and the generation of sAPPα. But the precise role of ADAMs in AD pathogenesis is not fully understood. ADAM10 seems to constitutively cleave APP while fine regulation is mediated by ADAM17 []. In the CSF of AD patients, ADAM10 is decreased [], which is correlated with the inhibition of the non-amyloidogenic pathway. Inversely, ADAM17 activity is increased both in early and advanced phases of AD [] that evidences more complex regulatory mechanisms. In fact, besides its neuroprotective role, ADAM17 is also highly activated by neuroinflammatory processes and promotes pro-inflammatory cytokines maturation in turn (for review, see []). These findings are relevant with our results in oAβ and CCoAβ groups. In these animals, we found decreased ADAM10 and sAPPα expressions and increased ADAM17 expression concomitantly with enhanced neuroinflammatory processes (Fig. 4h). So, we could imagine that ADAM10 participated to APP non-amyloidogenic cleavage more than ADAM17 that could be regulated by pro-inflammatory processes. To summarize, we found that both oAβ and chronic CORT consumption altered APP processing balance by increasing the amyloidogenic (C99, BACE1, and PS1) and inhibiting the non-amyloidogenic (ADAM10 and IDE) pathways (Fig. 3j). It is interesting to note that the main difference observed between chronic CORT and oAβ groups is their inflammatory phenotypes. Such difference could participate to explain the exacerbation of AD-related markers in CCoAβ animals (Fig. 4h).
Effects on Inflammatory Processes
Neuroinflammation is a key feature of AD pathology. Activated microglia and astrocytes trigger the overproduction of pro-inflammatory cytokines, which promotes Aβ accumulation and thus neurodegeneration []. As we already observed [-], we confirmed in this study that oAβ25–35 induced neuroinflammation, since it increased GFAP and Iba1, but also ADAM17, which is highly activated by neuroinflammatory processes and, as discussed above, promotes pro-inflammatory cytokines maturation in turn (for review, see []) (Fig. 4h). Inversely, chronic CORT consumption favored the anti-inflammatory pathways (Fig. 4h). It was well known that stress and GC influence substantially the level and the quality of the immune response. The activation of GR-annexin A1 pathway inhibits the pro-inflammatory response, thus mediating the major immunosuppressive effects of GC [, ]. Here, we observed the decrease of GFAP and Iba1 levels in the hippocampus, associated with the increase of GR, p(S211)GR, annexin A1 and the anti-inflammatory cytokine IL-10 expressions. Same results were obtained in recent research that investigated the effects of chronic stress on astroglial cells. Both chronic variable stress and chronic restraint stress increased GC plasma levels while decreasing GFAP expression by around 20% in astrocytes [-]. Such variation in astrocytes was sufficient to trigger depressive and anxiety-like behaviors [-]. The authors found that this modification is not due to glial cell loss, but could be due to an important remodeling of astrocytes morphology and network. This decrease of astrocyte plasticity led to a loss of their trophic functions on neurons, particularly on synaptic formation, LTP induction, and growth factor expression [, , ]. Based on our results, these alterations could be linked to synaptic and memory deficits induced by the chronic CORT consumption. Moreover, a negative glucocorticoid-responsive element on the promoter of GFAP was identified, indicating that GC inhibits directly the transcription of GFAP []. So, it is not surprising that in these conditions, chronic CORT seemed to be highly immunosuppressive (Fig. 4h). To reinforce this view, these results were also correlated with ADAM17 decreased expression. As mentioned, ADAM17 expression and activity are under the control of pro-inflammatory processes [].
Otherwise, we observed attractive results in CCoAβ animals. We found in this group that GC plasma levels were higher than in control group, but not as well as chronic CORT consumption or oAβ groups. Such result could be explained by the atrophy of adrenals and the impairment of HPA axis, indicating that this endocrine system was exhausted in CCoAβ animals. In accordance with our results, other groups demonstrated that chronic GC overexposure might blunt stress-induced CORT secretion [, ]. These findings were obtained with animals pretreated with GC and then submitted to behavioral stressors. This observation could be extended to cellular stressors such as oAβ25–35 in our study. These results in CCoAβ animals could also explain the incapacity of GC to restrain inflammatory processes induced by oAβ. Indeed, in these animals, we observed a potentiation of pro-inflammatory (GFAP, Iba1, ADAM17) and an inhibition of anti-inflammatory (IL-10) markers (Fig. 4h). This incapacity of GC to regulate neuroinflammatory processes could partly explain the exacerbation of AD hallmarks evidenced here. Such phenomenon was already described in chronically stressed animals injected with lipopolysaccharide [-]. Thus, in accordance with our results, it appears that chronic stress, as well as chronic CORT consumption, primes the neuroinflammatory response to a subsequent pro-inflammatory stimulus as oAβ25–35 or lipopolysaccharide. Therefore, the neuroimmune context is more responsive to inflammation, also favoring GC insensitivity or reducing the HPA response [-].
