Introduction
Inflammatory bowel disease (IBD) includes Crohn’s disease (CD) and ulcerative colitis (UC), two heritable chronic inflammatory disorders of the gastrointestinal (GI) tract, characterized by progressive mucosal inflammation and injury, associated with morbidity, surgery, and malignancy. CD and UC differ in their location and behavior, with CD manifesting as inflammation anywhere along the GI tract in a discontinuous pattern, as compared to UC which is confined to segments or the whole colon in a continuous pattern. Furthermore, CD with perianal disease prognosticates more severe disease activity and poor response to conventional therapies. Current treatment strategies for IBD relying on medications have demonstrated mixed success in patient tolerability and outcomes,. Recently, cellular therapy with mesenchymal stromal cells (MSCs) has shown promise in dampening the inflammatory microenvironment and improving morbidity in various diseases, but little is known about their biological properties and full potential for IBD treatment.
Mesenchymal stromal cells are tissue-resident, multipotent, non-hematopoietic cells that play an important role in regulating inflammation, repair, and regeneration through an intricate cell-to-cell crosstalk involving contact-dependent and paracrine mechanisms. The proliferative capacity of these cells coupled with their expression of growth factors and immunomodulatory molecules have made MSCs a promising candidate for cellular therapy in various diseases. In fact, allogenic bone marrow (BM)-derived MSCs have been extensively studied and approved for the treatment of acute graft-versus-host disease in several countries. Furthermore, recent advances with allogenic adipose-derived stem cells showed improved remission rates of complex CD perianal fistulas in a double-blind randomized, placebo-controlled trial conducted by the ADMIRE-CD Study Group, eventually resulting in European Medicines Agency approval of Alofisel®. These developments in therapy highlight the evolving therapeutic potential of MSCs in the management of IBD.
Thus, we are driven by the hypothesis that autologous treatment will provide the most biologically appropriate source of MSCs for mucosal healing while minimizing the adverse effects of IBD treatment. To this end, we investigated the feasibility of isolating and expanding ex vivo MSCs from endoscopic ileal and rectal biopsies obtained from IBD patients and characterized the proliferative fitness, immunophenotype, gene expression, and secretome profiles in comparison with non-IBD controls (Ctrl). We demonstrate that the IBD intestinal MSCs can be isolated and expanded ex vivo and exhibit a similar proliferative capacity to Ctrl. Furthermore, intestinal MSCs from IBD and Ctrl shared overall similar immunophenotypic, transcriptomic, and secretomic profiles, particularly after IFN-γ priming—highlighting the potential for autologous intestinal MSCs as a cellular therapy for IBD.
Materials and Methods
Patient Selection and Biopsy Collection
Recruited patients were consented before colonoscopy at the Children’s Healthcare of Atlanta/Emory University Hospitals. Patients undergoing endoscopy for abdominal pain with grossly normal endoscopy and histology were included as Ctrl. In total, 37 mucosal biopsy samples were obtained from both the terminal ileum and rectum of established or newly-diagnosed IBD patients, and Ctrl, and the tissues were identified as inflamed or noninflamed by both endoscopy and histology (Table 1). The study design and protocols were approved by the Emory University Institutional Review Boards (IRB) committee. Collected biopsies were placed in ice-cold RPMI 1640 with 10% fetal bovine serum (FBS), penicillin/streptomycin (pen/strep; Thermo Fisher Scientific), and Normocin (Invivogen) and immediately transported to the lab for processing.
Isolation and Doubling Time Determination of MSCs Cultured from Intestinal Biopsy
Within 2 hours of collection, mucosal biopsies were mechanically dissociated and plated in 60-mm tissue culture plates with α-MEM (Corning), supplemented with 10% human platelet lysate (Mill Creek Life Sciences), pen/strep, and Normocin. After 3 days following MSC attachment, tissue debris and nonadherent cells were discarded and the adherent cells were subsequently maintained in α-MEM with 5% human platelet lysate, pen/strep, and Normocin for 2 more weeks until visible colonies had formed (passage 0). Subsequently, MSCs were detached with TrypLE (Thermo Fisher Scientific) and replated at a seeding density of 5 × 103 cells/cm2. Media was changed every other day, and at 90% confluence, MSCs were dissociated with TrypLE and counted with a hemocytometer and trypan blue staining to determine their viability and rate of doubling using the following formula:
Downstream experiments were conducted on MSCs between passages 3-5.
Immunophenotyping of MSCs by Flow Cytometry
Mesenchymal stromal cells (MSCs) were immunophenotypically characterized with high-dimensional fluorescence-activated cell sorting (FACS). Briefly, TrypLE detached, Fc-blocked MSCs (1 × 106 cells) were stained with the following mouse antihuman fluorochrome-conjugated monoclonal antibodies from BD Biosciences with the clone in parentheses: CD73(AD2), CD90(5E10), CD105(266), CD106(51-10C9), CD146(P1H12), CD34(581), and CD45(HI-30). For experiments with 48 hours of IFN-γ priming (50 ng/mL),, MSCs were also stained for: CD80(L307.4), PD-L1(MIH1), PD-L2(MIH18), HLA-DR(G46-6), and HLA-ABC(G46-2.6). Unstained MSCs served as negative controls and BM-derived MSCs served as positive controls for CD34. Data were acquired on a BD FACSymphony A5 and analyzed with FlowJo 10.7.1.
Immunofluorescence Microscopy of MSCs
Mesenchymal stromal cells were recovered from liquid nitrogen storage, passaged, plated on glass coverslips, and grown to 50% confluence before 4% formaldehyde fixation and 0.5% Triton X-100 permeabilization. Cells were blocked in 5% BSA and then stained overnight with a combination of mouse anti-α-SMA (clone 1A4, Thermo Fisher Scientific) or mouse anti-CD90 BUV395 (BD Biosciences) and rabbit anti-vimentin (Cell Signaling Technology) antibodies. Signals were detected and visualized using AlexaFluor488 goat anti-mouse and goat anti-rabbit AlexaFluor555 on an Olympus FV1000 confocal microscope. Images were minimally processed by ImageJ.
RNA Isolation and Bulk RNA Sequencing Analysis of MSCs with IFN-γ Treatment
Total RNA was extracted from 48-hour IFN-γ-treated or untreated MSCs (2 × 105) using an AllPrep DNA/RNA extraction micro kit (Qiagen). RNA quantity and integrity were determined on a Qubit 2.0 Fluorometer (Life Technologies) and Agilent TapeStation 4200 (Agilent Technologies), respectively, using only RNA with RIN >7 for downstream applications. RNA sequencing libraries were prepared using the NEBNext Ultra RNA library prep kit for Illumina following the manufacturer’s instructions. Libraries were loaded in a flow cell and clustered on an Illumina HiSeq instrument (4000 or equivalent) and the samples were sequenced using a 2 × 150 bp Paired End configuration at Genewiz, LLC. Raw files were converted to fastq files and demultiplexed using Ilumina’s bcl2fastq 2.17. Reads were mapped to human genome (hg38), using STAR alignment package. Prcomp was used to perform Principal component (PC) analysis to identify the PCs, and Corrplot was used to identify whether those PCs were correlated with age, gender, tissue site, disease type, and inflammation status. Differential expression analysis between diseases-, tissues-, and treatment- conditions were tested using DESeq package in R by adjusting for age, gender, and inflammation status as covariates.
Single-Cell Preparation and Sequencing
Single-cell RNA sequencing (ScRNA-seq) was performed with rectum-derived MSCs according to the manufacturer’s instructions (10X Genomics). Briefly, frozen vials containing 1 million cells were thawed, washed/filtered, and resuspended in PBS (Corning) with 0.04% BSA (Thermo Fisher Scientific), and showed more than 90% viability (quantified with propidium iodide staining). Approximately, 7500 suspended cells (target 5000) were mixed with reverse transcription-PCR (RT-PCR) master mix and encapsulated in partitioning oil within a 10X Genomics Chromium Controller.
