Introduction
Interstitial lung disease (ILD) comprises a heterogeneous group of over 200 entities that cause inflammation and/or fibrosis of lung tissue []. Accurate diagnosis of the specific ILD including identification of an underlying connective tissue disease (CTD) has major management and prognostic implications []. CTD-ILD patients overall have favourable outcomes compared to idiopathic interstitial pneumonia (IIP). Furthermore, the mainstay of treatment in CTD-ILD is immunosuppression (IS), which is associated with increased mortality in the most common form of IIP, idiopathic pulmonary fibrosis (IPF) [].
It is well recognised that there is a subgroup of ILD patients who have autoimmune features, not meeting the diagnostic criteria for a defined CTD. The term “interstitial pneumonia with autoimmune features” (IPAF) was proposed in 2015 to allow study of a well-defined cohort []. To date, IPAF cohorts from America, Europe, and Asia have demonstrated heterogeneous populations [-]. It remains unclear if IPAF is a distinct disease entity and whether the heterogeneity is a true reflection of the IPAF population, a product of the current IPAF classification criteria, variable study designs, or likely a combination of all []. IPAF studies in geographically diverse populations are required to inform further revisions of the IPAF criteria. To date, no studies have described an IPAF cohort in Australia. We studied an Australian IPAF cohort and compared their clinical and prognostic features to well-defined control populations: IIP, CTD-ILD, and unclassifiable ILD.
Methods
Study Design and Patients
Consecutive patients referred to the Royal Prince Alfred Hospital ILD clinic (June 1, 2014, to June 30, 2016) were screened for inclusion. All patients were discussed at the ILD-multidisciplinary team meeting (ILD-MDM), attended by respiratory physicians, rheumatologists, a pathologist, and radiologist with ILD expertise. Patients with a consensus diagnosis of IIP, CTD-ILD, and unclassifiable ILD were included, and patients meeting the IPAF criteria were identified [, ]. Patients with no identifiable ILD, insufficient data for analysis, and other ILDs were excluded. Ethical approval was granted by the Sydney Local Health District Human Ethics Committee (protocol numbers X15-0458 and LNR/15/RPAH/609).
Data Collection
Baseline clinical data were obtained from medical records. Disease duration was defined as time from the first onset of symptoms to the first clinic assessment. All patients underwent routine extended autoantibody screening at the first clinic visit including autoantibodies to anti-nuclear antigens (ANAs), extractable nuclear antigens (ENAs; Sjögren’s syndrome-related antibody A[SSA]/Ro-60, Ro-52, SSB/La, RNP, Scl70, Sm, centromere, PCNA, and ribosomal-P), myositis antigens (Mi-2, Ku, PM-Scl100, PM-Scl75, Jo-1, SRP, PL-7, PL-12, EJ and OJ), double-stranded deoxyribonucleic acid (dsDNA), rheumatoid factor (RF), cyclic citrullinated peptide (CCP), anti-neutrophil cytoplasmic antigens (ANCA), myeloperoxidase, and proteinase 3.
Positive ANA and RF were defined according to the IPAF criteria []. Myositis autoantibodies (MA) were identified and reported as per protocol (Euroimmun Myositis Profile 3, DL 1530-1601-3 G). Anti-Ro52, anti-Mi2, and anti-SRP were considered as meeting serological criteria. Patients with positive anti-tRNA synthetase autoantibodies, amyopathic disease, and no other features diagnostic of anti-synthetase syndrome were classified as IPAF for this study.
Physiology, Radiology, and Histopathology
Lung function (forced expiratory volume in one second [FEV1], forced vital capacity [FVC], and diffusing capacity for carbon monoxide [DLCO]) was performed within 6 months of the first clinic visit. Severe lung disease was defined as an FVC <50% and/or DLCO <35% [, ]. Composite physiologic index (CPI) and ILD-gender-age-physiology (ILD-GAP) stage were calculated [, ].
The radiological pattern on high-resolution computed tomography (HRCT) and histopathology were assessed by a specialist radiologist and pathologist at ILD-MDM or specialist respiratory assessment and report if not available. The IPAF multicompartment criterion “unexplained vasculopathy” was defined as pulmonary hypertension (PH) on an echocardiogram (systolic pulmonary arterial pressure >35 mm Hg above the right atrial pressure) or right heart catheterisation (mean pulmonary arterial pressure ≥25 mm Hg) with an FVC >70% predicted []. “Unexplained airway disease” was defined as an FEV1/FVC ratio <70% with no history of asthma, chronic obstructive airway disease, or smoking.