Furthermore, additional effects of chronic CORT consumption and oAβ25–35 injection were evidenced in the hippocampus on annexin A1. As previously mentioned this protein is activated by pro-inflammatory mediators and is an important anti-inflammatory actor mediating GC and GR effects [, ]. However, no study identified a glucocorticoid response element on the promoter region of annexin A1 gene, and its transcription seems to be mediated by pro-inflammatory cytokines and oxidative stress [, ]. Here, surprisingly, even if GR signaling pathways were impaired, annexin A1 levels were highly increased in CCoAβ animals. Taken together, we can envisage that this increase of annexin A1 could be a compensatory mechanism to counteract exacerbated pro-inflammatory processes. Further investigations are needed to assess if the GR-annexin A1 axis is still functional, even in a context of GR overactivation.
We focused on GR as we previously observed and established its central role in AD pathophysiology [, , ]. In stress conditions, and a fortiori in pathological conditions, GR seem to be more involved than MR []. However, a recent work demonstrated that hippocampal MR are overexpressed and overactivated by oxidative stress. This leads to microglia activation, pro-inflammatory mediators’ secretion, and downregulation of anti-inflammatory factors []. Thus, we cannot totally exclude the involvement of MR in our study. Further investigations are needed to decipher the precise role of MR between chronic stress, high levels of GC, and the pathophysiology of AD.
Conclusions
All these results evidenced the crucial role played by GC and GR and brought new information on how chronic CORT consumption could accelerate the development of AD by generating a deleterious inflammatory environment. It also reinforces the idea making chronic GC overexposure, as well as anxiety, as main risk factors in AD. Despite physiological anti-inflammatory properties of GC, prior chronic exposure to CORT markedly potentiates neuroinflammation associated with a subsequent innate immune system challenge induced by oAβ injection. The direct consequence of this exacerbated neuroinflammation is the aggravation of synaptic and memory deficits (Graphical Abstract).
It also highlights the therapeutic potential of new GR modulators, called sGRm, which are in capacity to re-establish GR functioning and signaling pathways and subsequent HPA axis physiological activity. These compounds represent a really attractive therapeutic approach in all stress-related disorders, pathologies displaying GC overproduction, or pathology needing an anti-inflammatory treatment [, , -].
Otherwise, the present study strongly argues in favor of the hypothesis suggesting that AD, as well as depression, could be a stress-related disorder []. Indeed, for years, clinical evidence established a strong correlation between elevated GC and higher risk to develop AD []. Thus, it was not surprising that recent clinical trials showed no difference in cognitive decline in AD patients prescribed with the GC receptor agonist, prednisone []. That is why this work also aims to alert about the risk to prescribe GC-based therapies in the elderly or in early AD patients.
Acknowledgements
The authors are thankful to Elisabeth HUETTER and the animal facility staff (CECEMA, University of Montpellier, France) for their daily assistance.
Statement of Ethics
Animal procedures were conducted in strict adherence to the European Union Directive of 2010 (2010/63/EU). The National French Animal Welfare Committee and the local committee at the University of Montpellier approved all protocols (authorization: CEEA-LR-12160). All efforts were made to minimize the number of animals used, potential pain, suffering, and distress.
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
Funding Sources
Parts of this study were supported by INSERM, University of Montpellier, and EPHE annual resources (France), by a grant from “France Alzheimer” and “Fédération pour la Recherche sur le Cerveau” (Grant AAP SM2016#1512 – LG), by a grant from the “Agence Nationale de la Recherche” (ANR) under the program “Investissements d’Avenir” (ANR-11-LABEX-0021-LipSTIC – CD). G. CANET and C. HERNANDEZ are supported by a PhD Grant from the University of Montpellier, France (CBS2 PhD program). G. CANET is also supported by another PhD Grant from the Center of Excellence in Neurodegenerative Disorders (CoEN, CHU Montpellier).
Author Contributions
G.C., C.Z., C.H., and A.B. performed experiments. L.G. designed the study, wrote the protocol, performed part of experiments, and analyzed the data (with the help of G.C., C.H., C.Z., and A.B.). L.G. and G.C. wrote the manuscript. C.Z., C.H., N.C., N.M., and C.D. corrected the manuscript. All authors read and approved the final manuscript. All funders had no role in data collection, analysis, or in the writing of the manuscript.