10X Library Preparation and Sequencing
scRNA-seq libraries were generated using a 10X Genomics 3’ single-cell gene expression chemistry. After reverse transcription, emulsified single-cell droplets were disrupted, and cDNA was pooled for PCR amplification. After Agilent Bioanalyzer2100 quality control check for a sufficient yield of intact cDNA, the cDNA obtained was fragmented, ligated, and prepped with Illumina adapters as per the manufacturer’s protocol to generate a single multiplexed library that was sequenced on Illumina’s Novaseq6000 at the Yerkes Primate Research Center Genomics Core at Emory University.
Single-Cell Cluster Analysis
Raw reads were trimmed, and the gene-barcode matrix was generated using Cell Ranger v6.1 software (10× Genomics). After filtration, samples were aligned to Grch38 human reference genome to generate UMI (Unique Molecular Identifiers) counts for each gene per cell. Each patient sample had at least 5000 cells with an average of 7500 features (unique genes)/cell. Cells with mitochondrial expression greater than 5% were removed from downstream analysis. We followed default Seurat v4.1 workflow to cluster cells from all the samples based on their gene expression profiles and highly variable gene lists. In total, the Ctrl sample contained n = 7286 cells, CD with n = 5532 cells, and UC with n = 7865 cells (Supplementary Table S1). Receiver operating characteristics (ROC) and model-based analysis of single cell transcriptomics (MAST) algorithms were used to identify cell-cluster and-type specific genes for each sample. Cell type annotations were based upon marker genes derived from earlier publications.
Secretome Analysis of MSCs Conditioned Media With and Without IFN-γ Treatment
After 3 days of incubation, the culture media was collected from MSC plates seeded for bulk RNA sequencing and stored at −80 °C until assayed. Subsequently, culture media supernatants cleared by centrifugation were analyzed by magnetic bead-based multiplex Luminex multi-analyte profiling (MAP) assay with the Cytokine 30-Plex Human Panel (Thermo Fisher Scientific) following the manufacturer’s instructions. Results were plotted as picograms per milliliter.
Statistical Analysis
Data were analyzed with GraphPad Prism 8.0 software. Two-way ANOVA with Šídák multiple comparisons was used to calculate the immunomodulatory expression of surface proteins and secretome analysis of MSCs.
Results
MSCs Derived from the Ileum and Rectum of IBD Patients Display Intact Phenotype and Normal Replication Kinetics
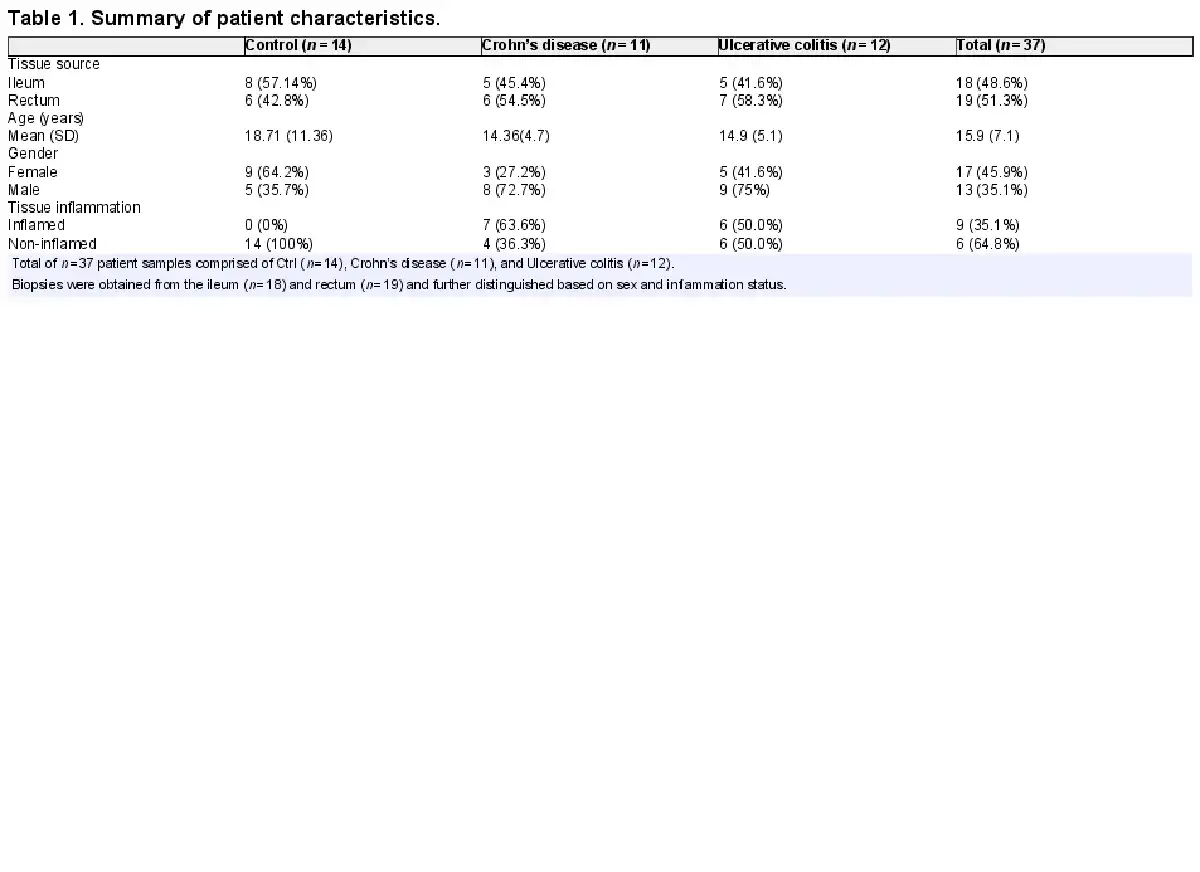
Mesenchymal stromal cells (MSCs) were isolated from the ileum and rectum of patients with CD, UC, and non-IBD control (Ctrl) from mucosal biopsies. Immunofluorescence microscopy analysis showed spindle-shaped morphology with a large nucleus, and a small cell body, along with distinct lamellipodia and filopodia and expressed MSC markers, CD90, and vimentin (Fig. 1A). Furthermore, immunophenotypic characterization with high-dimensional flow cytometry confirmed that expanded MSCs express CD105, CD73, and CD90 but not CD45, HLA-DR, or CD34 (Fig. 1B) in accordance with the minimal criteria pertaining to cell surface markers for human MSCs set-forth by the International Society for Cellular Therapy. Similar cell surface expression patterns of these molecules were noted regardless of tissue site (ileum versus rectum) and disease condition (Fig. 1B; Supplementary Fig. S1A). Additional MSC cell surface proteins noted to be expressed were: CD146, a multipotency/stemness marker, CD106, an immunoregulatory cell adhesion molecule, and MHC-I. Importantly, these cells from IBD and Ctrl demonstrated multipotency as they could be differentiated into osteoblasts, chondrocytes, and adipocytes albeit to different extents between the disease condition (Supplementary Fig. S1B), a finding consistent with previous studies. Altogether, these results are consistent with the identity of these cells being intestinal MSCs based on morphology, expression of established MSC phenotypic markers, and multipotency. Importantly, no major phenotypic differences were noted between MSCs found in the ileum and rectum nor between those from IBD and Ctrl.