Outcomes
Follow-up data (vitality, treatment, transplantation, and progress lung function) were censored on July 31, 2019. Disease progression was defined as a decrease in FVC ≥10% and/or DLCO ≥15% from baseline, sustained until censoring. Transplant-free survival (TFS) was defined as time from the first visit to death, transplant, or censoring. Progression-free survival (PFS) was defined as time to disease progression, death, transplant, or censoring.
Statistical Analysis
Comparison between groups was performed using two-tailed Student’s t, χ2, or Fisher’s exact test, as appropriate. The unadjusted and adjusted Cox proportional regression analysis or log-rank test of equality, as appropriate, was used to identify predictors of survival. Statistical analysis was performed using STATA statistical software (STATA/SE version, 14.0 for Windows, College Station, TX, USA).
Results
Study Population
291 patients were screened, and 228 met the inclusion criteria. ILD diagnoses determined by ILD-MDM included IIP (n = 123), CTD-ILD (n = 68), and unclassifiable ILD (n = 37). Of these, 36 patients met the criteria for IPAF. Specific ILD diagnoses are shown in online suppl. Supplementary Figure 1; for all online suppl. material, see http://www.karger.com/doi/10.1159/000515396.
Baseline Characteristics
The overall mean age was 69.7 years, 55.7% were male, and 50.9% were ever-smokers with a mean disease duration at presentation of 3.4 years (Table 1). Physiological impairment was mild (mean FVC 75.1%, DLCO 59.4% predicted) and did not differ between ILD groups.
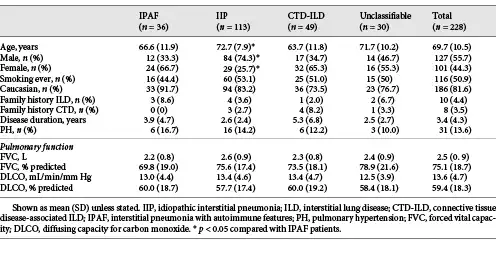
IPAF patients were younger than IIP patients (66.6 vs. 72.7 years, p = 0.008) with more female predominance (66.7 vs. 25.7%, p < 0.001). Age and gender were similar between IPAF and CTD-ILD, and unclassifiable ILD patients. The disease duration in IPAF did not differ from other ILD groups but was longer in CTD-ILD than IIP and unclassifiable ILD.
Dyspnoea was the most common presenting symptom overall (62.1%) and within each ILD group, except CTD-ILD where 51% presented with CTD-related manifestations (online suppl. Supplementary Table 1). CTD-related manifestations were the presenting symptoms in 36.1% of IPAF patients and more common than IIP (p < 0.001) and unclassifiable ILD (p = 0.001). Comorbidities are shown in online suppl. Supplementary Figure 2. PH prevalence did not differ between groups (Table 1).
IPAF Domains and Criteria
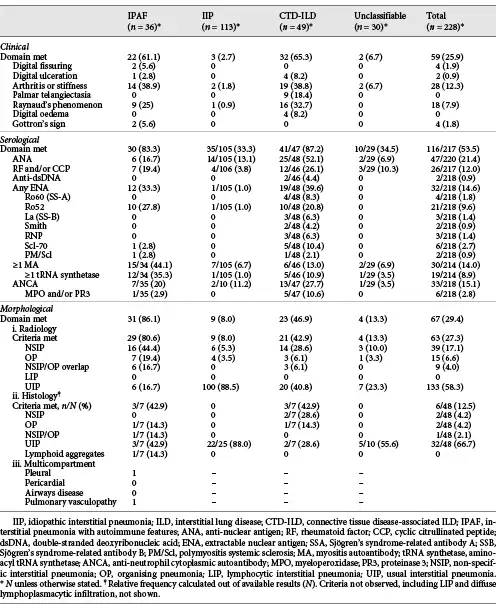
Thirty-six patients met the IPAF criteria. Morphological and serological domains were the most common (Table 2) and combined were the most common method by which the IPAF criteria were met (38.9%, n = 14). 30.6% (n = 11) met all 3 domains, 16.7% (n = 6) clinical and morphological, and 13.9% (n = 5) clinical and serological domains.
Clinical Criteria
Overall, 25.9% (n = 59) of all patients met the clinical IPAF criteria. The most common clinical features were inflammatory arthritis and/or early morning stiffness (12.3% overall, 38.9% IPAF, 38.8% CTD-ILD), and Raynaud’s phenomenon (7.9% overall, 25% IPAF, 32.7% CTD-ILD). Distribution of clinical features was similar between IPAF and CTD-ILD patients.