Data Availability Statement
All data generated or analyzed during this study are included in this article and its online supplementary material files. Further inquiries can be directed to the corresponding author.
References
- 1. Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010 Jan 28;362(4):329–44. http://dx.doi.org/10.1056/NEJMra0909142.
- 2. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016 Jun 8;8(6):595. http://dx.doi.org/10.15252/emmm.201606210.
- 3. Canet G, Chevallier N, Zussy C, Desrumaux C, Givalois L. Central role of glucocorticoid receptors in alzheimer's disease and depression. Front Neurosci. 2018 Oct 16;12(12):739–9. http://dx.doi.org/10.3389/fnins.2018.00739.
- 4. Canet G, Hernandez C, Zussy C, Chevallier N, Desrumaux C, Givalois L. Is AD a stress-related disorder? Focus on the HPA axis and its promising therapeutic targets. Front Aging Neurosci. 2019 Sep 27;11:269. http://dx.doi.org/10.3389/fnagi.2019.00269.
- 5. Mejía S, Giraldo M, Pineda D, Ardila A, Lopera F. Nongenetic factors as modifiers of the age of onset of familial Alzheimer's disease. Int Psychogeriatr. 2003 Dec;15(4):337–49. http://dx.doi.org/10.1017/s1041610203009591.
- 6. Herbert J, Lucassen PJ. Depression as a risk factor for Alzheimer’s disease: genes, steroids, cytokines and neurogenesis – What do we need to know?Front Neuroendocrinol. 2016 Apr;41:153–71.
- 7. Hartmann A, Veldhuis JD, Deuschle M, Standhardt H, Heuser I. Twenty-four hour cortisol release profiles in patients with Alzheimer’s and Parkinson’s disease compared to normal controls: ultradian secretory pulsatility and diurnal variation. Neurobiol Aging. 1997 May;18(3):285–9.
- 8. Csernansky JG, Dong H, Fagan AM, Wang L, Xiong C, Holtzman DM, et al. Plasma cortisol and progression of dementia in subjects with Alzheimer-type dementia. Am J Psychiatry. 2006 Dec;163(12):2164–9.
- 9. Hoogendijk WJ, Meynen G, Endert E, Hofman MA, Swaab DF. Increased cerebrospinal fluid cortisol level in Alzheimer's disease is not related to depression. Neurobiol Aging. 2006 May;27(5):780–2. http://dx.doi.org/10.1016/j.neurobiolaging.2005.07.017.
- 10. Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985 Dec;117(6):2505–11. http://dx.doi.org/10.1210/endo-117-6-2505.
- 11. Roozendaal B. 1999 Curt P. Richter award. Glucocorticoids and the regulation of memory consolidation. Psychoneuroendocrinology. 2000 Apr;25(3):213–38. http://dx.doi.org/10.1016/s0306-4530(99)00058-x.
- 12. Jankord R, Herman JP. Limbic regulation of hypothalamo-pituitary-adrenocortical function during acute and chronic stress. Ann N Y Acad Sci. 2008 Dec;1148:64–73. http://dx.doi.org/10.1196/annals.1410.012.
- 13. McEwen BS. Central effects of stress hormones in health and disease: understanding the protective and damaging effects of stress and stress mediators. Eur J Pharmacol. 2008 Apr 7;583(2–3):174–85. http://dx.doi.org/10.1016/j.ejphar.2007.11.071.
- 14. Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer's disease. J Neurosci. 2006 Aug 30;26(35):9047–56. http://dx.doi.org/10.1523/JNEUROSCI.2797-06.2006.
- 15. Rothman SM, Herdener N, Camandola S, Texel SJ, Mughal MR, Cong WN, et al. 3xTgAD mice exhibit altered behavior and elevated Aβ after chronic mild social stress. Neurobiol Aging. 2012 Apr;33(4):830.e1–2.
- 16. Han B, Yu L, Geng Y, Shen L, Wang H, Wang Y, et al. Chronic stress aggravates cognitive impairment and suppresses insulin associated signaling pathway in APP/PS1 mice. J Alzheimers Dis. 2016 Aug 8;53(4):1539–52. Ramirez M, editor.
- 17. Zussy C, Brureau A, Delair B, Marchal S, Keller E, Ixart G, et al. Time-course and regional analyses of the physiopathological changes induced after cerebral injection of an amyloid β fragment in rats. Am J Pathol. 2011 Jul;179(1):315–34.
- 18. Zussy C, Brureau A, Keller E, Marchal S, Blayo C, Delair B, et al. Alzheimer’s disease related markers, cellular toxicity and behavioral deficits induced six weeks after oligomeric amyloid-β peptide injection in rats. PLoS One. 2013;8(1):e53117.