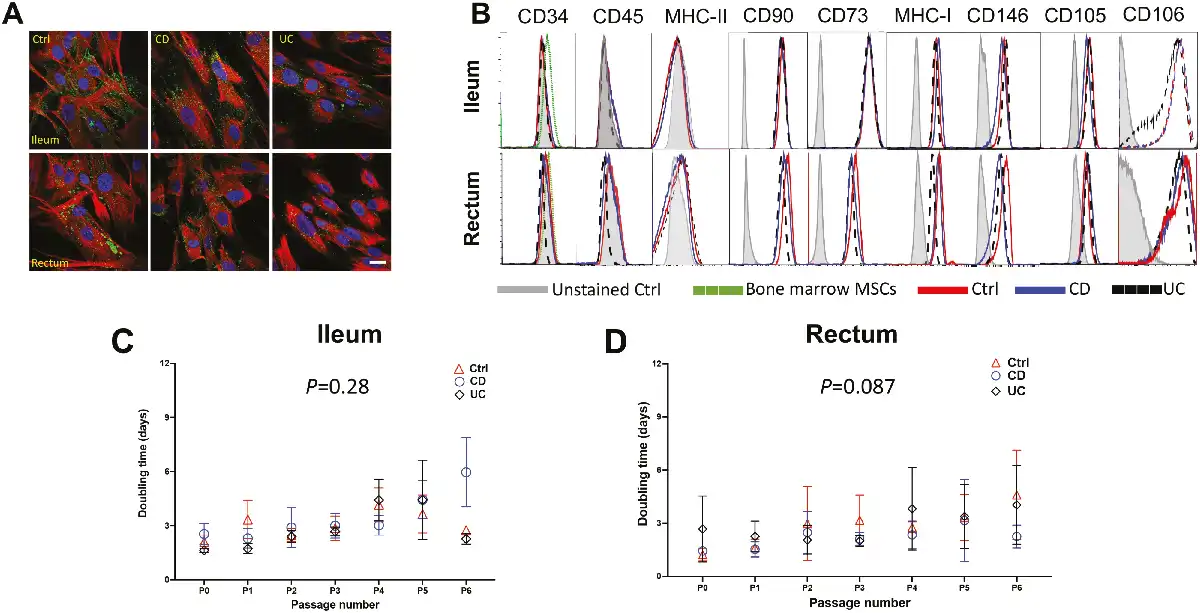
Figure 1
MSCs derived from ileum and rectum of CD, UC, and non-IBD control patients demonstrate similar surface immunophenotype and growth kinetics. (A): Representative immunofluorescent microscopic images of cultured cells after staining for MSC markers, CD90 (green color) and vimentin (red color), demonstrating characteristic spindle-shaped morphology with a large nucleus, a small cell body, and several small lamellipodia. Data representative of 2 independent experiments with at least 3 samples per group (scale bar = 10 µm). (B): Flow cytometric histograms for MSC cell surface markers pre-gated on live cells after doublet discrimination. Data representative of 3 independent experiments with 4-6 patients per group. Population doubling time of MSCs derived from (C) ileum and (D) rectum from control, CD and UC patients. Data representative of 5-6 patients per group.
Next, we analyzed the proliferative capacity of intestinal MSCs from IBD and Ctrl. In defining senescence to be the time point at which the cell number yielded from trypsinization was equal to or less than the cell number seeded in the absence of overt cell death, the mean number of passages to senescence between Ctrl and IBD MSCs were similar, ranging from 6-10 passages for ileum (P > .05; Supplementary Fig. S1C) and 6-11 passages for rectum (P > .05; Supplementary Fig. S1D). There was no significant difference in number of passages to senescence between the ileum and rectum (P = .318; Supplementary Fig. S1E). Furthermore, MSCs from Ctrl, CD, and UC exhibited similar cell number doubling time per passage from the ileum (Fig. 1C) and rectum (Fig. 1D). Thus, these data demonstrate the feasibility of autologous MSC expansion ex vivo from Ctrl and IBD patients, as these cells exhibit similar growth kinetics with no significant differences in time to senescence nor proliferative capacity.
Consensus Transcriptomic Profiles with Minimal Variations in Intestinal MSCs Irrespective of IBD Phenotypes
Despite intestinal MSCs from CD and UC exhibiting normal phenotypic and growth characteristics, there remains the possibility for substantial differences between them at the molecular level. To examine the extent to which overall transcriptional patterns were grossly influenced by MSC origin, we first decomposed the full transcriptome of bulk RNA-seq data derived from passaged MSCs into Principal Component (PC) to ask what extent PC analysis values associated with key aspects of MSC cell origin. Unsurprisingly, effects of age and sex are detectable, with age demonstrating weak negative correlations to PC2 (10.74%) and PC3 (5.22%) while sex had a weak correlation in PC6 (2.85%) but stronger with PC8 (2.61%; Fig. 2A). Whereas the presence of inflammation had a large detectable effect, strongly correlating with both PC1 (19.25%) and PC5 (3.47%; Fig. 2A), tissue site and disease condition both demonstrated weaker correlations accounting for less than 15% of the total variance. Thus, while differences in broad-scale patterns of transcription are detectable in the first several PCs, disease condition accounts for less than 10% of the total variance.
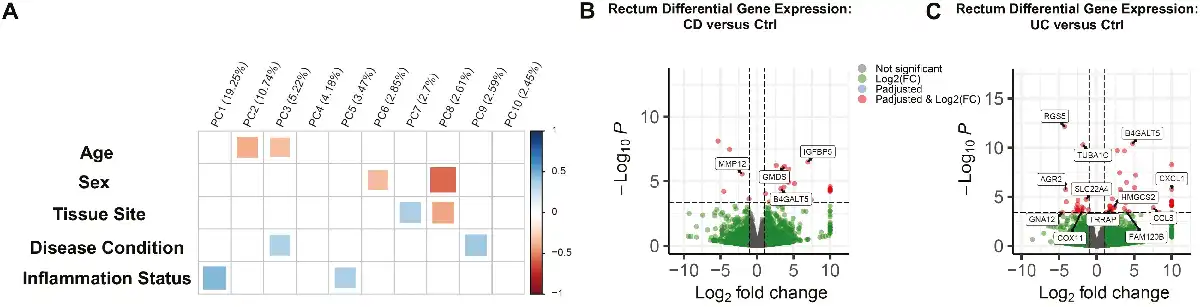
Figure 2
Consensus bulk RNA transcriptomic profiles with minimal variations in ileal and rectal MSCs irrespective of IBD phenotypes. (A) Principal component (PC) analyses of MSCs obtained from Ctrl (n = 6), CD (n = 6), and UC (n = 7) patients based on age, sex (male or female), tissue site (ileum or rectum), disease condition (control, CD, or UC), and inflammation status (inflamed or uninflamed). Volcano plot of differentially expressed genes based on log 2-fold change and adjusted P < .05 in (B) CD and (C) UC MSCs relative to Ctrl (n = 6) MSCs in the rectum with IBD-associated genes highlighted.
Next, to assess whether the disease state affects the expression of key genes associated with MSC biology, we compared both CD and UC to Ctrl and asked if there were any genes differentially expressed at false discovery rate (FDR) < .05. For CD, no significant genes were detected in ileal MSCs, while in the rectum 13 genes were detected—only 4 of which were previously published to be associated with IBD: B4GALT5,GMDS,IGFBP5, and MMP12 (Fig. 2B). Importantly, none of these differentially expressed genes were associated with known MSC biology. Similarly, MSCs from UC demonstrated a significant decrease in FSIP1 in the ileum while 51 genes were differentially expressed in the rectum (Fig. 2C). Of these 51 genes, 11 genes have been previously associated with IBD, and one of them, RGS5, a pericyte marker, was decreased in UC MSCs (Fig. 2C). Pathway analyses of the differentially expressed genes in both CD and UC failed to identify any specific biological pathways. These data indicate that the transcriptomes of MSCs from IBD and Ctrl patients are broadly similar but do share differences in a subset of IBD-related genes, particularly in the rectum, which are unlikely to have any gross effect on MSC development.