Serological Criteria
Overall, ANA was the most common autoantibody detected (21.4%, n = 47), followed by ANCA (15.1%, n = 33). ANA was more frequent in CTD-ILD (52.1%) than IIP (13.1%, p < 0.001), IPAF (16.7%, p = 0.001), and unclassifiable ILD (6.9%, p < 0.001). In contrast, MA were the most common autoantibodies in the IPAF cohort (44.1%, n = 15), compared with CTD-ILD (13.0%, p = 0.002), unclassifiable ILD (6.9%, p = 0.001), and IIP (6.7%, p < 0.001).
Morphological Domain
Non-specific interstitial pneumonia (NSIP) on HRCT was the most common pattern in IPAF (44.4%), in contrast to all other ILD groups, including CTD-ILD where usual interstitial pneumonia (UIP) predominated (Table 3). NSIP was more common in IPAF than IIP (5.3%, p < 0.001) and unclassifiable ILD (10.0%, p = 0.002) but no different to CTD-ILD (28.6%, p = 0.13). Histopathology was available in 48 patients (Table 2).
Treatment
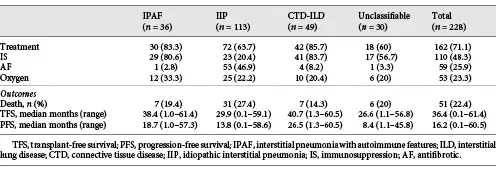
In all, 71.1% of patients received pharmacological ILD-targeted therapy (Table 3). ILD treatment was more common in CTD-ILD and IPAF than IIP (85.7 and 83.3% vs. 63.7%, p = 0.005 and 0.03, respectively).
48.3% of all patients received IS, including 83.7% CTD-ILD, 80.6% IPAF, 56.7% unclassifiable ILD, and 20.4% IIP patients. IS was similar in CTD-ILD and IPAF patients (p = 0.71), and more common in all groups than IIP (p < 0.001).
Antifibrotic (AF) treatment (pirfenidone or nintedanib) was recorded in 59 patients, 89.8% of whom had IPF. Non-IPF patients who received AFs included 4 CTD-ILD, 1 unclassifiable ILD and 1 IPAF patients. All AFs were accessed through special access schemes and clinical trials, with regulatory approval and reimbursement not available for wider prescription during the study.
Outcomes
There were 51 deaths and 3 transplants (2 IIP and 1 IPAF) during a median follow-up time of 36.4 months (range 4 days to 61.4 months; Table 4).
Transplant-Free Survival
IIP was associated with shorter TFS than CTD-ILD (p = 0.01), remaining significant after adjustment for age and FVC. There was a trend towards shorter TFS in IPAF than CTD-ILD and longer TFS than IIP, not meeting statistical significance (unadjusted Kaplan-Meier curve for TFS by ILD group shown in Fig. 1).
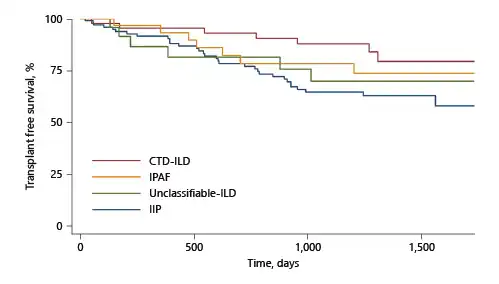
Fig. 1
Unadjusted Kaplan-Meier survival curves (TFS) by ILD group. ILD, interstitial lung disease; TFS, transplant-free survival; CTD, connective Q21 tissue disease.
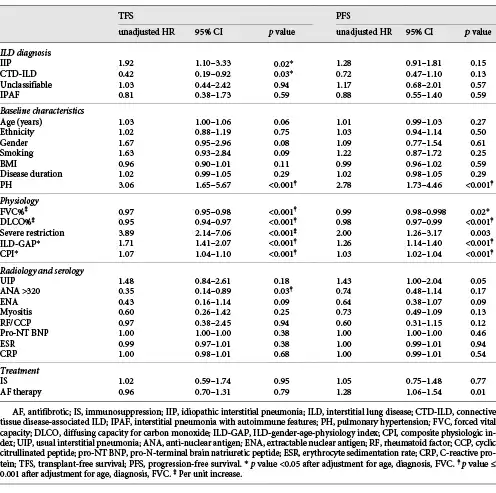
In univariate Cox analysis, shorter TFS was associated with the presence of PH, lower FVC, lower DLCO, severe restriction, and higher CPI or ILD-GAP score (Table 5). Elevated ANA demonstrated favourable TFS. These remained significant after adjustment for ILD diagnosis, age, and FVC.