- 19. Pineau F, Canet G, Desrumaux C, Hunt H, Chevallier N, Ollivier M, et al. New selective glucocorticoid receptor modulators reverse amyloid-β peptide-induced hippocampus toxicity. Neurobiol Aging. 2016;45:109–22.
- 20. Canet G, Pineau F, Zussy C, Hernandez C, Hunt H, Chevallier N, et al. Glucocorticoid receptors signaling impairment potentiates amyloid‐β oligomers‐induced pathology in an acute model of Alzheimer’s disease. FASEB J. 2020 Jan;34(1):1150–68.
- 21. Brureau A, Zussy C, Delair B, Ogier C, Ixart G, Maurice T, et al. Deregulation of hypothalamic-pituitary-adrenal axis functions in an Alzheimer’s disease rat model. Neurobiol Aging. 2013 May;34(5):1426–39.
- 22. Catania C, Sotiropoulos I, Silva R, Onofri C, Breen KC, Sousa N, et al. The amyloidogenic potential and behavioral correlates of stress. Mol Psychiatry. 2009 Jan;14(1):95–105.
- 23. Baglietto-Vargas D, Medeiros R, Martinez-Coria H, LaFerla FM, Green KN. Mifepristone alters amyloid precursor protein processing to preclude amyloid beta and also reduces tau pathology. Biol Psychiatry. 2013 Sep;74(5):357–66. http://dx.doi.org/10.1016/j.biopsych.2012.12.003.
- 24. Notarianni E. Hypercortisolemia and glucocorticoid receptor-signaling insufficiency in Alzheimer's disease initiation and development. Curr Alzheimer Res. 2013 Sep;10(7):714–31. http://dx.doi.org/10.2174/15672050113109990137.
- 25. Lanté F, Chafai M, Raymond EF, Salgueiro Pereira AR, Mouska X, Kootar S, et al. Subchronic glucocorticoid receptor inhibition rescues early episodic memory and synaptic plasticity deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology. 2015 Jun;40(7):1772–81.
- 26. Gourley SL, Wu FJ, Kiraly DD, Ploski JE, Kedves AT, Duman RS, et al. Regionally specific regulation of ERK MAP kinase in a model of antidepressant-sensitive chronic depression. Biol Psychiatry. 2008 Feb 15;63(4):353–9.
- 27. David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I, et al. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron. 2009 May 28;62(4):479–93.
- 28. van Donkelaar EL, Vaessen KRD, Pawluski JL, Sierksma AS, Blokland A, Cañete R, et al. Long-term corticosterone exposure decreases insulin sensitivity and induces depressive-like behaviour in the C57BL/6NCrl mouse. PLoS One. 2014;9(10):e106960.
- 29. Magariños AM, Orchinik M, McEwen BS. Morphological changes in the hippocampal CA3 region induced by non-invasive glucocorticoid administration: a paradox. Brain Res. 1998 Nov 2;809(2):314–8.
- 30. Luine VN, Spencer RL, McEwen BS. Effects of chronic corticosterone ingestion on spatial memory performance and hippocampal serotonergic function. Brain Res. 1993 Jul 9;616(1–2):65–70. http://dx.doi.org/10.1016/0006-8993(93)90193-q.
- 31. Conrad CD, MacMillan DD, Tsekhanov S, Wright RL, Baran SE, Fuchs RA. Influence of chronic corticosterone and glucocorticoid receptor antagonism in the amygdala on fear conditioning. Neurobiol Learn Mem. 2004 May;81(3):185–99. http://dx.doi.org/10.1016/j.nlm.2004.01.002.
- 32. Dobarro M, Orejana L, Aguirre N, Ramírez MJ. Propranolol reduces cognitive deficits, amyloid β levels, tau phosphorylation and insulin resistance in response to chronic corticosterone administration. Int J Neuropsychopharmacol. 2013 Jul;16(6):1351–60. http://dx.doi.org/10.1017/S1461145712001393.
- 33. Kaneko I, Morimoto K, Kubo T. Drastic neuronal loss in vivo by beta-amyloid racemized at Ser(26) residue: conversion of non-toxic [D-Ser(26)]beta-amyloid 1–40 to toxic and proteinase-resis-tant fragments. Neuroscience. 2001 Jul;104(4):1003–11.
- 34. Kubo T, Nishimura S, Kumagae Y, Kaneko I. In vivo conversion of racemized beta-amyloid ([D-Ser 26]A beta 1–40) to truncated and toxic fragments ([D-Ser 26]A beta 25–35/40) and fragment presence in the brains of Alzheimer’s patients. J Neurosci Res. 2002 Nov;70(3):474–83.