Single-cell RNA Sequencing Identifies Cell Subtype Heterogeneity of the MSC Compartment
While broad-scale differences in MSCs were not pronounced by disease state given the heterogeneity of the intestinal stromal compartment, cell-specific differences may be observable. To this end, scRNA-seq was used to characterize the cellular composition of rectal MSCs in IBD and Ctrl. After controlling for mitochondrial gene content and cell-cycle genes (Supplementary Fig. S2A-S2F), highly variable genes determined by PC analysis were used in parallel with previously published MSC markers to annotate clusters in UMAP projections. We observed n = 629 markers for Ctrl (Supplementary Fig. S3A), n = 516 markers for CD (Supplementary Fig. S3B), and n = 440 markers for UC (Supplementary Fig. S3C) data sets. Cells highly expressing genes COL3A1, POSTN, PDGFRA, and COL5A1 were annotated as fibroblasts,, whereas a combination of TAGLN, ACTG2, PDGFRB, MYH11, and DES expression defined myofibroblasts,,. Furthermore, cycling stromal cells were characterized by high expression of TYMS, TUBA1B, HIST1H4C, and MKI67, villus tip telocytes were annotated by IFI27, TM4SF1, and CRIP1, while crypt telocytes were annotated by SFRP1, and CD248 expression (Supplementary Fig. S3). Of the 6 distinct cell types annotated using scRNA-seq, the Ctrl data set comprised all 6 while both CD and UC were void of crypt and villus tip telocytes (Fig. 3A). Myofibroblasts were the most abundant cell sub-type in all the samples (Fig. 3B), and this finding was confirmed with immunofluorescence confocal microscopy showing α-SMA (myofibroblast marker) positive cells at approximately 60% abundance per high power field (Fig. 3C and 3D). Taken together, scRNA-seq revealed that MSCs from the rectum maintained the transcriptomic signature of multiple stromal subtypes, predominately fibroblast and myofibroblasts, with an absence of crypt and villus telocytes in CD and UC.
Figure 3
Single-cell RNA sequencing identifies cell subtype heterogeneity of the rectal MSC compartment. (A) Uniform manifold approximation and projection (UMAP) plot showing the visualization of cells colored by cluster identity. (B) Bar diagram represents the cell type proportion of all data sets. (C): immunostaining for alpha-SMA (green) and vimentin (red), mesenchymal markers in the cultured MSCs (scale bar = 10 µm). (D): Bar diagram represents the quantification of alpha-SMA positive cells in the culture.
IFN-γ Activates Broad and Shared Transcriptomic Responses in Intestinal MSCs Derived from IBD and Ctrl
Priming of MSCs to augment the immunomodulating properties and therapeutic output has a long history with multiple approaches. IFN-γ priming of MSCs potently induces their immunosuppressive properties, and as such, we used this treatment to look for similar and undiscovered transcriptional changes across ileal and rectal MSCs cultured from IBD and Ctrl biopsies. We found that any transcriptional profiles correlating with age and tissue site were no longer present after 48-hours IFN-γ priming, and the variance associated with disease state was reduced from PC3 (5.22%) and PC9 (2.59%) to PC5 (3.47%) and PC7 (2.7%; Figs. 2A and 4A). In addition, inflammation status became associated with PC6 (2.85%) after IFN-γ treatment from PC1 (19.25%) and PC5 (3.47%; Figs. 2A and 4A). Consequently, these data suggest that IFN-γ priming of intestinal MSCs augments their therapeutic fitness by decreasing the transcriptomic variance associated with age, disease condition, tissue specificity, and inflammation status.
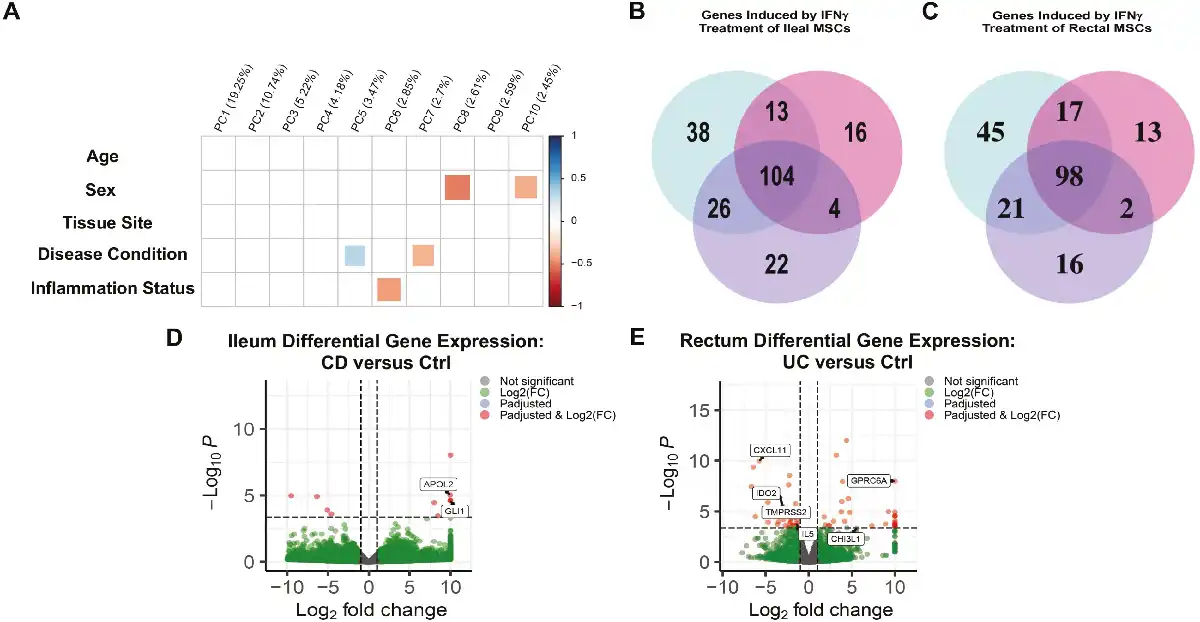
Figure 4
IFN-γ priming of MSCs neutralizes baseline transcriptomic differences and induces shared gene expression pathways. (A): Principal component analyses of MSCs after IFN-γ priming for Ctrl (n = 6), CD (n = 5) and UC (n = 7) patients based on age, sex (male or female), tissue site (ileum or rectum), disease condition (control, CD, or UC), and inflammation status (inflamed or uninflamed). Number of common and unique IFN-γ induced genes expressed for Ctrl, CD, and UC MSCs in the (B) ileum and (C) rectum. Volcano plot of differentially expressed genes of in (D) ileal CD and (E) rectal UC MSCs relative to Ctrl MSCs with IBD-associated genes highlighted.
At the level of individual gene expression, we first asked if treatment by IFN-γ resulted in differential gene expression (DGE) within Ctrl, CD, and UC, and found 181, 137, and 156 ileal genes differentially expressed by IFN-γ treatment for Ctrl, CD, and UC, respectively (Fig. 4B, Supplementary Table S2). Importantly, the majority of 104 IFN-γ induced genes were shared between all 3 groups, demonstrating that IFN-γ effects are largely the same regardless of the disease condition. PD1 signaling, IFN-γ signaling, and IBD were the most common biological pathways in the ileum associated with shared IFN-γ induced genes across the phenotypes (Supplementary Table S3). In the rectum, IFN-γ priming induced 181 genes in Ctrl, 130 genes in CD, and 137 genes in UC relative to their untreated counterparts (Fig. 4C, Supplementary Table S4). Like the ileum, the majority of the differentially expressed genes (98 genes) in the rectum were shared, and pathway analyses revealed these shared genes were also implicated in PD-1 signaling, IFN-γ signaling, and IBD pathway (Supplementary Table S5). Taken together, IFN-γ priming induced gene expression of immunomodulatory genes in MSCs regardless of disease state while homogenizing their transcriptomic signatures.