Progression-Free Survival
Progression on lung function was demonstrated in 49.1% of all patients, with no difference in PFS between ILD groups (Table 4). Predictors of shorter PFS included PH, severe restriction, increased ILD-GAP score or CPI, and AF therapy. Higher FVC and DLCO were associated with more favourable PFS. Except for severe restriction and AF therapy, these remained significant on multivariable analysis. No serological markers were predictive of progression. These findings were unchanged when considering IPAF patients as per their initial ILD-MDM diagnosis.
Prognosis in IPAF Patients
Among IPAF patients, factors associated with worse TFS in univariate analysis included severe restriction (HR = 7.54, 95% CI 1.64–34.72, p = 0.01) and higher GAP index (HR 1.82, 95% CI 1.16–2.83, p = 0.009). Neither remained significant when adjusted for age and FVC. Predictors of PFS in IPAF included disease duration (HR = 1.13, 95% CI 1.02–1.25, p = 0.02) and PH (HR = 4.23, 95% CI 1.45–12.33, p = 0.008), remaining significant following multivariable adjustment.
Discussion
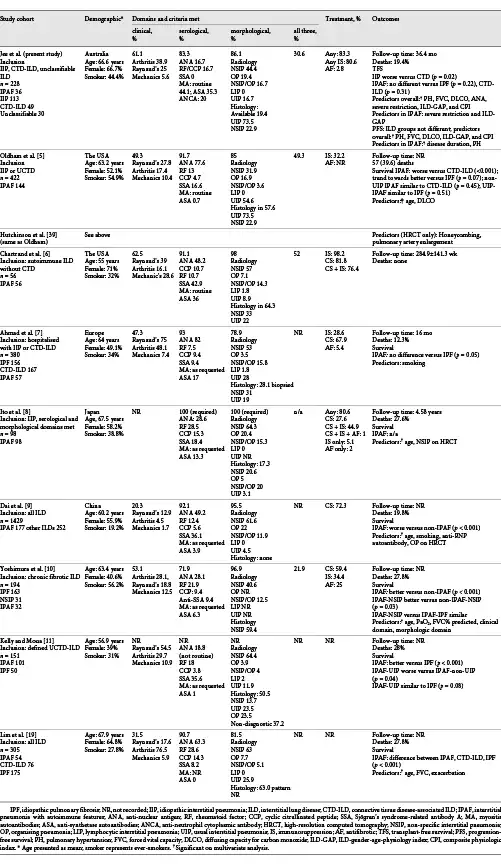
In this study, we describe a cohort of 228 well-defined ILD patients by ILD-MDM including IIP, CTD-ILD, and unclassifiable ILD, with 36 meeting the IPAF criteria. In the first described IPAF population in Australia, we demonstrate a heterogeneous phenotype that overlaps considerably with CTD-ILD and no definitive prognostic influence over ILD-MDM diagnosis. Differences between our study and international IPAF cohorts (detailed summary in Table 5) highlight areas for focus in future revisions of the IPAF classification as we seek to understand the clinical utility of identifying IPAF as a separate entity.
Supporting the importance of serology in the assessment of ILD patients, autoantibodies were detectable in 87.2% of CTD-ILD and 83.3% of IPAF patients. This is consistent with studies demonstrating the serological domain contributing to the IPAF criteria in 71.9–93% of each cohort [-, ]. However, there is no consensus about which autoantibodies to test when screening ILD patients, and the inclusion of anti-synthetase autoantibodies in the IPAF criteria has been contentious []. Our institution performs routine MA testing in all ILD patients, given the high mortality risk associated with IIM-ILD [, ]. Anti-synthetase autoantibodies, a subset of MA, were the most common autoantibody in our IPAF cohort (frequency 35.3%) – similar to Chartrand et al. who also performed routine testing (36%) []. Studies without routine testing reported substantially lower frequencies (1.0–17%), highlighting the importance of standardising testing practices to understand the true prevalence and implications of MA in ILD without defined CTD [-, ].