- 35. Gruden MA, Davidova TB, Malisauskas M, Sewell RD, Voskresenskaya NI, Wilhelm K, et al. Differential neuroimmune markers to the onset of Alzheimer’s disease neurodegeneration and dementia: autoantibodies to Abeta((25–35)) oligomers, S100b and neurotransmitters. J Neuroimmunol. 2007 May;186(1):181–92.
- 36. Fung J, Frost D, Chakrabartty A, McLaurin J. Interaction of human and mouse Abeta peptides. J Neurochem. 2004 Dec;91(6):1398–403. http://dx.doi.org/10.1111/j.1471-4159.2004.02828.x.
- 37. Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by beta-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993 Apr;13(4):1676–87. http://dx.doi.org/10.1523/jneurosci.13-04-01676.1993.
- 38. Pike CJ, Walencewicz-Wasserman AJ, Kosmoski J, Cribbs DH, Glabe CG, Cotman CW. Structure-activity analyses of beta-amy-loid peptides: contributions of the beta 25–35 region to aggregation and neurotoxicity. J Neurochem. 1995 Jan;64(1):253–65.
- 39. Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neuro-toxic effects of amyloid beta protein: reversal by tachykinin neuro-peptides. Science. 1990 Oct;250:279–82.
- 40. Yamada K, Nabeshima T. Animal models of Alzheimer's disease and evaluation of anti-dementia drugs. Pharmacol Ther. 2000 Nov;88(2):93–113. http://dx.doi.org/10.1016/s0163-7258(00)00081-4.
- 41. Varadarajan S, Kanski J, Aksenova M, Lauderback C, Butterfield DA. Different mechanisms of oxidative stress and neurotoxicity for Alzheimer’s Abeta(1–42) and A beta(25–35). J Am Chem Soc. 2001 Jun;123(24):5625–31.
- 42. Buttner-Ennever J. The rat brain in stereotaxic coordinates, 3rd edn. by George Paxinos and Charles Watson. 1996. J Anatomy. 1997 Aug;191(2):315–7.
- 43. Naert G, Zussy C, Tran Van Ba C, Chevallier N, Tang YP, Maurice T, et al. Involvement of endogenous brain-derived neurotrophic factor in hypothalamic-pituitary-adrenal axis activity. J Neuroendocrinol. 2015 Nov;27(11):850–60.
- 44. Deacon RM, Rawlins JN. T-maze alternation in the rodent. Nat Protoc. 2006 Jun;1(1):7–12. http://dx.doi.org/10.1038/nprot.2006.2.
- 45. Wang Z, Frederick J, Garabedian MJ. Deciphering the phosphorylation “code” of the glucocorticoid receptor in vivo. J Biol Chem. 2002 Jul 19;277(29):26573–80. http://dx.doi.org/10.1074/jbc.M110530200.
- 46. Kirschke E, Goswami D, Southworth D, Griffin PR, Agard DA. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell. 2014 Jun 19;157(7):1685–97. http://dx.doi.org/10.1016/j.cell.2014.04.038.
- 47. Lackie RE, Maciejewski A, Ostapchenko VG, Marques-Lopes J, Choy WY, Duennwald ML, et al. The Hsp70/Hsp90 chaperone machinery in neurodegenerative diseases. Front Neurosci. 2017 May 16;254(11):1–23.
- 48. Perretti M, D'Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. 2009 Jan;9(1):62–70. http://dx.doi.org/10.1038/nri2470.
- 49. Zub E, Canet G, Garbelli R, Blaquiere M, Rossini L, Pastori C, et al. The GR-ANXA1 pathway is a pathological player and a candidate target in epilepsy. FASEB J. 2019 Dec;33(12):13998–4009.
- 50. Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999 Dec 9;402(6762):615–22. http://dx.doi.org/10.1038/45159.
- 51. Lesort M, Jope RS, Johnson GV. Insulin transiently increases tau phosphorylation: involvement of glycogen synthase kinase-3beta and Fyn tyrosine kinase. J Neurochem. 1999 Feb;72(2):576–84. http://dx.doi.org/10.1046/j.1471-4159.1999.0720576.x.
- 52. Chen Y, Wang B, Liu D, Li JJ, Xue Y, Sakata K, et al. Hsp90 chaperone inhibitor 17-AAG attenuates a -induced synaptic toxicity and memory impairment. J Neurosci. 2014 Feb 12;34(7):2464–70.
- 53. Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol. 2013 Nov;132(5):1033–44. http://dx.doi.org/10.1016/j.jaci.2013.09.007.
- 54. Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. 2015 Apr;148:114–31. http://dx.doi.org/10.1016/j.pharmthera.2014.11.016.