Lastly, we used DGE analyses to identify individual genes that may be expressed at significantly different levels between Ctrl and IBD intestinal MSCs after IFN-γ priming. In the ileum, CD demonstrated differential expression of only 3 genes relative to Ctrl. Two of the 3 genes, APOL2 and GLI1, are known to harbor genetic variants associated with the risk of developing CD, (Fig. 4D; Supplementary Table S6). In the rectum, CD had decreased expression for ABCA6 and GPR20—neither of which have been previously associated with IBD pathogenesis (Supplementary Table S6). Importantly, none of the differentially expressed genes in the ileum nor rectum have been reported to play a role in MSC biology. For UC, ileal MSCs expressed higher levels of 2 genes relative to Ctrl with no known IBD nor MSC biology associations (SYCE3 and MOB3A; Supplementary Table S7), while UC from the rectum differentially-expressed 26 genes of which only 6 genes (CXCL11, IDO2, TMPRSS2, IL5, CHI3L1, and GPRC6A) have been implicated in IBD (Fig. 4C; Supplementary Table S7). Thus, majority of the differentially-expressed genes between IFN-γ primed MSCs from CD and UC relative to Ctrl are not associated with IBD pathogenesis nor MSC biology and may simply represent personalized heterogeneity.
IFN-γ-Mediated Upregulation of Immunomodulatory Cell Surface Proteins and Intact Secretome of Intestinal MSCs
Previous studies have indicated that MSCs may regulate T-cell activation by expression of co-stimulatory molecules CD80, PD-L1, and PD-L2, yet little is known about changes in cell-surface proteins of IBD-derived MSCs after IFN-γ priming and whether their response differs from Ctrl. Thus, we investigated the expression levels of immunomodulatory proteins on intestinal MSCs by flow cytometry after IFN-γ priming. In both the ileum and rectum, MSCs from all 3 groups did not demonstrate any induction of CD80 in response to IFN-γ (Fig. 5A-5D). However, like Ctrl, an increase in the number of cells expressing PD-L1 and PD-L2 was observed in both ileal and rectal MSCs from CD and UC after IFN-γ priming (Fig. 5A and 5B). The proportion of MSCs expressing MHC-II was low in both the ileum and rectum for all 3 groups as expected given their low immunogenicity but was significantly increased with IFN-γ treatment (Fig. 5A-5D). Furthermore, the expression of cell-surface immunomodulatory proteins on intestinal MSCs isolated from IBD and Ctrl yielded similar IFN-γ-mediated induction of PD-L1, PD-L2, and MHC-II but not CD80 (Fig. 5C and 5D).
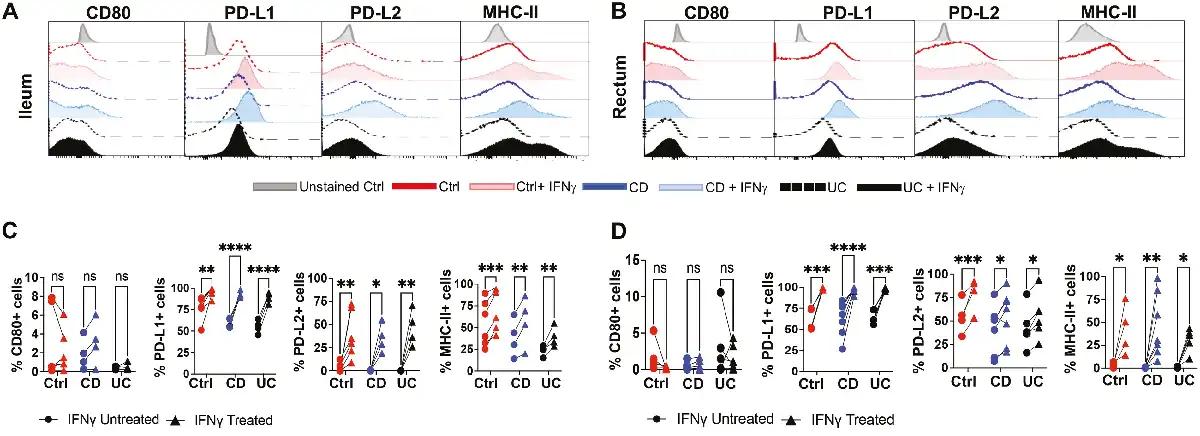
Figure 5
IFN-γ induced cell surface expression of immunomodulatory proteins on MSCs. Representative flow cytometric histograms for expression of CD80, PD-L1, PD-L2, and MHC-II on MSCs in the (A) ileum and (B) rectum after 48 hours of IFN-γ stimulation. Quantitation of changes in cells expressing CD80, PD-L1, PD-L2, and MHC-II in (C) ileum and (D) rectum before and after IFN-γ stimulation. Data representative of 2 independent experiments with 4-7 patients per group. Two-way, paired ANOVA with Sidak’s multiple comparison’s test. *P < .05, **P < .01, ***P < .001, ****P < .0001, ns for not significant.
Nonetheless, secretome production from intestinal MSCs in response to IFN-γ priming was not as robust and consistent amongst the groups (Fig. 6) as was observed for surface marker expression. A large portion of the analytes on the 30-plex Luminex panel was not detected in the conditioned media (Supplementary Tables S8 and S9). In the ileum, multiplex secretome analyses revealed increased production of the chemokines MCP-1 and CXCL9 in all 3 groups with IFN-γ treatment though only an increased trend for CXCL9 was observed for UC (Fig. 6A). Ileal Ctrl MSCs demonstrated distinct increases in IL-6 and CCL11 but a decrease in IL-8 production in response to IFN-γ (Fig. 6A). In contrast, rectal MSCs from the 3 groups failed to show any significant change in IL-6, CCL11, or IL-8, but higher CXCL10 and CXCL9 were observed with IFN-γ priming (Fig. 6B). Lastly, Ctrl MSCs increased the production of MCP-1 with IFN-γ treatment in the rectum while CD and UC MSCs did not, which differed from the ileum (Fig. 6B). Interestingly, remarkable patient-specific differences in magnitude and direction of IFN-γ response for several molecules including IL8, IL6, CXCL10, and MCP1 were observed. Thus, intestinal MSCs show a heterogenous response in secretome output after IFN-γ priming that may be an effect of intra-individual variability.
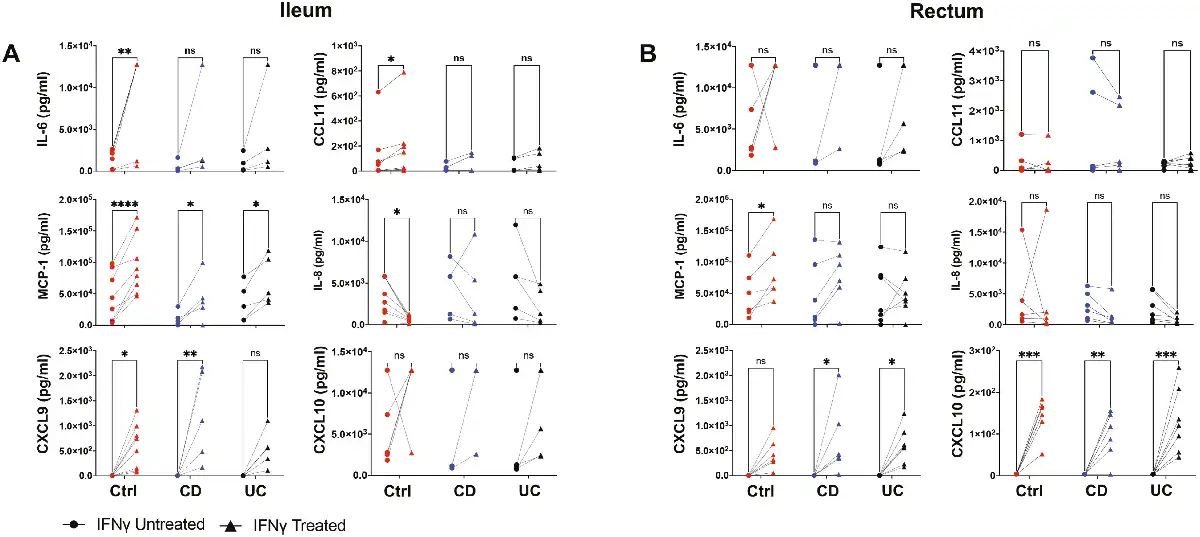
Figure 6
Secretome analysis of MSCs conditioned media. Thirty-plex secretome analysis was performed on (A) ileal and (B) rectal MSC conditioned media collected with and without IFN-γ treatment for 48 hours. Plots show only analytes with signal significantly greater than background media from control, CD, and UC samples. Data representative of 2 independent experiments with 4-7 samples per group. Two-way paired ANOVA and Sidak’s multiple comparison tests. *P < .05, **P < .01, ***P < .001, ****P < .0001, ns for not significant.