Our study also suggests that the decision to test for specific MA testing cannot be directed by broader screening autoantibodies and clinical features. Our IPAF and CTD-ILD cohorts had similar clinical phenotypes but significantly different serological profiles, with IPAF patients demonstrating a higher prevalence of MA and lower prevalence of ANA. The poor sensitivity of ANA for myositis has previously been reported []. Fidler et al. similarly found that ANA, RF, anti-cyclic citrullinated peptide, and clinical features were not predictive of MA in idiopathic ILD [, ]. A thorough assessment of accuracy, cost-effectiveness, and the impact of positive MA in patients with well-defined idiopathic ILD is required before routine MA testing can be clinically recommended.
The exclusion of anti-Ro52 autoantibodies and ANCA from the IPAF criteria has been debated [-]. We have demonstrated that anti-Ro52 and ANCA were the second and third most common autoantibodies overall and within our IPAF cohort. Both autoantibodies have demonstrated important associations with ILD and poor outcomes [, -]. From these data, we believe that the inclusion of anti-Ro52 and ANCA in future IPAF revisions may help define a more distinct “overlap” ILD with important prognostic implications.
The natural history and prognosis of IPAF remain elusive. To our knowledge, our study is the first to analyse PFS in an IPAF cohort. We confirmed the favourable prognosis of CTD-ILD compared with IIP [-]. However, meeting the IPAF criteria was not predictive of survival outcomes, perhaps limited by numbers. Survival in other IPAF studies has been mixed, unsurprising given the heterogeneity in patient cohorts [, , -].
Importantly, we found that PH and disease severity (FVC, DLCO, ILD-GAP, and CPI) were strong predictors of TFS and PFS, independent of age, lung function, and ILD diagnosis. AF therapy was not associated with outcome variables on multivariable analyses, perhaps reflecting limitations of access to therapy at the time of study. Within IPAF, ILD-GAP was a significant predictor of TFS, supporting prior IPAF studies where the individual measures comprising ILD-GAP predicted mortality and the hypothesis that ILD-GAP may have utility beyond IPF [, -, ]. Radiological UIP was not prognostic in our IPAF cohort, acknowledging a lower prevalence of UIP may limit this assertion. The variable prevalence and prognostic impact of UIP across IPAF cohorts warrant prospective investigation, with recognised prognostic and treatment implications in other ILDs [, , , ]. Positive ANA was associated with more favourable TFS, with wider literature on survival and ANA inconclusive [, ]. Taken together, a pragmatic approach may be to consider IPAF patients based on their working MDM diagnosis, with prevalent PH and disease severity holding greatest prognostic importance.
There remains no guidance for treating patients fulfilling the IPAF criteria. Our IPAF cohort was predominantly treated with IS, similar to our CTD-ILD patients, perhaps reflecting their similar phenotypes and low prevalence of UIP. Conversely, only 32.2% of the Oldham cohort was treated with IS, with over half demonstrating UIP and a phenotype more similar to IPF []. The impact of IS in non-IPF UIP is uncertain, but caution is often applied, given the known harms in IPF []. AFs may have therapeutic benefit in IPAF, extrapolating from recent positive trials in systemic sclerosis-associated, unclassifiable, and progressive-fibrosing ILD [-]. The latter studies included IPAF patients, including one of our patients, and we await subgroup analysis.
Clinical characteristics of our IPAF cohort were similar to prior studies and overlapped considerably with CTD-ILD (female predominant, non-smokers, mean age 66.6 years). Nearly two-thirds of our IPAF cohort demonstrated clinical CTD manifestations, similar to Chartrand et al. [, , , , ] (62.5%) but higher than other studies (20–53.1%). Rheumatologists are integral to the assessment and management of our ILD patients, participating in the ILD-MDM and a combined ILD rheumatology clinic, potentially contributing to the higher recognition of clinical features.
Our study has limitations. The retrospective nature and the small number of IPAF patients limit causal associations and assessment of the treatment effect. Missing data may have resulted in misclassification. The anti-MDA5 autoantibody was not routinely tested during the study. The low prevalence of lung biopsy limits robust assessment of histopathology but reflects clinical guidelines where surgery is reserved for patients who are surgically fit, and there remains considerable diagnostic uncertainty despite ILD-MDM, owing to procedural risks including up to 1.7% mortality [, ]. We included consecutive all comers to the clinic to reduce inherent selection bias of a single-centre study. Despite the small number of IPAF patients, clinical characteristics were similar to larger, international IPAF cohorts. Strengths include that serological and pulmonary function tests were all performed in the same laboratory with standardised methods, internal validation, and controls. As one of the largest dedicated ILD clinics in Australia, our cohort represents a diverse ILD population.