- 55. Desrumaux C, Pisoni A, Meunier J, Deckert V, Athias A, Perrier V, et al. Increased amyloid-β peptide-induced memory deficits in phospholipid transfer protein (PLTP) gene knockout mice. Neuropsychopharmacol. 2013 Apr;38(5):817–25.
- 56. Lahmy V, Meunier J, Malmström S, Naert G, Givalois L, Kim SH, et al. Blockade of Tau hyperphosphorylation and Aβ1-42 generation by the aminotetrahydrofuran derivative ANAVEX2-73, a mixed muscarinic and σ1 receptor agonist, in a nontransgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology. 2013 Aug;38(9):1706–23.
- 57. Qian M, Shen X, Wang H. The distinct role of ADAM17 in APP proteolysis and microglial activation related to Alzheimer's disease. Cell Mol Neurobiol. 2016 May;36(4):471–82. http://dx.doi.org/10.1007/s10571-015-0232-4.
- 58. Murray F, Smith DW, Hutson PH. Chronic low dose corticosterone exposure decreased hippocampal cell proliferation, volume and induced anxiety and depression like behaviours in mice. Eur J Pharmacol. 2008 Mar 31;583(1):115–27. http://dx.doi.org/10.1016/j.ejphar.2008.01.014.
- 59. Lupien SJ, de Leon M, de Santi S, Convit A, Tarshish C, Nair NP, et al. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat Neurosci. 1998 May;1(1):69–73.
- 60. McEwen BS. Glucocorticoids, depression, and mood disorders: structural remodeling in the brain. Metabolism. 2005 May;54(5):20–3. http://dx.doi.org/10.1016/j.metabol.2005.01.008.
- 61. Savas M, Vinkers C, Rosmalen J, Hartman C, Wester VL, van den Akker ELT, et al. Systemic and local corticosteroid use is associated with reduced executive cognition, and mood and anxiety disorders. Neuroendocrinology. 2019 Jun 21;110:282–91.
- 62. Magariños AM, Verdugo JM, McEwen BS. Chronic stress alters synaptic terminal structure in hippocampus. Proc Natl Acad Sci USA. 1997 Dec 9;94(25):14002–8.
- 63. Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2012 Jan;13:22–37. http://dx.doi.org/10.1038/nrn3138.
- 64. Kott JM, Mooney-Leber SM, Shoubah FA, Brummelte S. Effectiveness of different corticosterone administration methods to elevate corticosterone serum levels, induce depressive-like behavior, and affect neurogenesis levels in female rats. Neuroscience. 2016 Jan 15;312:201–14. http://dx.doi.org/10.1016/j.neuroscience.2015.11.006.
- 65. Chami L, Checler F. BACE1 is at the crossroad of a toxic vicious cycle involving cellular stress and β-amyloid production in Alzheimer's disease. Mol Neurodegener. 2012;7(1):52. http://dx.doi.org/10.1186/1750-1326-7-52.
- 66. Sambamurti K, Kinsey R, Maloney B, Ge YW, Lahiri DK. Gene structure and organization of the human beta-secretase (BACE) promoter. FASEB J. 2004 Jun;18(9):1034–6. http://dx.doi.org/10.1096/fj.03-1378fje.
- 67. Johnson GL, Nakamura K. The c-jun kinase/stress-activated pathway: regulation, function and role in human disease. Biochim Biophys Acta. 2007 Aug;1773(8):1341–8. http://dx.doi.org/10.1016/j.bbamcr.2006.12.009.
- 68. Martisova E, Solas M, Gerenu G, Milagro F I, Campion J, Ramirez M J. Mechanisms involved in BACE upregulation associated to stress. CAR. 2012 Aug 1;9(7):822–9.
- 69. Piccini A, Borghi R, Guglielmotto M, Tamagno E, Cirmena G, Garuti A, et al. β-amyloid 1-42 induces physiological transcriptional regulation of BACE1: BACE1 as paradigm of Aβ42 different effects. J Neurochem. 2012 Sep;122(5):1023–31.
- 70. Guglielmotto M, Monteleone D, Giliberto L, Fornaro M, Borghi R, Tamagno E, et al. Amyloid-β42 activates the expression of BACE1 through the JNK pathway. JAD. 2011 Dec 12;27(4):871–83.
- 71. Galliher-Beckley AJ, Cidlowski JA. Emerging roles of glucocorticoid receptor phosphorylation in modulating glucocorticoid hormone action in health and disease. IUBMB Life. 2009 Oct;61(10):979–86. http://dx.doi.org/10.1002/iub.245.