Discussion
Despite the development of BM-derived, autologous and adipose-derived, allogenic MSC therapies as options for IBD treatment, difficulties with side-effects and low success rates have demonstrated the need for more targeted, effective, and less immunogenic therapies. Herein, we are driven by the hypothesis supported by clinical trial data that the use of patient-derived, autologous intestinal MSCs could have a tremendous benefit in treating IBD and other related inflammatory diseases,. Our characterization of intestinal MSCs derived from IBD and Ctrl patient biopsies demonstrates the feasibility, potential functionality, and new biological features associated with intestinal MSCs. Using a combination of high-dimensional flow cytometry, microscopy, RNA-seq, and Luminex MAP, we have shown that intestinal MSCs derived from IBD patients appear overall normal, showing similar growth patterns, surface markers, transcriptomes, secretomes, and response to IFN-γ priming, yet some unique intra-individual and IBD-specific characteristics were detectable.
An overarching concern in autologous treatment is a reintroduction of cells harboring contributors to disease. MSCs isolated from the colon of patients with IBD showed a decrease in their differentiation potential and clonogenic properties. In our study, osteocyte differentiation was specifically decreased in IBD MSCs while adipocyte and chondrocyte differentiation remained intact. None of the few differentially expressed genes between Ctrl and IBD MSCs were related to osteocyte differentiation. The underlying mechanism for the observed defect in IBD MSC osteocyte differentiation is unknown and additional studies are needed especially because there is a higher prevalence of osteoporosis in IBD relative to the general population. Whereas we observed similar differentiation properties of IBD MSCs, it is not clear whether these or other minor differences in gene expression, being individual or IBD specific, would have a negative impact on the efficacy of autologous MSC treatment for IBD. Taken together, our results here have shown that MSCs derived from ileal and rectal tissue, regardless of IBD phenotype, display plastic adherence under standard xenofree cell culture conditions, multipotency, and also express canonical MSC markers that collectively satisfy the minimal criteria recommended by the International Society of Cell Therapy guidelines. Analysis by scRNA-seq showed that the predominant MSC subtypes, were present in the IBD but lacked the minor cell types of crypt and villus tip telocytes that were observed in Ctrl. Indeed, with such a small sample size in our single-cell experiments, future studies are warranted to understand the functional context of variations in the subpopulations and their effects on the potency of autologous intestinal MSCs as cellular therapeutics. Yet overall, there were no critical differences in MSC phenotype between IBD and Ctrl irrespective of tissue origin consistent with findings for BM-MSC from IBD patients.
Nonetheless, the lack of cellular reference standards for MSC comparison has made describing the potency and functionality of patient-derived MSCs as autologous cellular pharmaceuticals challenging,. A major limitation of this study is the lack of direct in vitro and in vivo immunosuppressive experiments. Considering the complexity of MSCs mechanism of action, defining a single effector pathway that is responsible for their cumulative pharmacological effect may be inaccurate, and utilization of universal cellular standards that depict the entire features of MSCs is difficult. The therapeutic benefit of MSCs has been linked to at least 3 major mechanisms of action based on their route of delivery, including differentiation into mesoderm tissues, efferocytosis-based polarization of macrophages, and direct contact and/or paracrine-mediated instruction of immune cells, but defining good molecular endpoints proves elusive. Thus, many studies have relied on the known immunosuppressive properties of MSCs after exposure to IFN-γ that primes the cells for therapeutic use,. In this strategy, immunomodulatory and regenerative effector molecules expressed and released from MSCs are assessed with and without IFN-γ and these comparative assessments can serve as the references and probes. In our study, the transcriptomes of intestinal MSCs were evaluated with and without IFN-γ activation, showing no major differences between Ctrl and IBD patients, along with consistent upregulation of close to 100 consensus genes irrespective of the disease status, demonstrating the fitness of culture-expanded intestinal MSCs in responding to IFN-γ. Not only did this serve as surrogate measures of intestinal MSCs functionality in our study, the immunomodulatory cell surface analysis we conducted upon IFN-γ priming demonstrated that these stimulated intestinal MSCs upregulate MHC-II, PD-L1, and PD-L2 but not co-stimulatory molecule CD80. The absence of co-stimulatory molecules and the presence of inhibitory ligands PD-L1 and PD-L2 suggest that cultured MSCs from the intestine may function as tolerogenic cells in promoting T-cell inhibition.
Although adipose-derived MSCs are topically administered to CD perianal fistulae for effective closure and healing, intravenous administration of MSCs is widely being used in clinical trials for IBD management. Intravenously administered MSCs are predominantly trapped in the lungs for reasons that are not clear but in some cases promote intestinal healing, suggesting that their paracrine effect from lungs post-infusion plays a role. To this end, our secretome analysis has demonstrated that biopsy-derived intestinal MSCs innately secrete molecules such as IL-8 and MCP-1, with studies showing these secretory molecules playing roles in leukocyte chemotaxis and macrophage polarization to M2 suppressor phenotype that modulates the immune response and regenerates damaged mucosa. In our study, cytokine priming of MSCs with IFN-γ also upregulated the innate secretion of MCP-1 and induced de novo secretion of CXCL9 and CXCL10, further demonstrating the immunoplasticity of intestinal MSCs in their secretome. While the potential exists for the emergence of malignant cellular transformation and unstable genomic aberrations in transplanted intestinal MSCs, prior studies have reported that culture-expanded MSCs from other tissue sources undergo cellular senescence and aneuploidy but never malignant transformation. In fact, under standard in vitro cell culture conditions, primary somatic cells undergo replication and subsequently enter cellular senescence, a phenomenon known as Hayflick limit. Our study has shown that intestinal MSCs comply with the Hayflick limit by undergoing replication senescence but not cellular transformation, thus elucidating an important safety insight when considering intestinal MSCs as a cellular pharmaceutical.
Conclusion
Collectively, our study has shown that the proliferative and immune effector pathways of biopsy-derived intestinal MSCs are functional and seemingly uncompromised, thus setting the platform for further development as an autologous cellular therapy for IBD. Future studies will focus on patient-specific differences and similarities in the surface markers and secretomes, their functionality and immunosuppressive properties, mode of mucosal delivery, and optimized cytokine/growth factor priming protocols that generate the most specific and effective healing properties.
Acknowledgments
This research was also supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of the National Institutes of Health, under grant numbers R01DK087694-10 and R01DK125936-01 to S.K and T32 DK 108735-5 to D.G. Graphical abstract was created with Biorender.
References
- 1. Torres J, Mehandru S, Colombel J-F, Peyrin-Biroulet L. Crohn’s disease. Lancet. 2017;389(10080):1741–1755.