In conclusion, our study has identified IPAF as a heterogeneous cohort with a significant overlap with CTD-ILD. Meeting the IPAF criteria was not in itself predictive of outcome, whereas disease severity, PH, and IPF diagnosis were all associated with worse outcomes. IPAF remains an important cohort of patients at the intersection of IIP and CTD-ILD, which warrants further study. Consideration should be given to the revision of the IPAF criteria to include anti-Ro52 autoantibodies and ANCA to serological criteria, given their prevalence in overlap CTDs, association with ILD, and prognostic implications.
Acknowledgements
The authors would like to acknowledge the support of the National Health and Medical Research Council (NHMRC) Centre of Research Excellence in Pulmonary Fibrosis (GNT1116371), supported by foundation partner Boehringer Ingelheim and program partners Roche and Galapagos.
Statement of Ethics
Research has been conducted in accordance with the World Medical Association Declaration of Helsinki. Ethical approval was granted by the Sydney Local Health District Human Ethics Committee (protocol numbers X15-0458 and LNR/15/RPAH/609).
Conflict of Interest Statement
There are no conflicts of interest relevant to this research. A.S.J. has received speaker’s fees from Boehringer Ingelheim. M.J.S.P. has received speaker’s fees from Boehringer Ingelheim. H.E.J. has received speaker’s fees from Roche and Boehringer Ingelheim. L.K.T. has received speaker’s fees from Boehringer Ingelheim, Roche, and Menarini; works in a unit that has received unrestricted educational grants from Boehringer Ingelheim and Roche; and has received advisory board fees from Roche. E.M.L. has received speaker’s fees, research support, and consultancy and advisory board fees from Actelion and speaker’s fees and research support from GlaxoSmithKline. A.K.Y.T. has received speaker’s fees from Roche and travel and conference support from Boehringer Ingelheim. T.J.C. has received research grants, advisory board fees, and speaker fees from Boehringer Ingelheim; research grants, advisory board, and speaker fees from Roche; research grants from Biogen, Galapagos, Actelion, and Avalyn; advisory board fees from BMS and Ad Alta; and is on trial steering committees for Roche, BMS and Promedior. J.F.B., S.A., and S.W. have no conflicts of interest to declare.
Funding Sources
A.S.J. would like to acknowledge funding support from the Lung Foundation Australia David Wilson PhD Scholarship.
Author Contributions
A.S.J. was responsible for writing the original draft, and data acquisition and curation, conceptualisation, formal analysis, investigation, and methodology of the study. M.J.S.P., H.E.J., E.M.L., A.K.Y.T., J.F.B., S.A., and S.W. were responsible for data acquisition, interpretation, and provision of resources. T.J.C. was responsible for conceptualisation, methodology, and supervision. All authors participated in revising and approving the final version of the manuscript.
References
- 1. Cottin V, Hirani NA, Hotchkin DL, Nambiar AM, Ogura T, Otaola M, et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27(150):180076.
- 2. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824.http://dx.doi.org/10.1164/rccm.2009-040GL.
- 3. Raghu G, Anstrom KJ, King TE, Lasky JA, Martinez FJIdiopathic Pulmonary Fibrosis Clinical Research Network. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366(21):1968–77http://dx.doi.org/10.1056/NEJMoa1113354.
- 4. Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46(4):976–87.http://dx.doi.org/10.1183/13993003.00150-2015.
- 5. Oldham JM, Adegunsoye A, Valenzi E, Lee C, Witt L, Chen L, et al. Characterisation of patients with interstitial pneumonia with autoimmune features. Eur Respir J. 2016;47(6):1767–75.http://dx.doi.org/10.1183/13993003.01565-2015.
- 6. Chartrand S, Swigris JJ, Stanchev L, Lee JS, Brown KK, Fischer A. Clinical features and natural history of interstitial pneumonia with autoimmune features: a single center experience. Respir Med. 2016;119:150–4.http://dx.doi.org/10.1016/j.rmed.2016.09.002.
- 7. Ahmad K, Barba T, Gamondes D, Ginoux M, Khouatra C, Spagnolo P, et al. Interstitial pneumonia with autoimmune features: clinical, radiologic, and histological characteristics and outcome in a series of 57 patients. Respir Med. 2017;123:56–62.http://dx.doi.org/10.1016/j.rmed.2016.10.017.