- 72. Cuadrado-Tejedor M, Ricobaraza A, Del Río J, Frechilla D, Franco R, Pérez-Mediavilla A, et al. Chronic mild stress in mice promotes cognitive impairment and CDK5-dependent tau hyperphosphorylation. Behav Brain Res. 2011 Jul;220(2):338–43.
- 73. Papadopoulou A, Siamatras T, Delgado-Morales R, Amin ND, Shukla V, Zheng YL, et al. Acute and chronic stress differentially regulate cyclin-dependent kinase 5 in mouse brain: implications to glucocorticoid actions and major depression. Transl Psychiatry. 2015 Jun;5(6):e578.
- 74. Lopes JP, Oliveira CR, Agostinho P. Neurodegeneration in an Aβ-induced model of Alzheimer’s disease: the role of Cdk5. Aging Cell. 2010 Feb;9(1):64–77.
- 75. Kurbatskaya K, Phillips EC, Croft CL, Dentoni G, Hughes MM, Wade MA, et al. Upregulation of calpain activity precedes tau phosphorylation and loss of synaptic proteins in Alzheimer’s disease brain. Acta Neuropathol Commun. 2016 Mar 31;4:34.
- 76. Jin N, Yin X, Yu D, Cao M, Gong CX, Iqbal K, et al. Truncation and activation of GSK-3β by calpain I: a molecular mechanism links to tau hyperphosphorylation in Alzheimer’s disease. Sci Rep. 2015 Feb 2;5:8187.
- 77. Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000 May 18;405(6784):360–4. http://dx.doi.org/10.1038/35012636.
- 78. Zalachoras I, Houtman R, Atucha E, Devos R, Tijssen AMI, Hu P, et al. Differential targeting of brain stress circuits with a selective glucocorticoid receptor modulator. Proc Natl Acad Sci USA. 2013 May 7;110(19):7910–5.
- 79. Meijer OC, Koorneef LL, Kroon J. Glucocorticoid receptor modulators. Ann Endocrinol. 2018 Jun;79(3):107–11. http://dx.doi.org/10.1016/j.ando.2018.03.004.
- 80. Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, et al. ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010 Sep 1;29(17):3020–32.
- 81. Sogorb-Esteve A, García-Ayllón MS, Gobom J, Alom J, Zetterberg H, Blennow K, et al. Levels of ADAM10 are reduced in Alzheimer’s disease CSF. J Neuroinflammation. 2018 Dec;15(1):213.
- 82. Jiang H, Hampel H, Prvulovic D, Wallin A, Blennow K, Li R, et al. Elevated CSF levels of TACE activity and soluble TNF receptors in subjects with mild cognitive impairment and patients with Alzheimer’s disease. Mol Neurodegeneration. 2011;6(1):69.
- 83. Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. 2011 Mar 15;335(1):2–13. http://dx.doi.org/10.1016/j.mce.2010.04.005.
- 84. Banasr M, Duman RS. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol Psychiatry. 2008 Nov;64(10):863–70. http://dx.doi.org/10.1016/j.biopsych.2008.06.008.
- 85. Tynan RJ, Beynon SB, Hinwood M, Johnson SJ, Nilsson M, Woods JJ, et al. Chronic stress-induced disruption of the astrocyte network is driven by structural atrophy and not loss of astrocytes. Acta Neuropathol. 2013 Jul;126(1):75–91.
- 86. Simard S, Coppola G, Rudyk CA, Hayley S, McQuaid RJ, Salmaso N. Profiling changes in cortical astroglial cells following chronic stress. Neuropsychopharmacology. 2018 Aug;43(9):1961–71. http://dx.doi.org/10.1038/s41386-018-0105-x.
- 87. Kajitani N, Hisaoka-Nakashima K, Morioka N, Okada-Tsuchioka M, Kaneko M, Kasai M, et al. Antidepressant acts on astrocytes leading to an increase in the expression of neurotrophic/growth factors: differential regulation of FGF-2 by noradrenaline. PLoS One. 2012 Dec 5;7(12):e51197. Hashimoto K, editor.
- 88. Laping NJ, Nichols NR, Day JR, Johnson SA, Finch CE. Transcriptional control of glial fibrillary acidic protein and glutamine synthetase in vivo shows opposite responses to corticosterone in the hippocampus. Endocrinology. 1994 Nov;135(5):1928–33. http://dx.doi.org/10.1210/endo.135.5.7956913.
- 89. Mishima Y, Shinoda Y, Sadakata T, Kojima M, Wakana S, Furuichi T. Lack of stress responses to long-term effects of corticosterone in Caps2 knockout mice. Sci Rep. 2015 Aug;5(1):8932. http://dx.doi.org/10.1038/srep08932.