- 2. Verstockt B, Noor NM, Marigorta UM, et al. Results of the Seventh Scientific Workshop of ECCO: precision medicine in IBD-disease outcome and response to therapy. J Crohn’s Colitis. 2021;15(9):1431–1442:jjab050.
- 3. Ungaro R, Mehandru S, Allen PB, Peyrin-Biroulet L, Colombel J-F. Ulcerative colitis. Lancet. 2017/04/29/ 2017;389(10080):1756–1770. https://doi.org/10.1016/s0140-6736(16)32126-2.
- Ashton JJ, Mossotto E, Ennis S, Beattie RM. Personalising medicine in inflammatory bowel disease—current and future perspectives. Trans Pediatr. 2019;8(1):56–69. https://doi.org/10.21037/tp.2018.12.03.
- 5. Ashton JJ, Ennis S, Beattie RM. Early-onset paediatric inflammatory bowel disease. Lancet Child Adol Health. 2017;1(2):147–158.
- 6. Wu X, Jiang J, Gu Z, et al. Mesenchymal stromal cell therapies: immunomodulatory properties and clinical progress. Stem Cell Res Ther. Aug 8 2020;11(1):345. https://doi.org/10.1186/s13287-020-01855-9.
- 7. Li C, Zhao H, Cheng L, Wang B. Allogeneic vs. autologous mesenchymal stem/stromal cells in their medication practice. Cell Biosci. Nov 2 2021;11(1):187. https://doi.org/10.1186/s13578-021-00698-y.
- 8. Martin I, Baldomero H, Bocelli-Tyndall C, et al. The survey on cellular and engineered tissue therapies in Europe in 2010. Tissue Eng Part A. Nov 2012;18(21-22):2268–2279. https://doi.org/10.1089/ten.TEA.2012.0169.
- 9. Johnson S, Hoch JS, Halabi WJ, et al. Mesenchymal stem/stromal cell therapy is more cost-effective than fecal diversion for treatment of perianal Crohn’s disease fistulas. Original research. Front Immunol. 2022-June-17 2022;13:859954. https://doi.org/10.3389/fimmu.2022.859954.
- 10. Panes J, Garcia-Olmo D, Van Assche G, et al. Expanded allogeneic adipose-derived mesenchymal stem cells (Cx601) for complex perianal fistulas in Crohn’s disease: a phase 3 randomised, double-blind controlled trial. Lancet. Sep 24 2016;388(10051):1281–1290. https://doi.org/10.1016/S0140-6736(16)31203-X.
- 11. Griffiths S, Baraniak PR, Copland IB, Nerem RM, McDevitt TC. Human platelet lysate stimulates high-passage and senescent human multipotent mesenchymal stromal cell growth and rejuvenation in vitro. Cytotherapy. 2013;15(12):1469–1483. https://doi.org/10.1016/j.jcyt.2013.05.020.
- 12. Chinnadurai R, Copland IB, Ng S, et al. Mesenchymal stromal cells derived from Crohn’s patients deploy indoleamine 2,3-dioxygenase-mediated immune suppression, independent of autophagy. Mol Ther. Jul 2015;23(7):1248–1261. https://doi.org/10.1038/mt.2015.67.
- 13. Krampera M, Galipeau J, Shi Y, Tarte K, Sensebe L; Therapy MSCCotISfC.Immunological characterization of multipotent mesenchymal stromal cells: the International Society for Cellular Therapy (ISCT) working proposal. Cytotherapy. Sep 2013;15(9):1054–1061. https://doi.org/10.1016/j.jcyt.2013.02.010.
- 14. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21.
- 15. Venables W, Ripley B. Modern Applied Statistics with S. 4th ed. Springer-Verlag; 2002.
- 16. Friendly M. Corrgrams: exploratory displays for correlation matrices. Am Stat. 2002;56(4):316–324. https://doi.org/10.1198/000313002533.
- 17. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):1–21.
- 18. Liang M, Chen C, Dai Y, Chang Y, Gao Y. Two closely spaced missense COL3A1 variants in cis cause vascular Ehlers‐Danlos syndrome in one large Chinese family. J Cell Mol Med. 2022;26(1):144–150.
- 19. Wnuk D, Lasota S, Paw M, Madeja Z, Michalik M. Asthma-derived fibroblast to myofibroblast transition is enhanced in comparison to fibroblasts derived from non-asthmatic patients in 3D in vitro culture due to Smad2/3 signalling. Acta Biochim Pol. 2020;67(4):441–448.
- 20. Hannan RT, Miller AE, Hung R-C, et al. Extracellular matrix remodeling associated with bleomycin-induced lung injury supports pericyte-to-myofibroblast transition. Matrix Biol Plus. 2021;10:100056. https://doi.org/10.1016/j.mbplus.2020.100056.
- 21. Zheng M, Sun X, Zhang M, et al. Variations of chromosomes 2 and 3 gene expression profiles among pulmonary telocytes, pneumocytes, airway cells, mesenchymal stem cells and lymphocytes. J Cell Mol Med. 2014;18(10):2044–2060. https://doi.org/10.1111/jcmm.12429.
- 22. Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–317. https://doi.org/10.1080/14653240600855905.
- 23. Russell KC, Phinney DG, Lacey MR, et al. In vitro high‐capacity assay to quantify the clonal heterogeneity in trilineage potential of mesenchymal stem cells reveals a complex hierarchy of lineage commitment. Stem Cells. 2010;28(4):788–798. https://doi.org/10.1002/stem.312.
- 24. Pepinsky B, Hession C, Chen L-L, et al. Structure/function studies on vascular cell adhesion molecule-1. J Biol Chem. 1992;267(25):17820–17826. https://doi.org/10.1016/s0021-9258(19)37117-0.
- 25. Grim C, Noble R, Uribe G, et al. Impairment of tissue-resident mesenchymal stem cells in chronic ulcerative colitis and Crohn’s disease. J Crohns Colitis. Aug 2 2021;15(8):1362–1375. https://doi.org/10.1093/ecco-jcc/jjab001.
- 26. Filimoniuk A, Blachnio-Zabielska A, Imierska M, Lebensztejn DM, Daniluk U. Sphingolipid analysis indicate lactosylceramide as a potential biomarker of inflammatory bowel disease in children. Biomolecules. 2020;10(7):1083. https://doi.org/10.3390/biom10071083.
- 27. Frenkel S, Bernstein CN, Sargent M, et al. Genome-wide analysis identifies rare copy number variations associated with inflammatory bowel disease. PLoS One. 2019;14(6):e0217846. https://doi.org/10.1371/journal.pone.0217846.
- 28. Zimmermann E, Li L, Hou Y, Mohapatra N, Pucilowska J. Insulin-like growth factor I and insulin-like growth factor binding protein 5 in Crohn’s disease. Am J Physiol Gastrointest Liver Physiol. 2001;280(5):G1022–G1029.
- 29. Nighot M, Ganapathy AS, Saha K, et al. Matrix Metalloproteinase MMP-12 promotes macrophage transmigration across intestinal epithelial tight junctions and increases severity of experimental colitis. J Crohn’s Colitis. 2021;15(10):1751–1765. https://doi.org/10.1093/ecco-jcc/jjab064.
- 30. Kinchen J, Chen HH, Parikh K, et al. Structural remodeling of the human colonic mesenchyme in inflammatory bowel disease. Cell. Oct 4 2018;175(2):372–386.e17. https://doi.org/10.1016/j.cell.2018.08.067.
- 31. Li R, Bernau K, Sandbo N, et al. Pdgfra marks a cellular lineage with distinct contributions to myofibroblasts in lung maturation and injury response. Elife. 2018;7:e36865.