- 8. Ito Y, Arita M, Kumagai S, Takei R, Noyama M, Tokioka F, et al. Serological and morphological prognostic factors in patients with interstitial pneumonia with autoimmune features. BMC Pulm Med. 2017;17(1):111.http://dx.doi.org/10.1186/s12890-017-0453-z.
- 9. Dai J, Wang L, Yan X, Li H, Zhou K, He J, et al. Clinical features, risk factors, and outcomes of patients with interstitial pneumonia with autoimmune features: a population-based study. Clin Rheumatol. 2018;37:2125–32.
- 10. Yoshimura K, Kono M, Enomoto Y, Nishimoto K, Oyama Y, Yasui H, et al. Distinctive characteristics and prognostic significance of interstitial pneumonia with autoimmune features in patients with chronic fibrosing interstitial pneumonia. Respir Med. 2018;137:167–75.http://dx.doi.org/10.1016/j.rmed.2018.02.024.
- 11. Kelly BT, Moua T. Overlap of interstitial pneumonia with autoimmune features with undifferentiated connective tissue disease and contribution of UIP to mortality. Respirology. 2018;23(6):600–5.http://dx.doi.org/10.1111/resp.13254.
- 12. Graney BA, Fischer A. Interstitial pneumonia with autoimmune features. Ann Am Thorac Soc. 2019;16:525–33.http://dx.doi.org/10.1016/b978-0-12-801238-3.11360-1.
- 13. Travis WD, Costabel U, Hansell DM, King TE Jr., Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–48.http://dx.doi.org/10.1164/rccm.201308-1483ST.
- 14. Latsi PI, du Bois RM, Nicholson AG, Colby TV, Bisirtzoglou D, Nikolakopoulou A, et al. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med. 2003;168(5):531–7.http://dx.doi.org/10.1164/rccm.200210-1245OC.
- 15. Egan JJ, Martinez FJ, Wells AU, Williams T. Lung function estimates in idiopathic pulmonary fibrosis: the potential for a simple classification. Thorax. 2005;60(4):270–3.http://dx.doi.org/10.1136/thx.2004.035436.
- 16. Ryerson CJ, Vittinghoff E, Ley B, Lee JS, Mooney JJ, Jones KD, et al. Predicting survival across chronic interstitial lung disease: the ILD-GAP model. Chest. 2014;145(4):723–8.http://dx.doi.org/10.1378/chest.13-1474.
- 17. Wells AU, Desai SR, Rubens MB, Goh NS, Cramer D, Nicholson AG, et al. Idiopathic pulmonary fibrosis: a composite physiologic index derived from disease extent observed by computed tomography. Am J Respir Crit Care Med. 2003;167(7):962–9.http://dx.doi.org/10.1164/rccm.2111053.
- 18. Augustine DX, Coates-Bradshaw LD, Willis J, Harkness A, Ring L, Grapsa J, et al. Echocardiographic assessment of pulmonary hypertension: a guideline protocol from the British Society of Echocardiography. Echo Res Pract. 2018;5(3):G11–24.http://dx.doi.org/10.1530/ERP-17-0071.
- 19. Lim JU, Gil BM, Kang HS, Oh J, Kim YH, Kwon SS. Interstitial pneumonia with autoimmune features show better survival and less exacerbations compared to idiopathic pulmonary fibrosis. BMC Pulm Med. 2019;19(1):120.http://dx.doi.org/10.1186/s12890-019-0868-9.
- 20. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44–68.http://dx.doi.org/10.1164/rccm.201807-1255ST.
- 21. Lega JC, Reynaud Q, Belot A, Fabien N, Durieu I, Cottin V. Idiopathic inflammatory myopathies and the lung. Eur Respir Rev. 2015;24(136):216–38.http://dx.doi.org/10.1183/16000617.00002015.
- 22. Lilleker JB, Vencovsky J, Wang G, Wedderburn LR, Diederichsen LP, Schmidt J, et al. The EuroMyositis registry: an international collaborative tool to facilitate myositis research. Ann Rheum Dis. 2018;77(1):30–9.http://dx.doi.org/10.1136/annrheumdis-2017-211868.
- 23. Aggarwal R, Dhillon N, Fertig N, Koontz D, Qi Z, Oddis CV. A negative antinuclear antibody does not indicate autoantibody negativity in myositis: role of anticytoplasmic antibody as a screening test for antisynthetase syndrome. J Rheumatol. 2017;44(2):223–9.http://dx.doi.org/10.3899/jrheum.160618.