- 90. de Pablos RM, Villarán RF, Argüelles S, Herrera AJ, Venero JL, Ayala A, et al. Stress increases vulnerability to inflammation in the rat prefrontal cortex. J Neurosci. 2006 May 24;26(21):5709–19. http://dx.doi.org/10.1523/JNEUROSCI.0802-06.2006.
- 91. Munhoz CD, Lepsch LB, Kawamoto EM, Malta MB, de Lima LS, Avellar MC, et al. Chronic unpredictable stress exacerbates lipopolysaccharide-induced activation of nuclear factor-kappaB in the frontal cortex and hippocampus via glucocorticoid secretion. J Neurosci. 2006 Apr 5;26(14):3813–20. http://dx.doi.org/10.1523/JNEUROSCI.4398-05.2006.
- 92. Espinosa-Oliva AM, de Pablos RM, Villarán RF, Argüelles S, Venero JL, Machado A, et al. Stress is critical for LPS-induced activation of microglia and damage in the rat hippocampus. Neurobiol Aging. 2011 Jan;32(1):85–102.
- 93. Liberman AC, Budziñski ML, Sokn C, Gobbini RP, Steininger A, Arzt E. Regulatory and mechanistic actions of glucocorticoids on T and inflammatory cells. Front Endocrinol. 2018 May 16;9:235. http://dx.doi.org/10.3389/fendo.2018.00235.
- 94. Frank MG, Hershman SA, Weber MD, Watkins LR, Maier SF. Chronic exposure to exogenous glucocorticoids primes microglia to pro-inflammatory stimuli and induces NLRP3 mRNA in the hippocampus. Psychoneuroendocrinology. 2014 Feb;40:191–200. http://dx.doi.org/10.1016/j.psyneuen.2013.11.006.
- 95. Frank MG, Watkins LR, Maier SF. Stress-induced glucocorticoids as a neuroendocrine alarm signal of danger. Brain Behav Immun. 2013 Oct;33:1–6. http://dx.doi.org/10.1016/j.bbi.2013.02.004.
- 96. Perretti M, Dalli J. Exploiting the annexin A1 pathway for the development of novel anti-inflammatory therapeutics: annexin A1 and endogenous anti-inflammation. British J Pharmacol. 2009 Oct;158(4):936–46.
- 97. Sheikh MH, Solito E. Annexin A1: uncovering the many talents of an old protein. Int J Mol Sci. 2018 Mar 31;19(4):1045. http://dx.doi.org/10.3390/ijms19041045.
- 98. Meyer M, Gonzalez Deniselle MC, Hunt H, de Kloet ER, De Nicola AF. The selective glucocorticoid receptor modulator CORT108297 restores faulty hippocampal parameters in wobbler and corticosterone-treated mice. J Steroid Biochem Mol Biol. 2014 Sep;143:40–8.
- 99. Brocca ME, Pietranera L, de Kloet ER, De Nicola AF. Mineralocorticoid receptors, neuroinflammation and hypertensive encephalopathy. Cell Mol Neurobiol. 2019 Aug;39:483–92. http://dx.doi.org/10.1007/s10571-018-0610-9.
- 100. Meyer M, Lara A, Hunt H, Belanoff J, de Kloet ER, Gonzalez Deniselle MC, et al. The selective glucocorticoid receptor modulator cort 113176 reduces neurodegeneration and neuroinflammation in wobbler mice spinal cord. Neuroscience. 2018 Aug;384:384–96.
- 101. Solomon MB, Wulsin AC, Rice T, Wick D, Myers B, McKlveen J, et al. The selective glucocorticoid receptor antagonist CORT 108297 decreases neuroendocrine stress responses and immobility in the forced swim test. Hormones Behav. 2014 Apr;65(4):363–71.
- 102. Atucha E, Zalachoras I, van den Heuvel JK, van Weert LTCM, Melchers D, Mol IM, et al. A mixed glucocorticoid/mineralocorticoid selective modulator with dominant antagonism in the male rat brain. Endocrinology. 2015 Nov;156(11):4105–14.
- 103. De Nicola AF, Meyer M, Guennoun R, Schumacher M, Hunt H, Belanoff J, et al. Insights into the therapeutic potential of glucocorticoid receptor modulators for neurodegenerative diseases. IJMS. 2020 Mar 20;21(6):2137.
- 104. Ouanes S, Popp J. High cortisol and the risk of dementia and Alzheimer’s disease: a review of the literature. Front Aging Neurosci. 2019 Mar;11(43):1–11.
- 105. Aisen PS. Anti-inflammatory therapy for Alzheimer’s disease: implications of the prednisone trial. Acta Neurol Scand Suppl. 2000;176:385–9.