- 32. Untergasser G, Gander R, Lilg C, et al. Profiling molecular targets of TGF-β1 in prostate fibroblast-to-myofibroblast transdifferentiation. Mech Ageing Dev. 2005;126(1):59–69. https://doi.org/10.1016/j.mad.2004.09.023.
- 33. Noronha NC, Mizukami A, Caliari-Oliveira C, et al. Priming approaches to improve the efficacy of mesenchymal stromal cell-based therapies. Stem Cell Res Ther. May 2 2019;10(1):131. https://doi.org/10.1186/s13287-019-1224-y.
- 34. Sheng H, Wang Y, Jin Y, et al. A critical role of IFNgamma in priming MSC-mediated suppression of T cell proliferation through up-regulation of B7-H1. Cell Res. Aug 2008;18(8):846–857. https://doi.org/10.1038/cr.2008.80.
- 35. Salvador-Martín S, Kaczmarczyk B, Álvarez R, et al. Whole transcription profile of responders to anti-TNF drugs in pediatric inflammatory bowel disease. Pharmaceutics. 2021;13(1):77.
- 36. Xie Z, Zhang M, Zhou G, et al. Emerging roles of the Hedgehog signalling pathway in inflammatory bowel disease. Cell Death Discov. 2021;7(1):1–9.
- 37. Dunn CM, Kameishi S, Grainger DW, Okano T. Strategies to address mesenchymal stem/stromal cell heterogeneity in immunomodulatory profiles to improve cell-based therapies. Acta Biomater. Oct 1 2021;133:114–125. https://doi.org/10.1016/j.actbio.2021.03.069.
- 38. Mittal SK, Cho W, Elbasiony E, et al. Mesenchymal stem cells augment regulatory T cell function via CD80-mediated interactions and promote allograft survival. Am J Transplant. Jun 2022;22(6):1564–1577. https://doi.org/10.1111/ajt.17001.
- 39. Davies LC, Heldring N, Kadri N, Le Blanc K. Mesenchymal stromal cell secretion of programmed death-1 ligands regulates T cell mediated immunosuppression. Stem Cells. Mar 2017;35(3):766–776. https://doi.org/10.1002/stem.2509.
- 40. Romieu-Mourez R, François M, Boivin M-N, Stagg J, Galipeau J. Regulation of MHC class II expression and antigen processing in murine and human mesenchymal stromal cells by IFN-γ, TGF-β, and cell density. J Immunol. 2007;179(3):1549–1558.
- 41. Cuende N, Rasko JEJ, Koh MBC, Dominici M, Ikonomou L. Cell, tissue and gene products with marketing authorization in 2018 worldwide. Cytotherapy. Nov 2018;20(11):1401–1413. https://doi.org/10.1016/j.jcyt.2018.09.010.
- 42. Panés J, García-Olmo D, Van Assche G, et al. Long-term efficacy and safety of stem cell therapy (Cx601) for complex perianal fistulas in patients with Crohn’s disease. Gastroenterology. 2018;154(5):1334–1342. e4.
- 43. Ankrum JA, Ong JF, Karp JM. Mesenchymal stem cells: immune evasive, not immune privileged. Nat Biotechnol. Mar 2014;32(3):252–260. https://doi.org/10.1038/nbt.2816.
- 44. Dhere T, Copland I, Garcia M, et al. The safety of autologous and metabolically fit bone marrow mesenchymal stromal cells in medically refractory Crohn’s disease - a phase 1 trial with three doses. Alimentary Pharmacol Therapeutics. Sep 2016;44(5):471–481. https://doi.org/10.1111/apt.13717.
- 45. Robb KP, Fitzgerald JC, Barry F, Viswanathan S. Mesenchymal stromal cell therapy: progress in manufacturing and assessments of potency. Cytotherapy. Mar 2019;21(3):289–306. https://doi.org/10.1016/j.jcyt.2018.10.014.
- 46. Moll G, Ankrum JA, Kamhieh-Milz J, et al. Intravascular mesenchymal stromal/stem cell therapy product diversification: time for new clinical guidelines. Trends Mol Med. Feb 2019;25(2):149–163. https://doi.org/10.1016/j.molmed.2018.12.006.
- 47. Lima CA, Lyra AC, Rocha R, Santana GO. Risk factors for osteoporosis in inflammatory bowel disease patients. World J Gastrointestinal Pathophysiol. 2015;6(4):210.
- 48. Huang Y, Li Q, Zhang K, et al. Single cell transcriptomic analysis of human mesenchymal stem cells reveals limited heterogeneity. Cell Death Dis. 2019;10(5):1–12.
- 49. Medrano-Trochez C, Chatterjee P, Pradhan P, et al. Single-cell RNA-seq of out-of-thaw mesenchymal stromal cells shows tissue-of-origin differences and inter-donor cell-cycle variations. Stem Cell Res Ther. 2021;12(1):1–14.
- 50. Hematti P. Characterization of mesenchymal stromal cells: potency assay development. Transfusion. Apr 2016;56(4):32S–35S. https://doi.org/10.1111/trf.13569.
- 51. de Wolf C, van de Bovenkamp M, Hoefnagel M. Regulatory perspective on in vitro potency assays for human mesenchymal stromal cells used in immunotherapy. Cytotherapy. Jul 2017;19(7):784–797. https://doi.org/10.1016/j.jcyt.2017.03.076.
- 52. Galipeau J, Krampera M, Leblanc K, et al. Mesenchymal stromal cell variables influencing clinical potency: the impact of viability, fitness, route of administration and host predisposition. Cytotherapy. May 2021;23(5):368–372. https://doi.org/10.1016/j.jcyt.2020.11.007.
- 53. Krampera M, Le Blanc K. Mesenchymal stromal cells: putative microenvironmental modulators become cell therapy. Cell Stem Cell. Oct 7 2021;28(10):1708–1725. https://doi.org/10.1016/j.stem.2021.09.006.
- 54. Galipeau J, Krampera M, Barrett J, et al. International Society for Cellular Therapy perspective on immune functional assays for mesenchymal stromal cells as potency release criterion for advanced phase clinical trials. Cytotherapy. Feb 2016;18(2):151–159. https://doi.org/10.1016/j.jcyt.2015.11.008.
- 55. Chinnadurai R, Garcia MA, Sakurai Y, et al. Actin cytoskeletal disruption following cryopreservation alters the biodistribution of human mesenchymal stromal cells in vivo. Stem Cell Rep. Jul 8 2014;3(1):60–72. https://doi.org/10.1016/j.stemcr.2014.05.003.
- 56. Giri J, Das R, Nylen E, Chinnadurai R, Galipeau J. CCL2 and CXCL12 derived from mesenchymal stromal cells cooperatively polarize IL-10+ tissue macrophages to mitigate gut injury. Cell Rep. 2020;30(6):1923–1934.e4. https://doi.org/10.1016/j.celrep.2020.01.047.
- 57. Tarte K, Gaillard J, Lataillade JJ, et al. Clinical-grade production of human mesenchymal stromal cells: occurrence of aneuploidy without transformation. Blood. Feb 25 2010;115(8):1549–1553. https://doi.org/10.1182/blood-2009-05-219907.
- 58. Chinnadurai R, Rajan D, Ng S, et al. Immune dysfunctionality of replicative senescent mesenchymal stromal cells is corrected by IFNgamma priming. Blood Adv. Apr 25 2017;1(11):628–643. https://doi.org/10.1182/bloodadvances.2017006205.
- 59. Sensebe L, Tarte K, Galipeau J, et al Limited acquisition of chromosomal aberrations in human adult mesenchymal stromal cells. Cell Stem Cell. Jan 6 2012;10(1):9–10; author reply 10-1. https://doi.org/10.1016/j.stem.2011.12.005.
- 60. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. Dec 1961;25:585–621. https://doi.org/10.1016/0014-4827(61)90192-6.