- 24. Fidler L, Doubelt I, Kandel S, Fisher JH, Mittoo S, Shapera S. Screening for myositis antibodies in idiopathic interstitial lung disease. Lung. 2019;197(3):277–84.http://dx.doi.org/10.1007/s00408-019-00212-9.
- 25. La Corte R, Lo Mo Naco A, Locaputo A, Dolzani F, Trotta F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity. 2006;39(3):249–53.http://dx.doi.org/10.1080/08916930600623791.
- 26. Marie I, Hatron PY, Dominique S, Cherin P, Mouthon L, Menard JF, et al. Short-term and long-term outcome of anti-Jo1-positive patients with anti-Ro52 antibody. Semin Arthritis Rheum. 2012;41(6):890–9.http://dx.doi.org/10.1016/j.semarthrit.2011.09.008.
- 27. Hozumi H, Oyama Y, Yasui H, Suzuki Y, Kono M, Karayama M, et al. Clinical significance of myeloperoxidase-anti-neutrophil cytoplasmic antibody in idiopathic interstitial pneumonias. PLoS One. 2018;13(6):e0199659.http://dx.doi.org/10.1371/journal.pone.0199659.
- 28. Hudson M, Pope J, Mahler M, Tatibouet S, Steele R, Baron M, et al. Clinical significance of antibodies to Ro52/TRIM21 in systemic sclerosis. Arthritis Res Ther. 2012;14(2):R50.http://dx.doi.org/10.1186/ar3763.
- 29. Shi J, Li S, Yang H, Zhang Y, Peng Q, Lu X, et al. Clinical profiles and prognosis of patients with distinct antisynthetase autoantibodies. J Rheumatol. 2017;44(7):1051–7.http://dx.doi.org/10.3899/jrheum.161480.
- 30. Alba MA, Flores-Suárez LF, Henderson AG, Xiao H, Hu P, Nachman PH, et al. Interstital lung disease in ANCA vasculitis. Autoimmun Rev. 2017;16(7):722–9.http://dx.doi.org/10.1016/j.autrev.2017.05.008.
- 31. Vij R, Strek ME. Diagnosis and treatment of connective tissue disease-associated interstitial lung disease. Chest. 2013;143(3):814–24.http://dx.doi.org/10.1378/chest.12-0741.
- 32. Song JW, Do KH, Kim MY, Jang SJ, Colby TV, Kim DS. Pathologic and radiologic differences between idiopathic and collagen vascular disease-related usual interstitial pneumonia. Chest. 2009;136(1):23–30.http://dx.doi.org/10.1378/chest.08-2572.
- 33. Park JH, Kim DS, Park IN, Jang SJ, Kitaichi M, Nicholson AG, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med. 2007;175(7):705–11.http://dx.doi.org/10.1164/rccm.200607-912OC.
- 34. Dinse GE, Parks CG, Weinberg CR, Meier HCS, Co CA, Chan EKL, et al. Antinuclear antibodies and mortality in the National Health and Nutrition Examination Survey (1999–2004). PLoS One. 2017;12(10):e0185977http://dx.doi.org/10.1371/journal.pone.0185977.
- 35. Selmi C, Ceribelli A, Generali E, Scirè CA, Alborghetti F, Colloredo G, et al. Serum antinuclear and extractable nuclear antigen antibody prevalence and associated morbidity and mortality in the general population over 15 years. Autoimmun Rev. 2016;15(2):162–6.http://dx.doi.org/10.1016/j.autrev.2015.10.007.
- 36. Maher TM, Corte TJ, Fischer A, Kreuter M, Lederer DJ, Molina-Molina M, et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med. 2019;8:P147–57.http://dx.doi.org/10.1183/13993003.congress-2019.rct1880.
- 37. Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, et al. Nintedanib for systemic sclerosis–associated interstitial lung disease. New Engl J Med. 2019;380:2518–28.http://dx.doi.org/10.1164/ajrccm-conference.2019.199.1_meetingabstracts.a7360.
- 38. Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, et al. Nintedanib in progressive fibrosing interstitial lung diseases. New Engl J Med. 2019;381:1718–27.http://dx.doi.org/10.1164/ajrccm-conference.2019.199.1_meetingabstracts.a5627.
- 39. Hutchinson JP, Fogarty AW, McKeever TM, Hubbard RB. In-Hospital mortality after surgical lung biopsy for interstitial lung disease in the United States. 2000 to 2011. Am J Respir Crit Care Med. 2016;193(10):1161–7.http://dx.doi.org/10.1164/rccm.201508-1632OC.